

Abstract
A combined approach of validated QSAR modeling and virtual screening was successfully applied to the discovery of novel tylophrine derivatives as anticancer agents. QSAR models have been initially developed for 52 chemically diverse phenanthrine-based tylophrine derivatives (PBTs) with known experimental EC50 using chemical topological descriptors (calculated with the MolConnZ program) and variable selection k nearest neighbor (kNN) method. Several validation protocols have been applied to achieve robust QSAR models. The original dataset was divided into multiple training and test sets, and the models were considered acceptable only if the leave-one-out cross-validated R2 (q2) values were greater than 0.5 for the training sets and the correlation coefficient R2 values were greater than 0.6 for the test sets. Furthermore, the q2 values for the actual dataset were shown to be significantly higher than those obtained for the same dataset with randomized target properties (Y-randomization test), indicating that models were statistically significant. Ten best models were then employed to mine a commercially available ChemDiv Database (ca. 500K compounds) resulting in 34 consensus hits with moderate to high predicted activities. Ten structurally diverse hits were experimentally tested and eight were confirmed active with the highest experimental EC50 of 1.8µM implying an exceptionally high hit rate (80%). The same ten models were further applied to predict EC50 for four new PBTs, and the correlation coefficient (R2) between the experimental and predicted EC50 for these compounds plus eight active consensus hits was shown to be as high as 0.57. Our studies suggest that the approach combining validated QSAR modeling and virtual screening could be successfully used as a general tool for the discovery of novel biologically active compounds.
Introduction
Natural products have been the major source of anticancer drugs. According to a recent review1 on New Chemical Entities (NCE), from 1981 to 2002, approximately 74% of anticancer drugs were either natural products, or natural product-based synthetic compounds, or their mimetics. (+)-(S)-Tylophorine (1) and its analogues are phenanthroindolizidine alkaloids, commonly referred to as tylophora alkaloids. They are a small group of alkaloids known for their profound cytotoxic activities2–5. Evaluation of these compounds in the antitumor screening at the National Cancer Institute (NCI) showed a uniform and potent inhibitory effect on the cell growth (GI50, ≅ 10−8 M) in all 60 cell lines, with notable selectivity toward several refractory cell lines, including melanoma and lung tumor cell lines6. Earlier studies in 1960s’ demonstrated that their mechanism of antitumor activity was due to irreversible inhibition of the protein synthesis at the elongation stage of the translation cycle7–10. In 1990s’, several key metabolic enzymes were reported as biological targets of tylophorine alkaloids including thymidylate synthase (TS)11 and dihydrofolate reductase (DHFR)12. In addition, these agents were found to induce cell apoptosis as well13. Most recently, Gao et al. demonstrated that tylophorine analogues had significant inhibitory effect on NF-kB mediated transcription14. These discoveries exemplified the great potential of developing tylophorine derivatives as a new class of antitumor drugs.
Although the phenanthroindolizidine alkaloid tylocrebrine (2) previously failed in clinical trials due to its CNS toxicity15, the very profound cytotoxicity of these alkaloids16, particularly against multidrug resistant cancer cells16, sparked our interest in additional studies of this class of potential anticancer agents. Recently, we have advanced a novel series of polar, water-soluble phenanthrene-based tylophorine derivatives (PBTs) (6, 18) with EC50 ≅10−7 M against the A549 human lung cancer cell line17. These compounds could possibly have lower or no CNS toxicity because their increased polarity should prevent them from penetrating the blood-brain barrier. Although the biological target of PBTs is unknown, several structure-activity trends have been observed17: 1) A planar phenanthrene system is required, but not sufficient for cytotoxic activity. 2) An N-hydrophilic substituent at the C-9 position is essential for the enhanced cytotoxicity and should be linked through a methylene rather than a carbonyl group. 3) The C-9 N-hydrophilic substituent is ideal for the introduction of a polar moiety. Analogs containing terminal carboxylic acid or hydroxymethyl groups are more favorable than those with methyl esters. 4) On the phenanthrene skeleton, a methoxyl substituent best fits both the steric and electronic requirements at the C-6 position and is preferred over benzyloxyl and hydroxyl groups. 5) Adding a methylenedioxyl ring at the 2, 3 positions of the planar phenanthrene system dramatically enhances the cytotoxic activity and leads to the most potent derivatives. The new PBT derivatives possess a novel structure and show remarkable EC50 values in the sub-micromolar range17, comparable to those of the front-line antineoplastic drugs, and suggesting that this new class of compounds may have a great potential as antitumor agents. The availability of experimental data on PBT derivatives afforded us an opportunity to apply advanced computational drug discovery approaches, in particular QSAR modeling, towards knowledge based accelerated discovery of novel anticancer agents.
Many different QSAR approaches have been developed during the past few decades18–21. Modern methods are characterized by the use of multiple descriptors of chemical structure combined with the application of both linear and non-linear optimization approaches, and a strong emphasis on rigorous model validation to afford robust and predictive QSAR models (see recent reviews22;23) The most important recent developments in the field have concurred with a substantial increase in the size of experimental datasets available for the analysis and an increased application of QSAR models as virtual screening tools to discover biologically active molecules in chemical databases and/or virtual chemical libraries23;24. The latter focus differs substantially from the traditional emphasis on developing so called explanatory QSAR models characterized by high statistical significance but only as applied to training sets of molecules with known chemical structure and biological activity. Our laboratory recently developed a robust computational QSAR modeling framework that combines various algorithms for model development (such as k nearest neighbor (kNN) QSAR approach with variable selection25), model validation, and model implementation for virtual screening24;26. This strategy was recently applied to several experimental datasets leading to the successful discovery of novel anticonvulsant agents26 and D1 dopaminergic antagonists27.
