


Abstract
Although the thermophilic bacterium Thermus aquaticus grows optimally at 70°C and cannot grow at moderate temperatures, its DNA polymerase I has significant activity at 20–37°C. This activity is a bane to some PCRs, since it catalyzes non-specific priming. We report mutations of Klentaq (an N-terminal deletion variant) DNA polymerase that have markedly reduced activity at 37°C yet retain apparently normal activity at 68°C and resistance at 95°C. The first four of these mutations are clustered on the outside surface of the enzyme, nowhere near the active site, but at the hinge point of a domain that has been proposed to move at each cycle of nucleotide incorporation. We show that the novel cold-sensitive mutants can provide a hot start for PCR and exhibit slightly improved fidelity.
INTRODUCTION
Two types of undesired DNA synthesis can occur during the PCR set-up: mispriming on less than specific sites in the template and/or the formation of a so-called primer dimer, where the primers are extended by serving each other as template. Each of these events, occurring in seconds at temperatures below the cycling conditions, can compromise the success, yield and/or the specificity of the PCR, particularly for amplifications involving high cycle numbers, high GC content of the amplicon or multiple primer pairs (multiplex PCR).
The most useful way to prevent this unwanted DNA synthesis and improve the PCR is a ‘hot start’, wherein any of several methods are used to reduce or prevent DNA synthesis before the reaction has been warmed to the normal DNA extension temperature (62–72°C) or higher. At high temperature the primers have their designed sequence specificity and cannot stably anneal to each other or to unwanted template sequences. In fact, many PCR analyses, especially the most demanding ones, benefit from a hot start. Such demanding PCRs are, for example, those with very low copy numbers of target and requiring many (between 35 and 45) cycles (such as 1 HIV genome per 10 000 cells) or ones where the DNA template is denatured in the DNA extraction procedure, so that the template is single-stranded during reaction set-up. In our experience, about 50% of all high cycle number PCRs show improved yield if a hot start is employed, and in some cases a hot start is absolutely critical. A seemingly obvious option is to add one or more components such as primer, enzyme, magnesium and/or dNTPs to the reactions only after they have heated up (manual hot set-up). Besides being tedious and prone to error, this manual method is very subject to contamination and cross-contamination of PCR samples. We use this method as a control, i.e. in Figures 2C (lanes 3 and 4), 3 and 4 (lanes 2).
Figure 2.
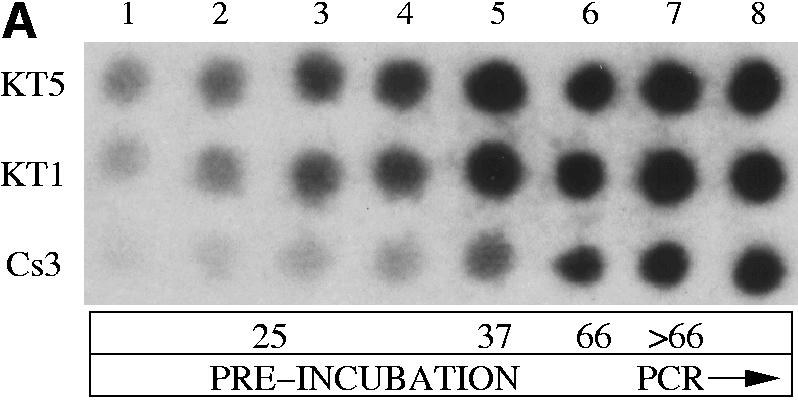
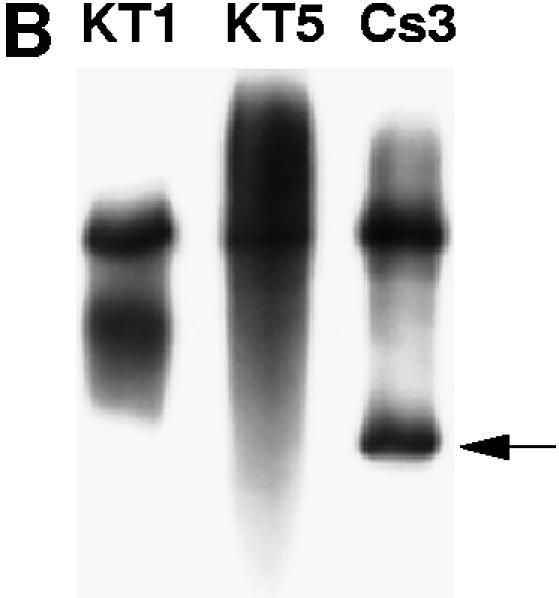
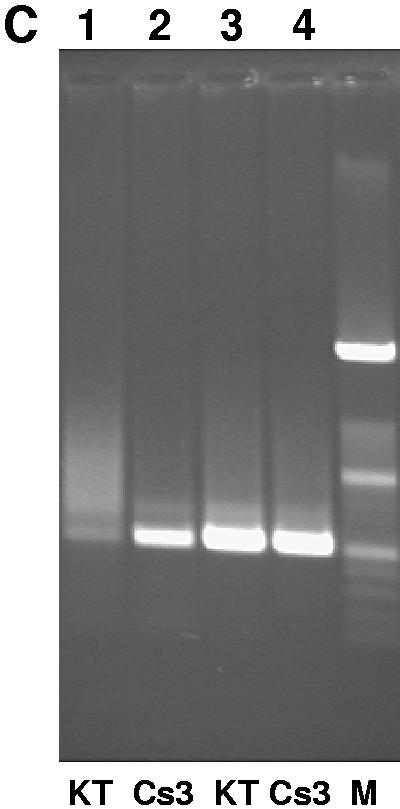
Amplification of 513 bp at the co-receptor CCR5 locus from human genomic DNA with Klentaq1 and the Cs3 triple mutant. (A) Relative activity of wild-type Klentaq and the Cs3 triple mutant during pre-PCR incubation as assayed by TCA insolubility. Each reaction contained 1 µCi of [32P]dCTP. Sixty minutes at 25°C was followed by 10 min at 37°C and a warming step of 15 min at 66°C. Parallel aliquots from the three reactions, catalyzed by Klentaq1, Klentaq or the Cs3 mutant enzyme, were taken at time points as follows: lane 1, 10 min; lane 2, 20 min; lane 3, 40 min; lane 4, 60 min (at 25°C); lane 5, additional 10 min at 37°C; lane 6, 15 min at 66°C; lanes 7 and 8, after three and six PCR cycles, respectively. The aliquots were dotted on filters, washed with 5% TCA and exposed to X-ray film. (B) After completion of the PCR (total 35 cycles), products from the very same radiolabeled reactions were electrophoresed in a 6% acrylamide/urea gel and autoradiographed on X-ray film. The correct amplified product (513 bp), shown by an arrow, appears only for Cs3. The larger, unintended products in all lanes are not reproducible and may have been caused somehow by the aliquot-taking for this experiment. (C) Non-radioactive amplicons catalyzed by Klentaq1 and the Cs3 triple mutant analyzed on a 1.4% agarose gel. Magnesium was added early to lanes 1 and 2 (before the 30°C preincubation for 30 min) and only at 68°C (manual hot start) to lanes 3 and 4.
