

Abstract
Communication between endothelial cells and cardiomyocytes regulates not only early cardiac development but also adult cardiomyocyte function, including the contractile state. In the normal mammalian myocardium, each cardiomyocyte is surrounded by an intricate network of capillaries and is next to endothelial cells. Cardiomyocytes depend on endothelial cells not only for oxygenated blood supply but also for local protective signals that promote cardiomyocyte organization and survival. While endothelial cells direct cardiomyocytes, cardiomyocytes reciprocally secrete factors that impact endothelial cell function. Understanding how endothelial cells communicate with cardiomyocytes will be critical for cardiac regeneration, in which the ultimate goal is not simply to improve systolic function transiently but to establish new myocardium that is both structurally and functionally normal in the long term.
Keywords: cell-cell interaction, regeneration, endothelial cell, heart failure
INTRODUCTION
Modern vascular biology has dispensed with the notion that endothelial cells provide a mere “passive lining” supportive role. We now recognize endothelial cells as dynamic regulators of vascular tone and growth of nearby cells. In fact, endothelial cells can participate in the development of many tissues, including the liver and pancreas (1). The myocardium is yet another example of a tissue in which many functions and responses have a surprising dependence on endothelial cells.
In the normal adult mammalian myocardium, there is at least one capillary next to every cardiomyocyte (Figure 1). The mass of cardiomyocytes in a mammalian heart is approximately 25 times that of cardiac endothelial cells, although the smaller endothelial cells outnumber cardiomyocytes by ∼3:1 (2). Because diffusion of a given factor is inversely proportional to the square of distance the factor must travel, a minimal endothelial-cardiomyocyte cell-to-cell distance is crucial for cardiomyocytes to obtain oxygen and nutrients. Cardiomyocytes are typically ∼10-100 μm in size, and the intercapillary distance in mammalian myocardium is ∼15-50 μm (2, 3). This intricate anatomical arrangement of cardiomyocytes within the capillary network not only allows for physiological transport but also for local communication between endothelial cells and cardiomyocytes.
Figure 1.
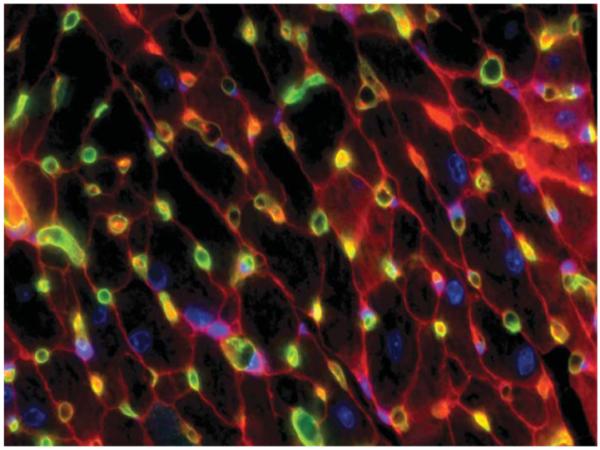
Endothelial-cardiomyocyte assembly in adult mouse myocardium. Normal adult mouse myocardium is stained with intravital perfusion techniques to demonstrate cardiomyocyte (outlined in red) and capillary (green; stained with isolectin-fluorescein) assembly. Nuclei are blue (Hoechst). Original magnification: 600X.
The ongoing molecular conversation between endothelial cells and cardiomyocytes is highly relevant to the recent excitement in promoting cardiac regeneration. The ultimate goal of myocardial regeneration is to rebuild a functional tissue that closely resembles mature myocardium, not just to improve systolic function transiently. Thus, regenerating myocardium will require rebuilding the vascular network along with the cardiomyocyte architecture. Here we review evidence demonstrating crucial molecular interactions between endothelial cells and cardiomyocytes. We first discuss endothelial-cardiomyocyte interactions during embryonic cardiogenesis, followed with morphological and functional characteristics of endothelial-cardiomyocyte interactions in mature myocardium. Finally, we consider strategies exploiting endothelial-cardiomyocyte interplay for cardiac regeneration.