In this paper, we discuss the application of the kNN QSAR method to a dataset of 52 PBTs with known EC50 values (Table 1, Table 2 & Table 3). The structures were characterized with MolConnZ descriptors28. The models developed for the PBT dataset have been extensively validated using several criteria of robustness and accuracy29. Several validated models with the high predictive power were used to mine the commercially available ChemDiv30 database resulting in 34 consensus hits with the moderate to high predicted activities. Ten structurally diverse hits were experimentally tested and eight compounds were confirmed active, with the most potent compound having EC50 of 1.8µM. The predictive power of these models were further confirmed by the high correlation coefficient between the predicted and actual cytotoxicity for an external set comprised of four new PBTs and the eight active ChemDiv hits, which were not included in the original 52 PBTs dataset. The correlation coefficient (R2) was as high as 0.57. The results of this study suggest that rigorously validated QSAR models could be successfully used as virtual screening tools for prioritizing untested compounds for experimental biological evaluation.
Table 1.
Structure and cytotoxic activity of PBTs compounds (1–38) used in model building.
 | |||
|---|---|---|---|
| Compound | R1 | R2 | EC50 (µM) |
| 3 | -CH3 | -CONH(CH2)4COOH | 73.3 |
| 4 | -CH3 | -CH2NH(CH2)4COOMe | 25.3 |
| 5 | -CH3 | -CH2NH(CH2)4COOH | 1.3 |
| 6 | -CH3 | -CH2NH(CH2)4CH2OH | 0.27 |
| 7 | -CH3 | -CONH(CH2)5COOH | 27 |
| 8 | -CH3 | - CH2NH(CH2)5COOMe | 18 |
| 9 | -CH3 | - CH2NH(CH2)5COOH | 0.8 |
| 10 | -CH3 | - CH2NH(CH2)5CH2OH | 0.2 |
| 11 | -CH3 |  |
5.3 |
| 12 | -CH3 |  |
73.8 |
| 13 | -CH3 |  |
2.1 |
| 14 | -CH3 |  |
0.7 |
| 15 | -CH3 |  |
0.5 |
| 16 | -CH3 |  |
0.16 |
| 17 | -CH3 |  |
0.23 |
| 18 | -CH3 |  |
0.08 |
| 19 | -CH3 |  |
33.8 |
| 20 | -CH3 |  |
65.2 |
| 21 | -CH3 | - CH2NH(CH2)10COOH | 3.2 |
| 22 | -CH3 | - CH2NH(CH2)10CH2OH | 2.6 |
| 23 | -CH2C6H5 | -CONH(CH2)5COOMe | 41.2 |
| 24 | -CH2C6H5 | -CONH(CH2)5COOH | 41.2 |
| 25 | -CH2C6H5 | -CH2NH(CH2)5COOH | 1.6 |
| 26 | -CH2C6H5 | -CH2NH(CH2)5CH2OH | 1.1 |
| 27 | -CH2C6H5 | -CH2NH(CH2)4COOMe | 17.0 |
| 28 | -CH2C6H5 | -CH2NH(CH2)4COOH | 2.2 |
| 29 | -CH2C6H5 |  |
42.6 |
| 30 | -CH2C6H5 |  |
32.1 |
| 31 | -CH2C6H5 |  |
4.4 |
| 32 | -CH2C6H5 |  |
1.8 |
| 33 | -CH2C6H5 |  |
3.2 |
| 34 | -CH2C6H5 |  |
1.3 |
| 35 | -CH2C6H5 | -CONH(CH2)5COOMe | 41.2 |
| 36 | -CH2C6H5 | -CONH(CH2)5COOH | 41.2 |
| 37 | -CH2C6H5 | -CH2NH(CH2)5COOH | 1.6 |
| 38 | -H |  |
39.7 |
| 39 | -H |  |
41.2 |
| 40 | -H |  |
39.7 |
Table 2.
Structure and cytotoxic activity of PBTs compounds (39–46) used in model building.
 | ||
|---|---|---|
| Compound | R | EC50 (µM) |
| 41 | -NH(CH2)10COOH | 13.0 |
| 42 | -NH(CH2)10CH2OH | 3.6 |
| 43 | -CH2NH(CH2)5COOH | 9.7 |
| 44 | -CH2NH(CH2)5CH2OH | 2.7 |
| 45 |  |
9.7 |
| 46 |  |
6.3 |
| 47 |  |
19.2 |
| 48 |  |
2.4 |
Table 3.
Structure and cytotoxic activity of PBTs compounds (47–52) used in model building.
| Compound | Structure | EC50 (µM) |
|---|---|---|
| 49 |  |
80 |
| 50 |  |
45.2 |
| 51 |  |
11.7 |
| 52 |  |
52.2 |
| 53 |  |
0.02 |
| 54 |  |
0.008 |
Materials and Methods
Chemistry and Biological Activity Data
All PBTs used in this study were synthesized and evaluated (Table 1, 2, 3 & 5) in one of our laboratories. The general synthetic procedure, biological activity, physical and spectral data have been reported previously17. The hit compounds identified by the means of database mining were purchased from ChemDiv, Inc. The human A549 lung cancer cell line was used for the cytotoxicity screening of both PBTs synthesized earlier as well as novel computational hits, employing a cell-based sulforhodamine B (SRB) microtitre plate assay31. The screening method was reported in detail elsewhere17.
Table 5.