Figure 3.

Hot start PCR performance of cold-sensitive mutants of Klentaq with various targets. Four targets were amplified in a test PCR assay (where amplification typically benefits from hot start): exon 21 and Alu7.2 repeats of the human dystrophin gene (DMD), the Cryptosporidium heat shock analog protein (Crypto HSP) and the HIV-1 gag gene (HIV gag). The expected amplified products of these targets were 323, 320, 360 and 114 bp, respectively. The following enzymes and reaction set-up conditions were used: lanes 1, Klentaq, with bench start (room temperature set-up); lanes 2, Klentaq, with manual hot start; lanes 3, Cs3C mutant, with bench start; lanes 4, Cs3AC mutant with bench start. Amplified products were resolved in ethidium stained 2% agarose gel, along with a molecular weight standard DNA ladder (M).
Figure 4.
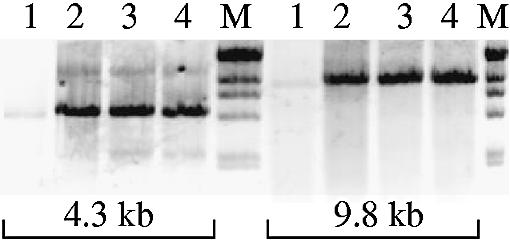
Hot start performance of LA versions of the Cs mutants in long PCR. Two targets of the human tPA gene, 4.3 and 9.8 kb, that typically require ‘hot start’, were amplified with Klentaq LA (without or with manual hot start, lanes 1 and 2, respectively) or with Cs3C LA or Cs3AC LA, without hot start (lanes 3 and 4, respectively). M, λ/HindIII DNA ladder. Amplified products were resolved in ethidium stained 1% agarose gels.
Several methods of achieving a hot start using room temperature set-up are, however, tedious, expensive and/or have limitations and shortcomings. One way of arranging a hot start is to physically separate the PCR components into two parts using paraffin wax. The wax melts at the first PCR heat step, causing mixing of all reaction components and activation of the DNA polymerase (1–4). A practical disadvantage of the wax method comes with the extra time needed to prepare the samples and to handle them after cycling, as the wax hardens.
Anti-Taq antibodies reduce the DNA polymerase activity but, being thermolabile, release the enzyme at PCR cycling temperatures to achieve a hot start (5–7). The antibodies available for this method are not very efficient and must therefore be used in a 5- or 10-fold molar excess or in a triple combination to effectively reduce the activity of the DNA polymerase. Some enzyme preparations, such as Amplitaq Gold (Perkin Elmer and Roche Molecular Systems) and Hotstart Taq (Qiagen) are chemically inactivated and can be reactivated by heating in a special preincubation step (8). The required reactivation conditions (5–15 min at 95°C and pH 8.3 or lower) are, however, harsh and could cause depurination and scission of DNA (9,10). For this reason, such a heating step is not compatible with effective long and accurate PCR (11–13). Similarly, one published hot start method explores heat activation of an affinity-immobilized Taq polymerase fusion (14). Some methods achieve a hot start with special primer design (15–17). All of these methods need only partially suppress DNA polymerase activity (80–95%; data not shown) to achieve functional hot starts. Full suppression of DNA polymerase activity is only achieved by physical or manual methods or by a magnesium precipitate hot start method (18).
Our approach was to identify cold-sensitive mutants of Taq DNA polymerases by screening a mutagenized library for thermostable DNA polymerase enzyme activity at two reaction temperatures. We show that such mutants have suppressed activity at room temperature to prevent undesired DNA synthesis, yet they are sufficiently active (and stable) at the higher temperatures of the PCR cycle. These temperature switch mutants provide hot start PCR without any special reaction conditions or additional components. Here we describe and functionally characterize some cold-sensitive mutants of the Klentaq enzyme, an N-terminal deletion of Taq DNA polymerase.
MATERIALS AND METHODS
Mutagenesis of the polymerase domain of Klentaq
Klentaq (Δ235 amino acids) (19) and Klentaq1 (Δ278 amino acids) (20) are different-sized N-terminal deletions of Thermus aquaticus DNA polymerase. All nucleotide or residue numbering is for the ORF (open reading frame) of the full-length (832 amino acids) wild-type enzyme. We exploited mutagenic PCR under error-prone amplification conditions, which consisted of normal PCR buffer KLA [50 mM Tris base, 16 mM (NH4)2SO4, 3.5 mM MgCl2, 0.1% Tween 20, pH 9.2] supplemented for mutagenicity (21) with additional metal ions to final concentrations of 0.5 mM MnSO4 + 7 mM MgCl2, and catalyzed by Klentaq1 (pure, no proof-reading supplement). To concentrate the mutagenesis on codons 543–824 of the polymerase domain, the left-hand PCR primer (KT85) was nucleotides 1599–1627, ending in codon 542. The right-hand primer (KT32) homology started one base past the last codon (832), matching the last 22 bases of the ORF (3′ end in codon 825). The template was pWB253 (19) when 12–20 cycles were employed, or genomic T.aquaticus DNA when 25–30 cycles were employed. After 12–30 cycles, the mutagenized PCR product was precipitated with PEG to remove primers and manganese and the resuspended pellet was used as the right-hand primer versus a primer (4478) (19) in the upstream kanamycin resistance gene NTPII on the template plasmid pWB302mk (22). High fidelity PCR using KlentaqLA (11) as enzyme was employed for this step of 24 cycles. To improve the final yield of mutagenized fragment, these amplifications were supplemented with primer KT32 for the last 12 cycles.