ENDOTHELIAL-CARDIOMYOCYTE INTERACTIONS IN CARDIAC DEVELOPMENT
In mice, endoderm-derived growth factors from the primitive cardiac crescent (∼E7.5) govern the formation of cardiac mesoderm, primary myocardium, and endocardium (4). Signals among endocardial cells, myocardial cells, and neural crest-derived cells control the endocardial endothelial cells undergoing the epithelial-to-mesenchymal transformation to form cardiac cushions and subsequent cardiac septa, valves, and the outflow tract (∼E10.5-12.5) (5, 6). Endocardial and myocardial cells may arise from the same cardiac mesodermal precursors and may thus share some of their specific lineage markers in early development (7). With formation of the primitive cardiac tube (∼E8.5), endocardial and myocardial cells become separated by a dense layer of extracellular matrix, the cardiac jelly (5, 6). However, these two distinct cardiac cell layers continuously and reciprocally communicate through paracrine signaling during further development. The interdependence of endothelial and myocardial cells for cardiac development has been demonstrated by cell-restricted gene inactivation experiments (Figure 2).
Figure 2.
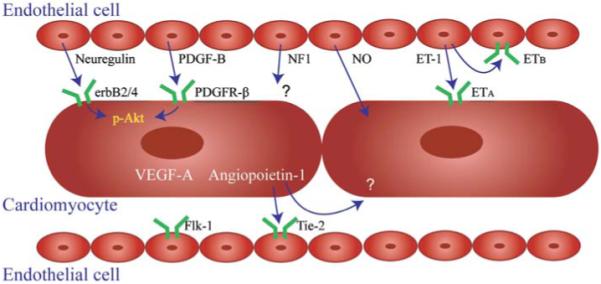
Endothelial-cardiomyocyte interactions through autocrine and paracrine signaling. Endothelial cells may secret signaling mediators modulating cardiomyocyte development (neuregulin, PDGF-B, and NF1), survival (neuregulin and PDGF-B), and contraction (NO and ET-1). Reciprocally, cardiomyocytes may also promote endothelial cell survival and assembly through VEGF-A and angiopoietin-1. Even more complicated is combined autocrine and paracrine signaling between endothelial cells and cardiomyocytes through ET-1 and angiopoietin-1. PDGF-B, platelet-derived growth factor-B; PDGFR-β, PDGF receptor-β; NF1, neurofibromatosis type 1; NO, nitric oxide; ET-1, endothelin-1; VEGF-A, vascular endothelial growth factor-A.
Signaling from Endothelial Cells to Cardiomyocytes in Development
NEUREGULIN-erbB
One well-characterized example of endothelial-cardiomyocyte interactions during early cardiogenesis is the neuregulin-erbB signaling pathway. Neuregulins are members of the epidermal growth factor family that are ligands for receptor tyrosine kinases of the erbB family. There are four neuregulins: neuregulin-1, -2, -3, and -4. During early cardiogenesis, endocardial endothelial cells produce neuregulin-1, and primary cardiomyocytes express the receptors erbB2 and erbB4. Paracrine signaling of the neuregulin-erbB2/4 system from endocardium to myocardium is essential for the formation of myocardial trabeculae and cardiac cushions (8). Mice lacking neuregulin or its receptors, erbB2 or erbB4, die in mid-embryogenesis because they lack myocardial trabeculae (9-11). Although the process of trabeculation is not well understood at the molecular level, cardiomyocyte proliferation and maturation are required. Neuregulin-1 can promote the proliferation, survival, and hypertrophic growth of cultured neonatal cardiomyocytes (12), and this may explain why this growth factor is essential for normal trabeculation.
NEUROFIBROMATOSIS TYPE 1
Another factor that can mediate endothelial-cardiomyocyte interactions during early cardiac development is neurofibromatosis type 1 (NF1). NF1 is a tumor-suppressor gene that downregulates Ras activity, and mutations in NF1 in humans may cause cardiovascular defects (13). In mice, conditional inactivation of NF1 in endothelial cells using the Cre-lox system causes cardiac developmental defects in the myocardium and endocardial cushions (14). In addition to causing myocardial thinning, endothelial-specific inactivation of NF1 leads to enlarged endocardial cushions, ventricular septal defects, and double-outlet right ventricle. Similar defects are present in Nf1-/- mice, but not in mice with cardiomyocyte-specific NF1 inactivation (14), demonstrating that NF1 signaling from endothelial cells causes defects in myocardial development.