Structure and cytotoxic activity for compounds (53–56) in the external set
 | ||||
|---|---|---|---|---|
| Compound | R | Actual Activity (EC50, µM) |
Actual Activity (−logEC50) |
Predicted Activity (−logEC50) |
| 55 |  |
0.22 | 6.65 | 6.81 |
| 56 | 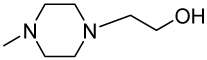 |
0.63 | 6.20 | 6.81 |
| 57 |  |
57.1 | 4.24 | 4.42 |
| 58 |  |
0.15 | 6.82 | 6.60 |
Generation of Molecular Descriptors
All chemical structures were generated using SYBYL 7.032. Molecular descriptors were calculated for each compound with the MolConnZ software version 4.0528;33. MolConnZ produced more than 400 descriptors; however, in our study, only 244 significant descriptors were used after removing those with zero variance. The descriptors were range-scaled prior to model generation because the absolute scales of different descriptors differed in some cases by orders of magnitude. Range scaling helps to avoid disproportional weightings of descriptors upon the Euclidean distance calculations in multidimensional descriptor space.
Dataset Division into Training and Test Sets
It is commonly accepted that the internal validation of QSAR models built from training sets is sufficient to confirm their predictive power34–38. However, previous studies in this as well as several other laboratories demonstrated that no correlation exists between leave-one-out (LOO) cross-validated R2 (q2) for the training set and the correlation coefficient R2 between the predicted and observed activities for the test set29;39. These findings indicated that in order to obtain QSAR models with high predictive ability, external validation was critical. Thus, a dataset of 52 compounds was divided into multiple chemically diverse training and test sets with a rational approach implemented in our group40 based on the Sphere Exclusion (SE) algorithm41. SE is a general procedure that is typically applied to molecules characterized by multiple descriptors of their chemical structures. The entire dataset can then be treated as a collection of points (each point corresponding to an individual compound) in the MolConnZ descriptor space. The goal of the SE method is to divide a dataset into two subsets (training and test sets) using a diversity sampling procedure40.
The SE algorithm used in this study included the following steps.40 The algorithm starts with the calculation of the distance matrix D between points representing compounds in the multidimensional descriptor space. Let Dmin and Dmax be the minimum and maximum elements of D, respectively. N probe sphere radii are defined by the following formulas: Rmin=R1=Dmin, Rmax=RN=Dmax/4, Ri=R1+(i-1)*(RN-R1)/(N-1), where i=2, …, N-1. Each probe sphere radius corresponds to one division into the training and test sets. Once the sphere size is defined the subsequent calculations include the following steps: (i) Select randomly a point in the MolConnZ descriptor space. (ii) Include it in the training set. (iii) Construct a probe sphere around this point. (iv) Select points from this sphere and include them alternatively into test and training sets. (v) Exclude all points within this sphere from further consideration. (vi) If no more compounds left, stop. Otherwise let m be the number of probe spheres constructed and n be the number of remaining points. Let dij (i=1,…,m; j=1,…,n) be the distances between the remaining points and probe sphere centers. Select a point corresponding to the lowest dij value and go to step (ii). The training sets were used to build models and the test sets were used for model validation.
kNN QSAR Method
Our implementation of this method25 employs the kNN pattern recognition principle42 and a variable selection procedure. Briefly, a subset of nvar descriptors (number of selected variables) is selected randomly. A model is built using this random descriptor selection with LOO cross-validation, where each compound is eliminated from the training set and its biological activity is predicted as the average activity of its k most similar molecules (usually k = 1–5). The value k is optimized during the model building process to give the best prediction for the training set. The similarity is characterized by the Euclidean distance between compounds in multidimensional space of selected descriptors. We used weighted molecular similarity27 to calculate the estimated activities ŷi of compounds excluded by LOO procedure using the following formula
| (1) |
where aj was the observed activity of the jth compound, and weights wij are defined as
| (2) |
and dij was the distance between compound i and its jth nearest neighbor. After each run, cross-validated q2 is calculated
| (3) |
where yi, and ȳ are the actual and average values of activity. The summation in (3) is performed over all compounds. A method of simulated annealing with the Metropolis-like acceptance criterion43 is used to sample the entire descriptor space to converge on the subset of the same size which afford the highest value of q2. The descriptor subsets of different sizes are optimized using this procedure to obtain a variety of models with acceptable q2 greater than a certain threshold (we used 0.5 as the default threshold value). The training set models with acceptable q2 are then validated on the test sets to select predictive models with R2 exceeding 0.6. Further details of the kNN method implementation, including the description of the simulated annealing procedure used for stochastic sampling of the descriptor space, were given elsewhere.28
In summary, the kNN QSAR algorithm generates both an optimal k value and an optimal nvar subset of descriptors that afford a QSAR model with the highest value of q2. Figure 1 shows both the overall flowchart of the current implementation of the kNN method (1a) and the predictive QSAR modeling workflow (1b).
Figure 1.

kNN QSAR modeling approach (a) and predictive QSAR modeling workflow (b).
Robustness and Predictive Power of QSAR Models
The robustness of the models was examined by comparing them to those obtained when using randomized activity of the training set (this procedure is commonly referred to as Y-randomization test)44. Briefly, the QSAR calculations were repeated with the randomized activities of the training sets. Then the q2 values for actual and random activities of training sets were compared to see whether there was a significant difference as expected for robust models. This test was applied to all data divisions considered in this study and it was repeated five times for each division.
To estimate the predictive power of a QSAR model, the following parameters were used29: (i) correlation coefficient R2 between the predicted and observed activities; (ii) coefficients of determination (predicted versus observed activities , and observed versus predicted activities ); (iii) slopes k and k' of regression lines (predicted versus observed activities, and observed versus predicted activities) through the origin. We concluded that a QSAR model had an acceptable predictive capability if the following conditions were satisfied29:
| (4) |
| (5) |
| (6) |
| (7) |
Applicability Domain of kNN QSAR Models
Following the procedures developed earlier,44;45 the distances (similarity) of compounds in our training set were compiled to produce an applicability domain threshold (i.e., similarity threshold), DT, calculated as follows:
| (8) |
where <d> is the average of Euclidean distances between k nearest neighbors of all compounds of the training set used in model derivation, σ is the standard deviation of these distances, and Z is the empirical parameter to control the significance level. The default value of Z was set to 0.5, which formally placed the boundary for the compounds to be predicted at one-half of the standard deviation (assuming a normal distribution of distances between k nearest neighbor compounds in the training set). Thus, if the distance of an external compound from at least one of its nearest neighbors in the training set exceeded this threshold, the prediction was considered unreliable.