Vector plasmid pWB302 and the mutagenized PCR product were processed and digested with restriction enzymes NcoI and HindIII and ligated as described (19), transformed into electrocompetent Escherichia coli X7029 (F– ΔlacX74) and plated on medium containing 50 µg/ml kanamycin. Single colonies were grown in rich medium in the wells of 96- or 384-well replica plates and stored at –80°C after mixing with an equal volume of 50% glycerol.
Screening the mutagenized library of Klentaq
Surviving DNA polymerase activity was evaluated by single colony assay. This assay is a simplification of one published by Sagner et al. (23). Library clones were transferred from 384-well plates, rejuvenated by growth without glycerol (single clone in each well) and inoculated in arrays on nitrocellulose filters using a multipin replicator. Two replicate filters were prepared for each plate to be screened. Bacteria were allowed to grow on the filters until small, thin colonies appeared (6–12 h). Filters were then moved to dry (no agar) plates, underlayed with PCR buffer supplemented with 0.2% Triton X-100, and heated for 15–20 min at 70°C. This heat step efficiently inactivates the bacterial DNA polymerase activity and eliminates the background of the assay. The two replicate filters of each original plate were then incubated in PCR buffer in the presence of 32P-labeled dCTP or dATP (10 µCi/µl) at 68°C for 10–15 min or at 37°C for 30–60 min. After the incorporation step, filters were washed with trichloroacetic acid (TCA) and exposed to X-ray film or a phosphorimager screen. A later, improved procedure was to incubate for the same 10 or 15 min time period at both temperatures. With either procedure, normalization by data processing or variant exposure of the filter images was carried out until the signal intensities produced by wild-type clones were approximately equalized between the two assay temperatures.
Sequence analysis of mutant Klentaq clones
ORFs were amplified and both DNA strands cycle sequenced with dye-ddNTPs and Klentaq1 carrying the FY mutation (24) using an ABI 377 DNA sequencer (Applied Biosystems). Data analysis and mutant identification was assisted by the programs phred, phrap and consed (25,26). Non-silent mutations found in the three cold-sensitive clones of the Klentaq library were individually introduced into the Klentaq or Klentaq1 molecule by using primer-directed mutagenesis, PCR, recloning and confirmatory DNA sequencing.
Purification and functional analysis of mutant Klentaq enzymes
Wild-type and mutant Klentaq enzymes were purified to homogeneity (with purity checked by SDS–PAGE) from 2- to 4-liter bacterial cultures, as described (20) except that the ammonium sulfate precipitations were eliminated. Details of the purification are posted in the Supplementary Material.
Incorporation assay of DNA polymerase activity
DNA polymerase activity of extracts or purified enzyme was assayed by incubating an appropriate range of enzyme dilutions with 250 µg/ml activated (partially DNase digested) calf thymus DNA (Sigma) in KLA PCR buffer, in the presence of 50 µM each dNTP and 1 µCi [α-32P]dCTP, at 37 or 68°C for 10 min. Aliquots of 5 µl of the samples were dotted on 3MM filter paper and washed for 3 × 15 min with 5% TCA/1% sodium pyrophosphate. After drying, filters were exposed to a phosphorimager screen and the intensities of the dots were quantitated with ImageQuant software. For Figure 2A (a PCR with primers and template as the DNA) exposure was to X-ray film.
Thermostability test
Aliquots of enzymes were heated in 20 µl of KLA buffer, pH 9.2, for up to 1 h at 95°C and then assayed as above.
PCR templates, primers and cycling conditions
Human genomic DNA was purchased from Novagen and Cryptosporidium DNA was kindly provided by Kansas State University (Cryptosporidium Research Program). The HIV gag gene control target was provided by Applied Biosystems.
A typical 50 µl PCR contained 1× KLA PCR buffer (50 mM Tris–HCl, pH 9.2, 16 mM ammonium sulfate, 2.5 mM magnesium chloride, 0.1% Tween 20) with addition of magnesium chloride and each dNTP (Pharmacia) to final concentrations of 3.5 mM and 250 µM each, respectively, and 0.05–0.1 µl (40–80 ng, 2–4 U) of Klentaq DNA polymerase or its mutants.
Primer sequences and buffer and cycling details are posted in Supplementary Material. Primers were purchased from Integrated DNA Technologies and were used at 10 pmol each per 50 µl reaction. As an example, 100 ng of mouse genomic DNA (Novagen) was amplified as follows: 94°C for 1 or 2 min, then 40 cycles of 94°C for 45 s, 66°C for 40 s and 68°C for 4 min, with the reactions containing 1.6 M betaine (Sigma). In comparative experiments, FastStart Taq (Roche) or Platinum Taq (Invitrogen) were used with the buffer, magnesium chloride concentration (2 mM) and enzyme amount (2.5 U) and required 95°C preincubation (for FastStart) recommended by the manufacturer. The initial 94°C step is not required by the enzymes in this paper; rather, it is an optional step which appears to denature high quality, high molecular weight genomic DNA more effectively so that it can function as single-stranded template for the first PCR cycle.
For performing manual hot start control reactions, magnesium chloride was omitted from the reaction set-up and added only after pre-heating the samples at 68°C for 5–10 min, immediately before cycling. Where indicated, PCRs were preincubated at 25 or 37°C before any heating or amplification. All PCR amplifications were performed in a Robocycler 40 (Stratagene).
Fidelity measurement
The quality of β-galactosidase gene products after amplification of a 4.8 kb lacZ-containing target for 16 PCR cycles was evaluated by cloning with selection for both ends of the PCR product as described (19). Where indicated, an improved template pWB407 was employed. PWB407 is an analogous and improved fidelity assay system using antibiotics for both of the terminal selection markers and the target size is only 3.8 kb (W.M.Barnes, in preparation, and Supplementary Material).