PLATELET-DERIVED GROWTH FACTOR-B
Platelet-derived growth factor (PDGF)-B and PDGF receptor-β are essential for mammalian cardiovascular development (15). During vasculogenesis, endothelial-derived PDGF-B recruits nearby pericytes to the nascent vascular structure (15); similarly, endothelial-derived PDGF-B may provide key signals to cardiomyocytes during development. Complete deletion of PDGF-B is lethal, whereas endothelial-restricted deletion of PDGF-B causes defects in the developing myocardium as well as vascular and glomerular abnormalities (16). Such cardiac defects include thinned myocardium, chamber dilation, hypertrabeculation, and septal abnormalities. Interestingly, thinning of the myocardium is no longer apparent at one month postnatally with endothelial-restricted deletion of PDGF-B. The Cre-lox endothelial-specific ablation method is only 60-90% efficient, which suggests either that postnatal PDGF-B from the minority of cells that did not delete the PDGF-B gene is sufficient to allow cardiomyocyte growth to compensate postnatally or that other growth factors drive this compensation (16).
Signaling from Cardiomyocytes to Endothelial Cells in Development
The examples of neuregulin-1, NF1, and PDGF-B demonstrate that signals from endothelial cells regulate the formation of primary myocardium. Similarly, signaling from myocardial cells to endothelial cells is also required for cardiac development. Two examples of myocardial-to-endothelial signaling are vascular endothelial growth factor (VEGF)-A and angiopoietin-1.
VASCULAR ENDOTHELIAL GROWTH FACTOR-A
VEGF-A is a key regulator of angiogenesis during embryogenesis. In mice, a mutation in VEGF-A causes endocardial detachment from an underdeveloped myocardium (17). A mutation in VEGF receptor-2 (or Flk-1) also results in failure of the endocardium and myocardium to develop (18). Furthermore, cardiomyocyte-specific deletion of VEGF-A results in defects in vasculogenesis/angiogenesis and a thinned ventricular wall (19), further confirming reciprocal signaling from the myocardial cell to the endothelial cell during cardiac development. Interestingly, this cardiomyocyte-selective VEGF-A-deletion mouse has underdeveloped myocardial microvasculature but preserved coronary artery structure, implying a different signaling mechanism for vasculogenesis/angiogenesis in the myocardium and in the epicardial coronary arteries.
Cardiomyocyte-derived VEGF-A also inhibits cardiac endocardial-to-mesenchymal transformation. This process is essential in the formation of the cardiac cushions and requires delicate control of VEGF-A concentration (20-22). A minimal amount of VEGF initiates endocardial-to-mesenchymal transformation, whereas higher doses of VEGF-A terminate this transformation (23). Interestingly, this cardiomyocyte-derived VEGF-A signaling for endocardial-to-mesenchymal transformation may be controlled by an endothelial-derived feedback mechanism through the calcineurin/NFAT pathway (24), demonstrating the importance of endothelial-cardiomyocyte interactions for cardiac morphogenesis.
ANGIOPOIETIN-1
Another mechanism of cardiomyocyte control of endothelial cells during cardiac development is the angiopoietin-Tie-2 system. Both angiopoietin-1 and angiopoietin-2 may bind to Tie-2 receptors in a competitive manner, but with opposite effects: Angiopoietin-1 activates the Tie-2 receptor and prevents vascular edema, whereas angiopoietin-2 blocks Tie-2 phosphorylation and increases vascular permeability. During angiogenesis/vasculogenesis, angiopoietin-1 is produced primarily by pericytes, and Tie-2 receptors are expressed on endothelial cells. Angiopoietin-1 regulates the stabilization and maturation of neovasculature; genetic deletion of angiopoietin-1 or Tie-2 causes a defect in early vasculogenesis/angiogenesis and is lethal (25, 26). Because in the early embryo the cardiac endocardium is one of the earliest vascular components (along with the dorsal aorta and yolk sac vessels) and the adult heart can be regarded as a fully vascularized organ, angiopoietin-Tie-2 signaling may also be required for early cardiac development. Indeed, mice with mutations in Tie-2 have underdeveloped endocardium and myocardium (27). These Tie-2 knockout mice display defects in the endocardium but have normal vascular morphology at E10.5, suggesting that the endocardial defect is the fundamental cause of death. In addition, a recent study showed that overexpression, and not deletion, of angiopoietin-1 from cardiomyocytes caused embryonic death between E12.5-15.5 due to cardiac hemorrhage (28). The mice had defects in the endocardium and myocardium and lack of coronary arteries, suggesting that, as with VEGF-A, a delicate control of angiopoietin-1 concentration is critical for early heart development.