Database Mining
A commercially available chemical database, Chemical Diversity (ChemDiv)30, containing ca. 500K compounds, was used for virtual screening. MolConnZ descriptors were generated for each compound in the databases and linearly normalized based on the maximum and minimum values of each descriptor in the training set27.
Ten best kNN models were used to predict activities of the database compounds that were within the applicability domain of each individual model. The results for each individual prediction exercise were then combined and the mean predicted activity was calculated for each compound that was within the applicability domain of multiple models. The number of models that predicted each compound in the database and the standard deviation of those predictions for each compound were also recorded. We selected a subset of compounds as hits that were predicted by at least 50% of the models and exhibited a small standard deviation across all models. We also performed an additional analysis as to whether the hits resulting from database mining possessed the features of PBTs essential for their activities. This additional consideration was considered important because it helped us identify novel scaffolds expected to have anticancer activities.
Results and Discussions
QSAR Models and Their Robustness
In the kNN QSAR method, nvar can be set to any value that is less than the total number of descriptors. Since the optimal number for nvar is not known a priori, multiple models have to be generated to examine the relationship between q2 and nvar. As previously discussed, Y-randomization is a widely used technique to ensure the robustness of a QSAR model46. It is expected that the resulting QSAR models from the randomized sets should generally have low training set fitness q2 and low test set R2. Sometimes, though infrequently, high training set q2 may be obtained due to a chance correlation or structural redundancy of the training set44. If all QSAR models obtained in the Y-randomization test exhibit relatively high training set q2 and test set R2 values, it implies that an acceptable QSAR model cannot be built for the given dataset by the current modeling method.
To compare results from the actual data set with those from data sets with randomized activity values, models with 10, 20, 30, 40 and 50 descriptors were generated. Figure 2 shows a plot of q2 vs. nvar for the actual and random data sets obtained with kNN calculations. Every q2 value was the average of 10 independent computations. Overall, we have obtained consistently much higher q2 values for the actual data set compared to those from Y-randomization. The q2 values for the real data set were in the range 0.60 to 0.7 while they were from −0.01 to 0.05 for the random data sets. This demonstrated that the high q2 values of the models for the real data sets were not due to chance correlations.
Figure 2.

Plots of q2 vs. the number of descriptors selected for the best kNN QSAR models for 52 PBTs. The results for both actual and random (with shuffled activity values) data sets are shown. Every q2 value is the average of 10 independent calculations. The black circles represent the actual data set, and the grey triangles represent the random data set.
kNN QSAR Model Validation
Generally, we accept models with q2 values for the training set greater than 0.5 and R2 values for predicted vs. actual activities of the test set compounds greater than 0.6.34 Table 4 presents 10 best models obtained from multiple kNN analyses. In order to estimate the statistical significance of the models, the original dataset of 52 compounds was divided into 50 training and test sets. Multiple kNN models with the high q2 values (greater than 0.5) were collected. However, similar to our previous observations29, no correlation was found between q2 and R2 (Figure 3) demonstrating that q2 alone does not serve as an estimate of the predictive power of kNN models. On the basis of our criteria, acceptable models with both high statistical significance (q2 > 0.5) and predictive power (R2 > 0.6) represented only a fraction of all models with q2 > 0.5 (Figure 3). So the aforementioned conditions (Eq. 4, 5, 6, and 7) are indeed very important. Based on all of these criteria, the best models were obtained for the test sets including 14 and 18 compounds, with the optimal number of descriptors of 15 and 20, respectively (Table 4).
Table 4.
Ten best kNN QSAR models that were used for database mining.
| Models | Test Sets | Training Sets | Number of Descriptors |
q2 | R2 |
|---|---|---|---|---|---|
| 1 | 8 | 44 | 15 | 0.52 | 0.75 |
| 2 | 11 | 41 | 20 | 0.53 | 0.8 |
| 3 | 12 | 40 | 10 | 0.72 | 0.71 |
| 4 | 12 | 40 | 15 | 0.72 | 0.72 |
| 5 | 14 | 38 | 10 | 0.56 | 0.77 |
| 6 | 14 | 38 | 20 | 0.51 | 0.81 |
| 7 | 14 | 38 | 15 | 0.58 | 0.81 |
| 8 | 14 | 38 | 15 | 0.54 | 0.79 |
| 9 | 18 | 34 | 20 | 0.59 | 0.81 |
| 10 | 20 | 32 | 15 | 0.55 | 0.73 |
Figure 3.

R2 vs. q2 for all selected models with q2>0.5. Grey triangle for models with R2<0.6 and black dots for models with R2>0.6.
Figure 4 shows the correlation coefficient between actual and calculated activity for the training and test sets with q2 = 0.59 and R2 = 0.81 respectively. Two outlier points (compound 20 and 54) were poorly predicted in the training set. The one in black circle represents antofine (Figure 4), a positional isomer of tylophorine isolated from Asclepiadaceae by Dr. T. S. Wu in Taiwan47. It was used as a reference compound when we screened PBTs. A possible explanation for this observation is that antofine lacks the chemical structure descriptors that are most meaningful and statistically significant in terms of correlation with biological activity: a N-hydrophilic substituent and free rotated C-9 methylene bond, which were proven to be required for the PBTs analogs activity in our previous study17. Studies on the mechanisms of action for antofine and PBTs are still ongoing, and it remains possible that they have different mechanisms of action. In regards to compound 20 (in grey circle, Figure 4), the terminal -Cl group may cause some solubility or cell membrane transportation problem that would result in diminished activity in our cell-based assay. After excluding these two outlying points, the q2 for training set increased to 0.69. Given the high predictive R2 (0.81), this model was considered acceptable for virtual screening.