Structure interpretation
The software for visualizing and modeling the mutable cold-sensitive residues was Rasmol 2.7.2 (27,28; http://www.rasmol.org) and Pymol (W.L.Delano, personal communication; http://www.pymol.org). The structures (29) 2KTQ (open conformation) and 3KTQ (closed conformation) were analyzed.
RESULTS
Screening of mutagenized Klentaq library by single colony DNA polymerase assay
Klentaq is an N-terminal deletion variant of Taq DNA polymerase that uses Met236 as a start codon (19). A mutagenized Klentaq library was generated by cloning the amplicon from low cycle number PCR of the polymerase domain of the Klentaq gene, amplified in the presence of manganese and excess magnesium (21). We found that under these conditions a low number of cycles, in the range 12–16, generates about 3–10 base changes per kb of amplified gene copy. The resultant mutagenized amplicon was then extended in a second, high fidelity PCR to cover the whole Klentaq ORF (22). Approximately 3800 mutant Klentaq clones, harbored in a lac promoter expression vector plasmid pWB302mk (22) were screened for cold-sensitive phenotype by an in-colony DNA polymerase assay. Duplicate filter replicates of library clones were incubated at 68 and 37°C (Fig. 1A and B). The rationale for this dual temperature screen is that the clones that exhibit a 37/68°C incorporation ratio lower than that of the wild-type would have a potential for cold-sensitive DNA polymerase activity. Figure 1 illustrates the performance of some Klentaq mutants in the initial screening step. The incorporation activity of several of the clones was hardly or not detectable at the low temperature, yet close to the wild-type enzyme activity at 68°C. We also detected several mutants with a temperature-sensitive phenotype (one visible in Fig. 1A), which was an indication of the general mutagenesis achieved in our library and detectable by our assay. Promisingly cold-sensitive candidates were picked and subjected to additional rounds of screening to demand reproducibly lower incorporation at 37 and 42°C, good incorporation at 68°C and resistance at 95°C.
Figure 1.



Primary screening of mutagenized library. Clones were arrayed in parallel with 9 mm spacing and the single colony assay was incubated at 37°C for 45 min (A) or 68°C for 10 min (B). Examples of cold-sensitive phenotype, temperature-sensitive phenotype and wild-type are labeled. Clone Cs3 is marked with an arrow in both panels. (C) Relative activity of cold-sensitive mutants versus wild-type Klentaq1, at low versus high temperature, was best demonstrated and confirmed by serial dilutions of purified enzymes. Cs3C and Cs3AC mutant Klentaq1 (singly and doubly mutated derivatives of Cs3) were tested in standard 50 µl DNA polymerase assays, in parallel with Klentaq1 enzyme (KT1). (D) The ratio of dCTP incorporation at 74 versus 37°C was calculated from the input data for (C).
Sequence analysis of cold-sensitive Klentaq clones
Table 1 presents DNA sequence data for the first three of the promising cold-sensitive isolates. Based on previous screening of the Klentaq library (for other features of the enzyme) and sequencing some 30 clones, we expected a maximum of about 10 base changes introduced per clone. Overlapping two-strand sequencing revealed several amino acid substitutions and a few silent mutations in each clone. All mutations were clustered in a relatively narrow portion of the polymerase domain (29,30–32) of the enzyme. Strikingly, three adjacent amino acids (Trp706, Ile707 and Glu708) were hit (and later found to be cold-sensitive when single) in three independent clones, implying early that this might be a hot-spot related to the cold sensitivity.
Table 1. DNA sequence changes found.
| Name | DNA change | Codon change | Amino acid change | Phenotype | Residue location | Side chain orientation |
|---|---|---|---|---|---|---|
| Cs1 | ||||||
| Cs1A | A1912T | ATC→TTC | I638F | Wild-type | ||
| Cs1B | G1974T | ATG→ATA | M658I | Wild-type | ||
| Cs1C | T2116A | TGG→AGG | W706R | Cold-sensitive | Surface | Inward |
| Cs2 | ||||||
| Cs2A | T2429G | GTG→GGG | V810G | Wild-type | ||
| Cs2B | A2419T | ATG→TTG | M807L | Wild-type | ||
| Cs2C | A2042G | GAG→GGG | E681G | Wild-type | ||
| Cs2D | G2124T | GAG→GAT | E708D | Cold-sensitive | Surface | Outward |
| Cs3 | ||||||
| (silent) | A1842T | ATA→ATT | I614I | Not determined | ||
| Cs3A | G1876A | GAG→AAG | E626K | Cold-sensitive | Surface | Outward |
| Cs3B | A2069G | CAG→CGG | Q690R | Could not PCR | ||
| Cs3C | A2119C | ATT→CTT | I707L | Cold-sensitive | Surface | Inward |
Assays at two temperatures
To further characterize some of the promising cold-sensitive mutants of Klentaq, we produced highly purified enzyme from several original mutant clones, single mutant clones and one doubly mutant clone using our standard procedure for preparing commercial quality Klentaq1 and aliquots were adjusted to a working concentration of 0.72 mg/ml (OD280 = 0.8). The enzyme was purified to >95% homogeneity, as judged by a single very predominant protein band in Coomassie stained gels (Supplementary Material). To confirm that the cold sensitivity was a property intrinsic to the enzymes, purified enzymes were assayed at two temperatures (74°C for 10 min or 37°C for 15 min) in a standard in vitro DNA polymerase assay of 50 µl volume. We found that at high enzyme levels, in both the colony assay and in vitro, the magnitude of the cold-sensitivity phenotype can be obscured due to saturation of the radioactive incorporation assay. We found that it was important to test several low enzyme levels to ensure lack of saturation of the assay and to normalize the data between enzyme preparations to adjust effectively for relative concentration. Typical assay results with purified Cs3A (single mutant) and Cs3AC (double mutant) in Klentaq1 are shown in Figure 1C and normalized by presenting the 74/37°C ratios in Figure 1D. Both presentations demonstrate a cold-sensitive phenotype for the mutant enzymes when compared to wild-type Klentaq1, since the activity of the mutants is reduced by a greater extent at 37°C. Similar results were obtained when the control was wild-type Klentaq (data not shown).