ENDOTHELIAL-CARDIOMYOCYTE INTERACTIONS IN NORMAL CARDIAC FUNCTION
Cardiac Endothelial Cells Regulate Cardiomyocyte Contraction
The vascular endothelium senses the shear stress of flowing blood and regulates vascular smooth muscle contraction. It is therefore not surprising that cardiac endothelial cells—the endocardial endothelial cells as well as the endothelial cells of intramyocardial capillaries—regulate the contractile state of cardiomyocytes. Autocrine and paracrine signaling molecules released or activated by cardiac endothelial cells are responsible for this contractile response (Figure 2).
NITRIC OXIDE
Three different nitric oxide synthase isoenzymes synthesize nitric oxide (NO) from l-arginine. The neuronal and endothelial NO synthases (nNOS and eNOS, respectively) are expressed in normal physiological conditions, whereas the inducible NO synthase is induced by stress or cytokines. Like NO in the vessel, which causes relaxation of vascular smooth muscle, NO in the heart affects the onset of ventricular relaxation, which allows for a precise optimization of pump function beat by beat. Although NO is principally a paracrine effector secreted by cardiac endothelial cells, cardiomyocytes also express both nNOS and eNOS. Endothelial expression of eNOS exceeds that in cardiomyocytes by greater than 4:1 (29). Cardiomyocyte autocrine eNOS signaling can regulate β-adrenergic and muscarinic control of contractile state (30).
Barouch et al. (31) demonstrated that cardiomyocyte nNOS and eNOS may have opposing effects on cardiac structure and function. Using mice with nNOS or eNOS deficiency, they found that nNOS and eNOS have not only different localization in cardiomyocytes but also opposite effects on cardiomyocyte contractility; eNOS localizes to caveolae and inhibits l-type Ca2+ channels, leading to negative inotropy, whereas nNOS is targeted to the sarcoplasmic reticulum and facilitates Ca2+ release and thus positive inotropy (31). These results demonstrate that spatial confinement of different NO synthase isoforms contribute independently to the maintenance of cardiomyocyte structure and phenotype.
As indicated above, mutation of neuregulin or either of two of its cognate receptors, erbB2 and erbB4, causes embryonic death during mid-embryogenesis due to aborted development of myocardial trabeculation (9-11). Neuregulin also appears to play a role in fully developed myocardium. In adult mice, cardiomyocyte-specific deletion of erbB2 leads to dilated cardiomyopathy (32, 33). Neuregulin from endothelial cells may induce a negative inotropic effect in isolated rabbit papillary muscles (34). This suggests that, along with NO, the neuregulin signaling pathway acts as an endothelial-derived regulator of cardiac inotropism. In fact, the negative inotropic effect of neuregulin may require NO synthase because l-NMMA, an inhibitor of NO synthase, significantly attenuates the negative inotropy of neuregulin (34).
ENDOTHELIN-1
Endothelin (ET)-1 is a 21-amino acid peptide produced and released by the endothelial cells with a direct vasoconstrictive effect. In the myocardium, endothelin-1 may act in both autocrine and paracrine manners; it binds to ETB receptors on cardiac endothelial cells and ETA receptors on cardiomyocytes. When endothelin-1 binds to ETB receptors, it results in the release of other signaling molecules (NO and prostaglandin I2) rather than vasoconstriction. By contrast, when binding to the ETA receptors on cardiomyocytes, endothelin-1 causes cardiomyocyte constriction, as observed when vascular smooth muscle cells are treated with endothelin-1 (35). These results imply that there may be a feedback mechanism between cardiac endothelial cells and cardiomyocytes for control of cardiomyocyte constriction through the endothelin-1 system.
There is now considerable evidence showing a role of the endothelin-1 system in the pathogenesis and progression of heart failure. Patients with heart failure exhibit increased plasma endothelin-1 levels and expression of myocardial endothelin receptors, and the extent of the increases correlate with disease severity (36). Indeed, it has been hypothesized that endothelin receptor antagonists may be a therapeutic approach for heart failure. In a double-blinded study, darusentan, an ETA-receptor antagonist, improved cardiac index in patients with heart failure (37). However, in other clinical trials endothelin antagonists resulted in no benefit to or worsening of patient condition (38, 39).