Figure 4.

Plot of actual vs. predicted activity for one of the best 10 models. This model has 18 compounds in the test set (grey triangles), and correspondingly 34 in the training set (black dots). Twenty descriptors were selected for the calculation. q2=0.59 and R2=0.81. The circled are two outliers, Antofine 54 (black circle) and Compound 20 (grey circle), respectively.
Interpreting Predictive QSAR Models
Upon the analysis of our QSAR models, a number of MolConnZ descriptors were found in most of the accepted models, suggesting that they played critical roles in defining antitumor activity of PBTs. These included molecular connectivity descriptors such as Chi indices, hydrogen bond counts, E-state descriptors, shape indices, etc. The results were consistent with our preliminary SAR observations described in the Introduction Section.
Based on the MolConnZ manual and personal communication with Dr. Lowell Hall, one of the principal developers of MolConnZ software, these selected descriptors were grouped into the following classes: 1) The high frequency of Chi indices, including dXvp7, dXvp10, nXp6, Xvp7, etc., suggested the importance of those structure features such as size, branching, cyclicity and so on. Our previous SAR study showed that the para-positioned functional groups and three intra-atomic distances were very important for the antitumor activity. These features defined the relative spatial dispositions of three significant atoms: the oxygen of C-9 chain terminal, the nitrogen atom, and a methoxyl oxygen of the phenanthrene ring. 2) Hydrogen bond donors/acceptors counts and E-states descriptors, such as nHBint9, SHBd, Hmax, and Hmin, indicated the importance of polar hydrogen atoms and hydrogen bond interactions during binding. This observation agreed with the finding that the presence of a hydrogen bond acceptor/donor group at C-9 chain terminus appeared essential for the cytotoxic activity, and analogs containing terminal carboxylic acid or hydroxymethyl groups were more favorable than those with methyl esters. 3) The appearance of atom type counts and E-states descriptors (SssCH2, nsssCH, and SsOH) demonstrated the high importance of electron accessibility for those atoms. Our studies indeed found that an N-hydrophilic substituent at the C-9 position was essential for enhanced cytotoxicity and should be linked through a methylene rather than a carbonyl group. 4) Several steric crowding related descriptors (n2Pag12 and Tm) occurred many times in our models. This point was reflected in our finding that, for example, a methoxyl substituent best fitted both the steric and electronic requirements at the C-6 position. 5) Several Kappa and complexity indices (ka1, ka2, tets1, tets2, graph complexity, etc.) were found in many models, suggesting the shape related features were important for the cytotoxicity of our PBTs. It was noted that a planar phenanthrene system was required, although not sufficient, for the cytotoxic activity. Also adding a methylenedioxyl ring at the 2, 3 positions of the planar phenanthrene system dramatically enhanced the cytotoxic activity and led to the most potent derivatives. 6) Last, but not the least, descriptors such as nCl and SsCl indicated the importance of a -Cl group to the activity. Inclusion of this group in structures led to the significant change of their activities (e.g. compound 20, 64 and 68).
Database Mining with Predictive QSAR Models
The ChemDiv database of over 500K compounds was screened with our ten best QSAR models within a defined applicability domain (i.e., similarity threshold). Formally, a QSAR model can predict the target property of any compound for which chemical descriptors have been calculated. Since the training set models are developed with the kNN QSAR approach by interpolating activities of the nearest neighbor compounds, the applicability domain should help avoid making predictions for compounds that differ substantially from the training set molecules44. We hypothesized that the higher the number of models with a stringent applicability domain that predict a compound's activity, the more likely the compound actually has the predicted activity. This postulate may also apply to the standard deviation of the predictions made for a single compound. The smaller the prediction variance across all models, the more confidence we have that the predicted biological activity for that compound is accurate. The predicted activities for each compound from those selected models were averaged to yield consensus values.
Thirty-four consensus hits were identified with moderate to high predicted activities. Among them, some compounds shared very similar core chemical structures while the others were quite different. Eventually, ten structurally diverse hits (59–68, Table 5.) with moderate to high predicted activity were purchased and screened using the same cell line and assay method as used for the PBTs screening. The chemical structure and experimental biological data are shown in Table 5. Eight (59, 61–62, 64–68) of ten compounds were confirmed to be active against A549 lung cancer cell line, and compound 68 (ChemDiv #: K915-0700) showed the best activity with EC50 of 1.8µM, comparable to the active PBTs. This hit (68) has a novel core structure which is different from either known PBTs or other anticancer drugs, and our screening established the anticancer activity for this structure for the first time.
Prediction of Anticancer Activity for an External Data Set
The accurate prediction of the bioactivity is a more challenging task compared to the relative ranking in virtual screening. A reliable and truly predictive QSAR model should be able to accurately predict activities of new compounds in external sets. To this end, the kNN QSAR models validated with the test sets were used to predict the activity of four new compounds (Table 5), which were not available prior to our QSAR studies of the 52 PBT derivatives dataset. Concurrently, the eight active hits (Table 6) from ChemDiv, which had moderate to high predicted activities, were also used to evaluate the accurate activity prediction capability of our models in a quantitative manner.
Table 6.