Thermostability test
In a preliminary test, the Cs3 mutant enzymes were treated with up to 1 h of 95°C incubation and then assayed for survival of activity, compared to Klentaq1. Survival was within the range 82–86% for each enzyme form, which was not judged to constitute any experimentally significant difference (data posted in the Supplementary Material).
Hot start utility of cold-sensitive Klentaq mutants
We tested the PCR performance of these mutants in a variety of relatively difficult, challenging amplifications, choosing targets that often give rise to non-specific products and benefit from hot start PCR. Examples of such targets are genes that encode the human HIV co-receptor CCR5, tissue plasminogen activator and dystrophin. Another example target for which PCR often fails under normal conditions is rDNA, the rRNA genes. Complications in the latter case stem from the high copy number of rDNA genes (which provides dispersed priming sites), high GC base composition (which leads to stable annealing of partial homology) and from the abundant inverted repeats (allowing one primer-dependent amplifications). To increase the challenge and better see the potential advantage of the cold-sensitive enzyme, in some of our tests we preincubated the PCRs at 25–42°C, temperatures at which, based on our screening results, one should expect lower activity with the mutants compared to the wild-type enzyme.
When the Cs3 mutant was used in such a test PCR to amplify the CCR5 locus from human genomic DNA, the mutant outperformed Klentaq1 enzyme under the test conditions, while both enzymes performed equally in a manual hot start PCR (warm addition of Mg; Fig. 2C). This result indicates that the cold sensitivity feature of the mutant can functionally provide the hot start effect. It also demonstrates that the mutant enzyme is thermostable enough to perform high cycle number PCR [35 cycles in this case, and at least 42 cycles in other tests (see below)].
As an extension to the initial radioactive screen, we performed PCR while including a 32P-labeled nucleotide in the reaction. Following preincubation at 25 and 37°C, the human CCR5 locus was amplified with Klentaq1, Klentaq and the Cs3 mutant. To measure incorporation of label during the preincubation step and early in the PCR, time point aliquots were taken from the PCR tube. After completion of the amplification, the labeled products were also gel analyzed. As shown in Figure 2A, Klentaq enzyme incorporates a detectable amount of the labeled nucleotide during the preincubation steps, while the Cs3 mutant is practically silent below 37°C. However, above 66°C, when cycling starts, the activities of the wild-type and the mutant enzyme tend to equalize. The gel image of the same reactions (Fig. 2B) shows that at the end of the PCR, the target band was successfully amplified only by the mutant.
To identify the amino acid changes found in the Cs1, Cs2 and Cs3 mutants which were truly responsible for the cold-sensitivity phenotype, and possibly identify some deleterious mutations which should remain wild-type for best utility, we reintroduced each mutation singly into wild-type Klentaq1 by primer-directed mutagenesis. Table 1 indicates which single mutations exhibited the cold-sensitive phenotype according to the two-temperature assay of DNA polymerase activity.
The three individual mutants, Cs3A, Cs3B and Cs3C (see Table 1), were tested again in CCR5 amplification of human DNA. Under the test PCR conditions, including a 25/37°C preincubation, the parent Cs3 mutant, as well as the Cs3A and Cs3C single mutants, outperformed Klentaq (data not shown). The Cs3B single mutant was unable to catalyze any amplification, indicating that this particular mutation is deleterious to the overall activity of the enzyme, and the original Cs3 clone mutations A and/or C must somehow be compensating for the negative effect of mutation B. The data showed that amino acid changes Glu626Lys and Ile707Leu are each individually cold sensitive when present in either the Klentaq or Klentaq1 molecule.
To further demonstrate the utility of the most promising mutants, we carried out additional tests with various genomic PCR targets: exon 21 and Alu7.2 repeats of the human dystrophin gene DMD, the Cryptosporidium heat shock analog protein and the HIV-1 gag gene. Under our test conditions specific and/or efficient amplification of these targets with the wild-type enzyme required a hot start. As shown in Figure 3, in all cases the Cs3C mutant enzyme outperformed Klentaq (lane 1 versus 3) and worked as well as the control reactions with a manual hot start (lanes 2). Identical results were obtained with the Cs3AC double mutant of Klentaq, where the two mutations A and C were co-introduced (lanes 4). In some reactions (Crypto HSP and HIV gag) the double mutant gave rise to a higher yield as compared to the single mutant, implying that the two mutations might act synergistically.
For longer PCR, we tested the cold-sensitive mutants as LA enzyme mixes, where DeepVent DNA polymerase was added as a proofreader to 2% (v/v). Such ‘LA’ (long and accurate) enzyme mixtures are generally more robust, have higher fidelity and can amplify long targets (11,12,20). The results of such a test are shown in Figure 4, where long amplicons were amplified by cold-sensitive mutant LA enzyme mixtures under conditions where the wild-type Klentaq1 depended on a hot start set-up. This demonstrates that in the LA version of the mutants their cold-sensitive phenotype is not compromised by the low level of active Deep Vent polymerase, thus extending the potential application of the cold-sensitive enzymes in hot start PCR.
We compared the performance of our cold-sensitive enzymes with some widely used commercial Taq polymerases formulated for hot start. Four targets of the mouse rRNA gene were amplified with the Cs3AC-LA double mutant enzyme, FastStart Taq (Roche) or Platinum Taq (Invitrogen). As shown in Figure 5, the mutant clearly outperformed the two commercial enzymes with all targets tested, which, when working at all, were dependent on a manual hot start set-up.
Figure 5.

Hot start PCR performance of Cs3AC LA mutant compared to FastStart Taq and Platinum Taq. Four targets (1–4) of mouse rRNA genes were amplified from 100 ng genomic DNA with FastStart Taq (Roche) or Platinum Taq (Invitrogen) with or without manual hot start (H.S.) or with Cs3AC LA enzyme (CS) without H.S. (details of targets and conditions are given in Materials and Methods). The expected amplified products of targets 1–4 were 998, 557, 1155 and 818 bp, respectively. PCR products were electrophoresed in ethidium stained 1.4% agarose gel, along with a λ/HindIII DNA ladder (M).