Structure and cytotoxic activity for the 10 computational hits (59–68) from ChemDiv database.
| Compound | Structure | ChemDiv ID # |
Actual Activity (EC50, µM) |
Actual Activity (−logEC50) |
Predicted Activity (−logEC50) |
|---|---|---|---|---|---|
| 59 |  |
1661-1313 | 10.9 | 4.96 | 5.86 |
| 60 |  |
2188–3298 | N/A | N/A | 4.99 |
| 61 |  |
3253–1073 | 19.3 | 4.71 | 4.52 |
| 62 |  |
3346–2033 | 42.6 | 4.37 | 4.47 |
| 63 |  |
3570-0022 | N/A | N/A | 5.76 |
| 64 |  |
4106-0061 | 7.9 | 5.10 | 5.83 |
| 65 |  |
6658-0014 | 11.1 | 4.96 | 6.56 |
| 66 |  |
C614-0150 | 17.3 | 4.76 | 4.97 |
| 67 |  |
K901-0689 | 72.4 | 4.14 | 5.01 |
| 68 |  |
K915-0700 | 1.8 | 5.74 | 5.21 |
As mentioned above, all of the external compounds displayed moderate to high predicted activity that ranged between 0.15 and 72.4µM (Table 5 & 6), while the original training set compounds (Table 1, 2 & 3) featured EC50 activity values that ranged from 0.08 to 80µM. Table 5 & 6 lists the average predicted activity values for the external data set obtained from the best kNN models. We intentionally selected a series of compounds that had a wide range of predicted activity, paralleling those used in the training and test sets during the model building. This hit selection strategy helped us confirm the predictive power of our models in a wide applicability domain. The resulting correlation coefficient R2 was as high as 0.57 (Figure 5). It was interesting to analyze the performance of QSAR models on the congeneric (with similar core structure) compounds and novel (core structure different) compounds. With four congeneric PBTs, the difference between the predicted and experimental activity (−logEC50) was about 0.2. For the eight structurally diverse hits (Table 6), the result was not as good as that for the congeneric compounds. Two reasons could be suggested and discussed: 1) The congeneric and the novel hit compounds had high structural dissimilarity. The models were based on the structure-activity of the training set (congeneric), so the selected descriptors were not sufficient to reliably and accurately predict the activity of external diverse structures (novel). This problem always existed in statistical modeling and the final results could be improved by using additional descriptors. 2) Since compounds were tested in whole cell based assay, activity could deviate from the predicted values due to different mechanisms of inhibition, transportation, metabolism, etc. Although the prediction accuracy of the activity for screening hits was not as good as that for the original congeneric (core structure similar) compounds, the high experimental hit rate (eight out of ten hits were active) and the capability of detecting novel active structures from a large chemical database confirmed that this method was a very useful and powerful tool for lead identification. The overall high correlation coefficient (R2=0.57) demonstrated that our QSAR models were very robust and predictive for most of the compound structures (both congeneric and novel) and could be used to diversify the chemical repertoire of anticancer agents.
Figure 5.

Plot of actual vs. predicted activity for the four designed compounds and eight computational hits. Compound 60 and 63 were shown to be inactive during experimental test, so the consensus EC50 values for only 12 out of 14 compounds were predicted based on the 10 best models with R2 = 0.57.
Conclusions
As part of our ongoing studies on plant-derived antitumor agents, we recently initiated the design and synthesis of new tylophorine analogs because of their profound anti-cancer activity. The structures of this class of compounds (PBTs) are still being optimized and their mechanisms of action are under investigation. Herein, we report the use of validated QSAR modeling analysis and database mining in advancing the discovery of novel antitumor agents. Using the QSAR modeling workflow we have developed robust models for a series of PBT derivatives with high internal and external prediction accuracy. These models were further exploited in database mining for new lead identification and computational hits have been tested experimentally yielding an exceptionally high confirmed hit rate of 80%. Those hits (such as compound 68) lacking a phenanthrine ring but having high activity (e.g., EC50=1.8µM) may in fact lead to a novel structural class of anticancer agents.
In principle, virtual screening with QSAR models could be regarded as a sophisticated form of similarity searching. Our results demonstrate that the use of multiple descriptors of chemical structures such as molecular topological indices in validated QSAR models could indeed afford the identification of novel compounds. These results are in agreement with earlier observations of Brown and Martin48 on the efficiency of 2D molecular descriptors in molecular similarity calculations leading to the identification of biologically active molecules. We place particular emphasis on the rigorous validation of QSAR models as well as conservative extrapolation limited to the applicability domain as two major factors that allow us to achieve the highest possible accuracy in predicted biological activity of compounds external to the training set. Furthermore, we select only consensus hits obtained with multiple validated QSAR models as opposed to the predictions based on a single best model. We believe that this approach should facilitate the design of new natural product analogs as well as the search for new structures with anti-cancer activity from large chemical databases. We further suggest that our recent successes in identifying novel active compounds using combined application of rigorous QSAR modeling and database mining for several classes of compounds such as anticonvulsants26, D1 antagonists27 and now, anti-tumor agents positions the QSAR – virtual screening (QSAR-VS) as a general methodology for computer aided drug discovery.
Chart 1.

Structures of tylophorine, tylocrebrine, and PBTs 6 & 18
Acknowledgment
This investigation was supported by grant CA17625 from National Cancer Institute awarded to K. H. Lee and by grants GM066940 and P20-RR20751 awarded to AT. We wish to thank Dr. Susan L. Morris-Natschke for her critical reading of the manuscript and Dr. Lowell Hall for his comments on the interpretation of MolConnZ descriptors. The authors dedicate this paper to Dr. Yvonne C. Martin who has been a source of inspiration and encouragement for many years to the senior author.
REFERENCES
- 1.Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat. Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 2.Gellert E, Rudzats R. The antileukemia activity of tylocrebrine. J Med. Chem. 1964;15:361–362. doi: 10.1021/jm00333a029. [DOI] [PubMed] [Google Scholar]
- 3.Rao KV, Wilson RA, Cummings B. Alkaloids of tylophora. 3. New alkaloids of Tylophora indica (Burm) Merrill and Tylophora dalzellii Hook f. J Pharm. Sci. 1971;60:1725–1726. doi: 10.1002/jps.2600601133. [DOI] [PubMed] [Google Scholar]
- 4.Pettit GR, Goswami A, Cragg GM, Schmidt JM, Zou JC. Antineoplastic agents, 103. The isolation and structure of hypoestestatins 1 and 2 from the East African Hypoestes verticillaris. J Nat. Prod. 1984;47:913–919. doi: 10.1021/np50036a001. [DOI] [PubMed] [Google Scholar]
- 5.Suffness M, Cordell GA. The Alkaloids, Chemistry and Pharmacology. New York: Academic Press; 1985. pp. 3–355. [Google Scholar]
- 6.The 60-cell line NCI test data, along with in vivo data can be accessed from the NSC numbers at the following web site http://dtp.nci.nih.gov/dtpstandard/dwindex/index.jsp. 2006
- 7.Donaldson GR, Atkinson MR, Murray AW. Inhibition of protein synthesis in Ehrlich ascites-tumour cells by the phenanthrene alkaloids tylophorine, tylocrebrine and cryptopleurine. Biochem. Biophys. Res. Commun. 1968;31:104–109. doi: 10.1016/0006-291x(68)90037-5. [DOI] [PubMed] [Google Scholar]
- 8.Huang MT, Grollman AP. Mode of action of tylocrebrine: effects on protein and nucleic acid synthesis. Mol. Pharmacol. 1972;8:538–550. [PubMed] [Google Scholar]
- 9.Grant P, Sanchez L, Jimenez A. Cryptopleurine resistance: genetic locus for a 40S ribosomal component in Saccharomyces cerevisiae. J Bacteriol. 1974;120:1308–1314. doi: 10.1128/jb.120.3.1308-1314.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta RS, Siminovitch L. Mutants of CHO cells resistant to the protein synthesis inhibitors, cryptopleurine and tylocrebrine: genetic and biochemical evidence for common site of action of emetine, cryptopleurine, tylocrebine, and tubulosine. Biochemistry. 1977;16:3209–3214. doi: 10.1021/bi00633a026. [DOI] [PubMed] [Google Scholar]
- 11.Rao KN, Bhattacharya RK, Venkatachalam SR. Inhibition of thymidylate synthase and cell growth by the phenanthroindolizidine alkaloids pergularinine and tylophorinidine. Chem Biol. Interact. 1997;106:201–212. doi: 10.1016/s0009-2797(97)00065-3. [DOI] [PubMed] [Google Scholar]
- 12.Rao KN, Venkatachalam SR. Inhibition of dihydrofolate reductase and cell growth activity by the phenanthroindolizidine alkaloids pergularinine and tylophorinidine: the in vitro cytotoxicity of these plant alkaloids and their potential as antimicrobial and anticancer agents. Toxicol. In Vitro. 2000;14:53–59. doi: 10.1016/s0887-2333(99)00092-2. [DOI] [PubMed] [Google Scholar]
- 13.Ganguly T, Khar A. Induction of apoptosis in a human erythroleukemic cell line K562 by tylophora alkaloids involves release of cytochrome c and activation of caspase 3 3. Phytomedicine. 2002;9:288–295. doi: 10.1078/0944-7113-00146. [DOI] [PubMed] [Google Scholar]
- 14.Gao W, Lam W, Zhong S, Kaczmarek C, Baker DC, Cheng YC. Novel mode of action of tylophorine analogs as antitumor compounds. Cancer Res. 2004;64:678–688. doi: 10.1158/0008-5472.can-03-1904. [DOI] [PubMed] [Google Scholar]
- 15.Suffness M, Douros JD. Anticancer Agents Based on Natural Product Models. London: Academic Press; 1980. pp. 465–487. [Google Scholar]
- 16.Staerk D, Lykkeberg AK, Christensen J, Budnik BA, Abe F, Jaroszewski JW. In vitro cytotoxic activity of phenanthroindolizidine alkaloids from Cynanchum vincetoxicum and Tylophora tanakae against drug-sensitive and multidrug-resistant cancer cells. J Nat. Prod. 2002;65:1299–1302. doi: 10.1021/np0106384. [DOI] [PubMed] [Google Scholar]
- 17.Wei L, Brossi A, Kendall R, Bastow KF, Morris-Natschke SL, Shi Q, Lee KH. Antitumor agents 251: synthesis, cytotoxic evaluation, and structure-activity relationship studies of phenanthrene-based tylophorine derivatives (PBTs) as a new class of antitumor agents. Bioorg. Med. Chem. 2006;14:6560–6569. doi: 10.1016/j.bmc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Hadjipavloulitina D, Hansch C. Quantitative Structure-Activity-Relationships of the Benzodiazepines - A Review and Reevaluation. Chem. Rev. 1994;94:1483–1505. [Google Scholar]
- 19.Hansch C, Muir RM, Fujita T, Maloney PP, Geiger E, Streich M. The Correlation of Biological Activity of Plant Growth Regulators and Chloromycetin Derivatives with Hammett Constants and Partition Coefficients. J. Am. Chem. Soc. 1963;85:2817–2824. [Google Scholar]
- 20.Klein TE, Huang C, Ferrin TE, Langridge R, Hansch C. Computer-Assisted Drug Receptor Mapping Analysis. Acs Symposium Series. 1986;306:147–158. [Google Scholar]
- 21.Kubinyi H. Quantitative Relationships Between Chemical-Structure and Biological-Activity. Chemie in Unserer Zeit. 1986;20:191–202. [Google Scholar]