Fidelity comparison
The results are enumerated in Table 2 and shown in Figure 6. Wild-type (full-length) Taq DNA polymerase was included as a benchmark to aid comparison with other published fidelity studies, which vary widely as to their conclusions. Mutations per 100 000 nucleotides incorporated were calculated from the equation used previously (19), and the error in these measurements is estimated at 20% of the number shown. The three largest contributions to this error are as follows. (i) The uncertainty in the fold amplification is about one doubling either way. It is also uncertain whether the last doubling should be counted (we did not count it; 19). (ii) Uncertainty in the colony counts is the square root of the count, by Poisson statistics. (iii) The identity of about 5% of the mutant colonies is uncertain, as they may not have actually been less than full blue or some slightly less blue colonies may not have been counted as mutant.
Table 2. Fidelity comparisons assayed after 16 cycles of PCR.
| Enzyme | Blue | White | Percent lacZ– | Doublings | Errors per 100 000 | Betaine | lacZ template |
|---|---|---|---|---|---|---|---|
| Taq | 209 | 206 | 50% | 9 | 18 | 0 | pWB407 |
| KT1 | 765 | 409 | 35% | 7 | 15 | 0 | pWB407 |
| Cs3C | 1150 | 338 | 23% | 8 | 8 | 0 | pWB407 |
| Cs3AC | 820 | 259 | 24% | 8 | 8 | 0 | pWB407 |
| Taq | 216 | 269 | 55% | 9 | 21 | 1.3 M | pWB407 |
| KT1 | 572 | 419 | 42% | 10 | 12 | 1.3 M | pWB407 |
| Cs3C | 389 | 139 | 26% | 10 | 7 | 1.3 M | pWB407 |
| Cs3AC | 1244 | 586 | 32% | 9 | 10 | 1.3 M | pWB407 |
| KT1 | 341 | 231 | 40% | 9 | 13 | 1.3 M | pWB305 |
| Cs3C | 266 | 84 | 24% | 9 | 7 | 1.3 M | pWB305 |
| Cs3AC | 499 | 156 | 24% | 9 | 7 | 1.3 M | pWB305 |
Figure 6.

Fidelity of PCR products catalyzed by wild-type and cold- sensitive variants of Taq DNA polymerase when they catalyzed the amplification of a lacZ gene (bracketed on each side by a selectable marker) for 16 cycles of PCR. The percentage of lacZ– clones was converted to errors per nucleotide incorporated as described (19), wherein the target size is assumed to be 1000 bp. The bars labeled pWB305 were determined as described (19) and the rest used plasmid pWB407. Except as noted for the top four bars, the PCR amplifications contained 1.3 M betaine. Taq, full-length, wild-type Thermus aquaticus DNA polymerase; KT1, N-terminal deletion Klentaq1; Cs3C, Klentaq1 I707L; Cs3AC, Klentaq1 I707L, E626K.
Taken together, the results demonstrate the practical benefit of cold-sensitive mutations that can cause Taq DNA polymerases to perform a hot start.
DISCUSSION
Taq polymerase belongs to the Pol I family of DNA polymerases which share structural homology and modular functional organization with E.coli Pol I and phage T7 DNA polymerases. In this group of enzymes the DNA polymerization domain resides at the C-terminus and it is preceded by a 3′→5′ exonuclease and sometimes a 5′-nuclease domain (27,28,31,32). The central domain of Taq exhibits some structural homology but zero 3′→5′ exonuclease activity and no sequence homology (28).
The crystal structure of Klentaq1 and the Klenow fragment of Pol I, which have both been engineered to lack the 5′-nuclease domain, reveals an architecture that has been likened to a human right hand. A fingers subdomain binds the incoming dNTP and interacts with the single-stranded DNA template, a thumb subdomain binds double-stranded DNA and a palm subdomain harbors the catalytic amino acids and also interacts with the incoming dNTP (29,31). In the presence of the dNTP (actually, a ddCTP in the study; 29), Klentaq1 adopts a ‘closed’ conformation in which the tips of the fingers move inward. Li et al. (29) propose that this movement occurs at each cycle of base incorporation. Although this closed conformation was not observed in a similar study of Bacillus DNA polymerase (33), an analogous closed conformation has been trapped in the crystal of an archebacterial DNA polymerase, 9° N 7 (34).
In our attempt to design a better enzyme for hot start PCR, we generated cold-sensitive mutants of Klentaq DNA polymerase which exhibit a decreased activity at low (reaction set-up) temperatures, but normal activity at the optimal cycling temperatures. These mutations were chosen arbitrarily as four of the more reproducible in phenotype among some 24 arising from the initial screen, and they are the only ones for which we have sequence data confirmed by single change mutant phenotype. All four of the cold-sensitive mutable residues that we have identified so far are in α-helices on the outside surface of the finger domain, seemingly quite far from any active site or conserved residues. The eventual description of how they work must explain their action at this distance from the active site, why their negative phenotype is overcome by increased temperature and how they can have their observable effect when they are merely conservative changes (I707L and E708D) or stick out into space, away from all other parts of the enzyme (E626K and E708D) (Fig. 7) or both. The mutation Cs1A is our most drastic mutation, since it replaces an internal tryptophan with a more hydrophilic arginine.
Figure 7.
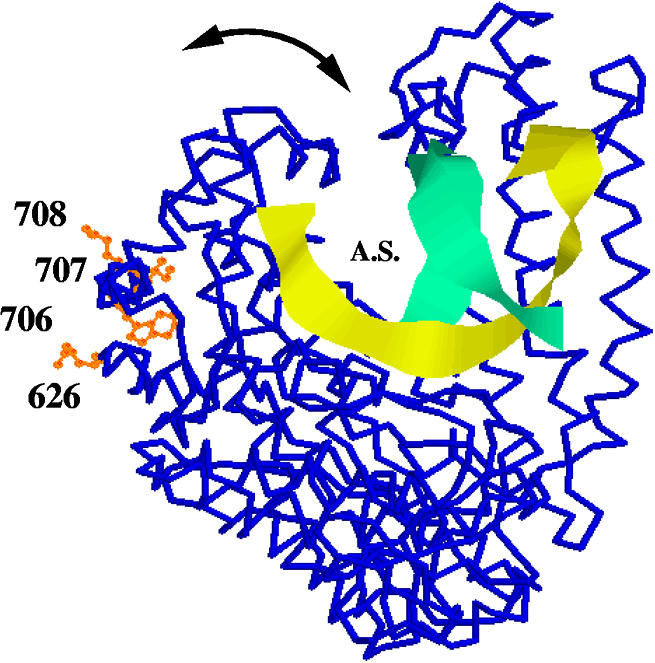
Looking directly through the axis of the P α-helix (residues 704–717) on the left, four amino acids mutable to cold-sensitive are rendered as orange ball and stick wild-type residues on the backbone structure (blue) of Klentaq1 and are labeled with their residue numbers. Depicted with the aid of Rasmol 2.7.2 is the closed conformation of protein database file 3KTQ (29). Also shown as yellow and green ribbons are the determined positions of a short portion of primer and template DNA. Direction of synthesis would be away from the viewer. The curved arrow indicates the proposed (29) range of motion of the fingertips. A.S., active site. Not shown is a molecule of ddCTP in the active site.