- 22.Tropsha A. Predictive QSAR (Quantitative Structure Activity Relationships) Modeling. In: Martin YC, editor. Comprehensive Medicinal Chemistry II. Elsevier; 2006. pp. 113–126. [Google Scholar]
- 23.Tropsha A. Application of Predictive QSAR Models to Database Mining. In: Oprea T, editor. Cheminformatics in Drug Discovery. Wiley-VCH; 2005. pp. 437–455. [Google Scholar]
- 24.Tropsha A, Cho SJ, Zheng W. "New Tricks for an Old Dog": Development and Application of Novel QSAR Methods for Rational Design of Combinatorial Chemical Libraries and Database Mining. In: Parrill AL, Reddy MR, editors. Rational Drug Design: Novel Methodology and Practical Applications. 1999. pp. 198–211. [Google Scholar]
- 25.Zheng WF, Tropsha A. Novel variable selection quantitative structure-property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci. 2000;40:185–194. doi: 10.1021/ci980033m. [DOI] [PubMed] [Google Scholar]
- 26.Shen M, Beguin C, Golbraikh A, Stables JP, Kohn H, Tropsha A. Application of predictive QSAR models to database mining: Identification and experimental validation of novel anticonvulsant compounds. J. Med. Chem. 2004;47:2356–2364. doi: 10.1021/jm030584q. [DOI] [PubMed] [Google Scholar]
- 27.Oloff S, Mailman RB, Tropsha A. Application of validated QSAR models of D1 dopaminergic antagonists for database mining. J. Med. Chem. 2005;48:7322–7332. doi: 10.1021/jm049116m. [DOI] [PubMed] [Google Scholar]
- 28.MolConnZ. [4.05] Quincy, MA: Hall Associates Consulting; 2002. [Google Scholar]
- 29.Golbraikh A, Tropsha A. Beware of q(2)! J. Mol. Graph. Model. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 30.ChemDiv. 2005 http://www.chemdiv.com.
- 31.Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl. Cancer Inst. 1990;82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 32.SYBYL. [Version 6.9] St. Louis, MO: Tripos, Inc.; 2002. [Google Scholar]
- 33.Kier LB, Hall LH. Molecular Connectivity in Chemistry and Drug Research. New York: Academic Press; 1976. [Google Scholar]
- 34.Benigni R, Giuliani A, Franke R, Gruska A. Quantitative structure-activity relationships of mutagenic and carcinogenic aromatic amines. Chem. Rev. 2000;100:3697–3714. doi: 10.1021/cr9901079. [DOI] [PubMed] [Google Scholar]
- 35.Oloff S, Zhang S, Sukumar N, Breneman C, Tropsha A. Chemometric analysis of ligand receptor complementarity: identifying Complementary Ligands Based on Receptor Information (CoLiBRI) J. Chem. Inf. Model. 2006;46:844–851. doi: 10.1021/ci050065r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trohalaki S, Gifford E, Pachter R. Improved QSARs for predictive toxicology of halogenated hydrocarbons. Comput. Chem. 2000;24:421–427. doi: 10.1016/s0097-8485(99)00093-5. [DOI] [PubMed] [Google Scholar]
- 37.Zhang S, Golbraikh A, Tropsha A. Development of quantitative structure-binding affinity relationship models based on novel geometrical chemical descriptors of the protein-ligand interfaces. J. Med. Chem. 2006;49:2713–2724. doi: 10.1021/jm050260x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang S, Golbraikh A, Oloff S, Kohn H, Tropsha A. A Novel Automated Lazy Learning QSAR (ALL-QSAR) Approach: Method Development, Applications, and Virtual Screening of Chemical Databases Using Validated ALL-QSAR Models. J Chem Inf. Model. 2006;46:1984–1995. doi: 10.1021/ci060132x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kubinyi H, Hamprecht FA, Mietzner T. Three-dimensional quantitative similarityactivity relationships (3D QSiAR) from SEAL similarity matrices. J. Med. Chem. 1998;41:2553–2564. doi: 10.1021/jm970732a. [DOI] [PubMed] [Google Scholar]
- 40.Golbraikh A, Tropsha A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Aided Mol. Des. 2002;16:357–369. doi: 10.1023/a:1020869118689. [DOI] [PubMed] [Google Scholar]
- 41.Snarey M, Terrett NK, Willett P, Wilton DJ. Comparison of algorithms for dissimilarity-based compound selection. J Mol. Graph. Model. 1997;15:372–385. doi: 10.1016/s1093-3263(98)00008-4. [DOI] [PubMed] [Google Scholar]
- 42.Sharaf MA, Illman DL, Kowalski BR. Chemometrics. New York: John Wiley & Sons; 1986. pp. 1–332. [Google Scholar]
- 43.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953:1087–1092. [Google Scholar]
- 44.Tropsha A, Gramatica P, Gombar VK. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003;22:69–77. [Google Scholar]
- 45.Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003;17:241–253. doi: 10.1023/a:1025386326946. [DOI] [PubMed] [Google Scholar]
- 46.Wold S, Eriksson L. Statistical Validation of QSAR Results. In: Waterbeemd Hvd., editor. Chemometrics Methods in Molecular Design. VCH; 1995. pp. 309–318. [Google Scholar]
- 47.Wu PL, Rao KV, Su C-H, Kuoh C-S, Wu T-S. Phenanthroindolizidine alkaloids and their cytotoxicity from the leaves of Ficus septica. Heterocycles. 2002;57:2401–2408. [Google Scholar]
- 48.Brown RD, Martin YC. An evaluation of structural descriptors and clustering methods for use in diversity selection. SAR QSAR. Environ. Res. 1998;8:23–39. doi: 10.1080/10629369808033260. [DOI] [PubMed] [Google Scholar]