The position of the cold-sensitive mutable residues near the base of the movable domain suggests to us that they may be located at a fulcrum which magnifies small effects. The α-helix known as ‘P’ (residues 704–717) may exert pressure on the finger domain, known as ‘Q’. We believe we can identify the residue closest to the cold-sensitive Leu707 as the Q domain phenylalanine at residue 749, which is oriented with its π electrons directed at the side chain of residue 707 (see Fig. 8A). Between the open and closed conformations, this phenylalanine moves sideways by 1.53 Å relative to residue 707. When 707 is a leucine, perhaps this movement is more difficult when an additional methyl group interacts, possibly more strongly, with the Phe749 π electrons, rather than just the terminal methyl group of the wild-type isoleucine (closest carbon–carbon distance to Phe749 = 3.8 Å). In Figure 8C we have conservatively modeled the mutant leucine as the rotomer most closely resembling the wild-type isoleucine. Depending on the rotamer adopted by the mutant leucine (and other possible longer range effects on the structure), the closest contact between a methyl carbon and a ring carbon of Phe749 could be 1.5 Å (not shown or allowed) or 2.5 Å (depicted in Fig. 8C), both closer than the wild-type. The result of altered interactions with the nearest residues might make this movement more difficult or it might tend to favor one of the conformers of the fingertips domain. If one of the conformers were more or less preferred at the restrictive temperature, we cannot say at this time which one it might be, but this question might be answerable by crystallography, since crystals are normally grown at the restrictive temperature.
Figure 8.

Close-up representation, from approximately the same point of view as Figure 7, of the region surrounding residues 707 (isoleucine, blue space-filled) and 749 (phenylalanine, orange space-filled). (A) Wild-type open configuration. (B) Wild-type closed configuration. (C) Model of mutant leucine at position 707 shows that it may now crowd Phe749 with one of its methyl groups (white arrow).
The fidelity of Taq variants, as measured by screening of cloned PCR products, has been shown to be altered and slightly improved by the N-terminal deletions (11,19). In an assay which enumerates blue and white (or light blue) colonies carrying clones of the amplified lacZ gene (19) we measured the apparent fidelity of two of our cold-sensitive mutants in Klentaq1, Cs3C and the double mutant Cs3AC, compared to otherwise wild-type Klentaq1. Since most of our other PCRs contained betaine and since this fidelity assay has not yet been applied in the presence of betaine, we measured fidelity with and without 1.3 M betaine. In our tests betaine did not have a consistent or significant effect on fidelity. Under all conditions, however, the cold-sensitive mutations produced fewer errors in the PCR products, compared to the parent Klentaq1. The measured increase in fidelity exhibited by the mutants was less than a factor of two, yet it was reproducible with two sizes of lacZ PCR target, with betaine and without betaine. As noted previously (19), the slight effect observed with this test may be due to an increased loss of strands with mismatched bases at their 3′-ends and is not necessarily an actual improvement in base pair discrimination. The practical result appears to be slightly improved fidelity.
Before this study, rational design of cold-sensitive mutations might have concentrated on the binding sites for magnesium or the substrate molecules in the palm or on the inside of the finger domain. After this study, it may be predicted that further cold-sensitive mutations will be located near or at the locations of the first four, on the surface of the enzyme finger domain, nowhere near the active site.
Even though they often share little homology, DNA polymerases of all four families, including reverse transcriptases, have been proposed to utilize the same fingers motion for the catalytic cycle (35,36). The cold-sensitive mutations have brought to our attention that Taq DNA polymerase can be made less active by a very subtle change at a surface-accessible, non-conserved location at the finger fulcrum point. If analogous cold-sensitive mutations can be isolated for HIV-1 reverse transcriptase, their location may spotlight a reasonable target for specific pharmaceutical inhibition.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Shannon Flynn, Brett Evans and Erika Vail for technical help in purification and testing cold-sensitive mutant enzymes and helpful discussions. This work was supported by STTR and SBIR grants 1 R41 GM6081001and 2 R44 GM6081002A1 from The National Institutes of Health. It should be noted that the authors are the inventors and providers of some of the subject technology for amplification and hot starts.
REFERENCES
- 1.Hebert B., Bergeron,J., Potworowski,E.F. and Tijssen,P. (1993) Increased PCR sensitivity by using paraffin wax as a reaction mix overlay. Mol. Cell. Probes, 7, 249–252. [DOI] [PubMed] [Google Scholar]
- 2.Kaijalainen S., Karhunen,P.J., Lalu,K. and Lindstom,K. (1993) An alternative hot start technique for PCR in small volumes using beads of wax-embedded reaction components dried in trehalose. Nucleic Acids Res., 21, 2959–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horton R.M., Hoppe,B.L. and Conti-Tronconik,B.M. (1994) AmpliGrease: ‘hot start’ PCR using petroleum jelly. Biotechniques, 16, 42–43. [PubMed] [Google Scholar]
- 4.Ramanujam R., Burdick,B.A., Landegren,U.D. and Sevigny,P. (1997) Method and preparation for sequential delivery of wax-embedded, inactivated biological and chemical reagents. US patent 5,599,660.
- 5.Scalice E.R., Sharkey,D.J. and Daiss,J.L. (1994) Monoclonal antibodies prepared against the DNA polymerase from Thermus aquaticus are potent inhibitors of enzyme activity. J. Immunol. Methods, 172, 147–163. [DOI] [PubMed] [Google Scholar]
- 6.Kellogg D.E., Rybalkin,I., Chen,S., Mukhamedova,N., Vlasik,T., Siebert,P.D. and Chenchik,A. (1994) TaqStart antibody: ‘hot start’ PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase. Biotechniques, 16, 1134–1137. [PubMed] [Google Scholar]
- 7.Sharkey D.J., Scalice,E.R., Christy,K.G., Atwood,S.M. and Daiss,J.L. (1994) Antibodies as thermolabile switches: high temperature triggering for the polymerase chain reaction. Biotechnology, 12, 506–509. [DOI] [PubMed] [Google Scholar]
- 8.Moretti T., Koons,B. and Budowle,B. (1998) Enhancement of PCR amplification yield and specificity using AmpliTaq Gold DNA Polymerase. Biotechniques, 25, 716–722. [PubMed] [Google Scholar]
- 9.Lindahl T. and Nyberg,B. (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry, 11, 3611–3618. [DOI] [PubMed] [Google Scholar]
- 10.Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 11.Barnes W.M. (1994) PCR amplification of up to 35 kb DNA with high fidelity and high yield from λ bacteriophage templates. Proc. Natl Acad. Sci. USA, 91, 2216–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng S., Fockler,C., Barnes,W.M. and Higuchi,R. (1994) Effective amplification of long targets from cloned inserts and human genomic DNA. Proc. Natl Acad. Sci. USA, 91, 5695–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnes W.M. (1994) Tips and tricks for long and accurate PCR. Trends Biochem. Sci., 19, 342. [DOI] [PubMed] [Google Scholar]
- 14.Nilsson J., Bosnes,M., Larsen,F., Nygren,P.A., Uhlen,M. and Lundeberg,J. (1997) Heat-mediated activation of affinity-immobilized Taq DNA polymerase. Biotechniques, 4, 744–751. [DOI] [PubMed] [Google Scholar]
- 15.Kaboev O.K., Luchkina,L.A., Tretiakov,A.N. and Bahrmand,A.R. (2000) PCR hot start using primers with the structure of molecular beacons (hairpin-like structure). Nucleic Acids Res., 28, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kainz P., Schmiedlechner,A. and Strack,H.B. (2000) Specificity-enhanced hot-start PCR: addition of double-stranded DNA fragments adapted to the annealing temperature. Biotechniques, 28, 278–282. [DOI] [PubMed] [Google Scholar]
- 17.Dang C. and Jayasena,S.D. (1996) Oligonucleotide inhibitors of Taq DNA polymerase facilitate detection of low copy number targets by PCR. J. Mol. Biol., 264, 268–278. [DOI] [PubMed] [Google Scholar]
- 18.Barnes W.M. and Rowlyk,K.R. (2002) Magnesium precipitate hot start method for PCR. Mol. Cell. Probes, 16, 167–171. [DOI] [PubMed] [Google Scholar]
- 19.Barnes W. (1992) The fidelity of Taq polymerase catalyzing PCR is improved by an N-terminal deletion. Gene, 112, 29–35. [DOI] [PubMed] [Google Scholar]
- 20.Barnes W.M. (1995) Thermostable DNA polymerase with enhanced thermostability and enhanced length and efficiency of primer extension. US patent 5,436,149.
- 21.Cadwell R.C. and Joyce,G.F. (1992) Randomization of genes by PCR mutagenesis. PCR Methods Appl., 2, 28–33. [DOI] [PubMed] [Google Scholar]
- 22.Barnes W.M. and Kermekchiev,M. (2001) Cold sensitive mutant DNA polymerases. US patent 6,214,557.
- 23.Sagner G., Ruger,R. and Kessler,C. (1991) Rapid filter assay for the DNA polymerase activity: direct identification of the gene for the DNA polymerase from Thermus aquaticus. Gene, 97, 119–123. [DOI] [PubMed] [Google Scholar]
- 24.Tabor S. and Richardson,C.C. (1995) A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc. Natl Acad. Sci. USA, 92, 6339–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewing B., Hillier,L., Wendl,M. and Green,P. (1998) Basecalling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res., 8, 175–185. [DOI] [PubMed] [Google Scholar]
- 26.Gordon D., Abajian,C. and Green,P. (1998) Consed: a graphical tool for sequence finishing. Genome Res., 8, 195–202. [DOI] [PubMed] [Google Scholar]
- 27.Blanco L., Bernad,A., Blasco,M. and Salas,M. (1991) A general structure for DNA-dependent DNA polymerases. Gene, 100, 27–38. [DOI] [PubMed] [Google Scholar]
- 28.Korolev S., Nayal,M., Barnes,W.M., DiCera,E. and Waksman,G. (1995) Crystal structure of the large fragment of Thermus aquaticus DNA polymerase I at 2.5 Å resolution: structural basis for thermostability. Proc. Natl Acad. Sci. USA, 92, 9264–9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y., Korolev,S. and Waksman,G. (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J., 17, 7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernstein H.J. (2000) Recent changes to RasMol, recombining the variants. Trends Biochem. Sci., 25, 453–455. [DOI] [PubMed] [Google Scholar]
- 31.Olis D., Brick,R., Hamlin,R., Xuong,N. and Steitz,T. (1985) Structure of the large fragment of Escherichia coli DNA polymerase complexed with dTMP. Nature, 313, 762–766. [DOI] [PubMed] [Google Scholar]
- 32.Joyce C. and Steitz,T. (1987) DNA polymerase I: from crystal structure to function via genetics. Trends Biochem. Sci., 12, 288–292. [Google Scholar]
- 33.Kiefer J.R., Mao,C., Braman,J.C. and Beese,L.S. (1998) Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature, 391, 304–307. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez A.C., Park,H.W., Mao,C. and Beese,L.S. (1999) Crystal structure of a pol alpha family DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. 9 degrees N-7. J. Mol. Biol., 299, 447–462. [DOI] [PubMed] [Google Scholar]
- 35.Huang H., Chopra,R., Verdine,G.L. and Harrison,S.C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science, 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- 36.Doublie S., Sawaya,M.R. and Ellenberger,T. (1999) An open and closed case for all polymerases. Structure, 7, R31–R35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}