ENDOTHELIAL-CARDIOMYOCYTE INTERACTIONS AND REGENERATION
Regeneration Occurs in Myocardium But Is Inadequate for Functional Improvement
In some nonmammalian higher organisms like amphibians and zebrafish, regeneration of the myocardium occurs after experimental myocardial injury (40, 41). In humans, however, cardiomyocytes that die following ischemia are not adequately replaced, resulting in loss of ventricular function (42). Recent evidence indicates that the adult heart is capable of some limited regeneration (43) and that adult mouse cardiomyocytes may be able to proliferate under specific conditions (44). The origins of regenerating cardiomyocytes are controversial. Studies have shown the presence of Y chromosome-positive bone marrow-derived cardiomyocytes in adult female hearts (45), indicating the possibility that bone marrow-derived precursors differentiate into cardiomyocytes or fuse with existing cardiomyocytes. Other studies suggest that resident cardiac stem cells are the source of regenerating myocardium (43, 46-48).
In part owing to the lack of understanding of how myocardium regenerates, many different cell types have been injected or infused into injured myocardium in an attempt to stimulate myocardial regeneration. Surprisingly, several different cell types have demonstrated the potential for benefit, including skeletal myoblasts, adult and neonatal cardiomyocytes, bone marrow stem cells, and embryonic stem cells (49). In some circumstances, there is poor survival of injected cells or incomplete differentiation of the implanted cells, and in others, lack of integration into the host myocardium (50). Bone marrow-derived progenitor cells have been shown to assist in vascularization of the myocardium following infarction and to improve cardiomyocyte survival (51, 52), although their capacity to transdifferentiate into cardiomyocytes is probably very limited (53).
Why does experimental cell therapy appear to benefit left ventricular function, even though most cells do not survive and even fewer appear to integrate functionally into the myocardium? There are many potential explanations (Figure 3), but one plausible theory is that injected cells stimulate angiogenesis, either as living cells or as they die. As discussed further below, endothelial cells can promote the survival of cardiomyocytes. Many types of injected cells, including endothelial progenitor cells, are rich sources of angiogenic factors (54). The potential for cell therapy to be an expensive method of introducing angiogenic factors suggests that similar benefits may be derived from injection of individual angiogenic factors alone, although the success of this approach to date has been limited (55).
Figure 3.
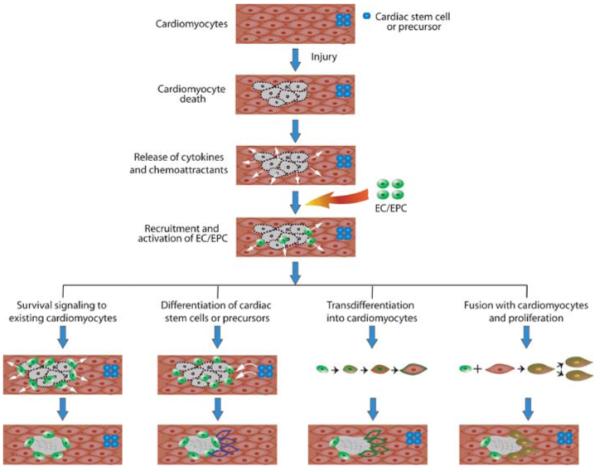
Mechanisms of myocardial regeneration through endothelial and endothelial progenitor cell therapies. After injury to a site, cardiomyocytes undergo apoptosis and necrosis and release cytokines and chemoattractants to recruit endothelial cells (EC) and endothelial progenitor cells (EPC) to the site. EC/EPC are activated and may promote myocardial regeneration through different mechanisms, including (a) releasing cardiomyocyte survival factors to protect fragile cardiomyocytes, (b) promoting differentiation of resident cardiac stem cells or progenitors for population into the injured area, (c) undergoing transdifferentiation into cardiomyocytes to replace dead cells, and (d) fusing with cardiomyocytes to facilitate cell proliferation for repair.
Endothelial Cells Promote Cardiomyocyte Survival
The theory that angiogenic stimulation can improve systolic function is supported by recent studies demonstrating that endothelial cells can promote cardiomyocyte survival. Kuramochi et al. (56) tested the hypothesis that reactive oxygen species regulate neuregulin-erbB signaling. They found that neuregulin is a prosurvival factor for cardiomyocytes via the phosphatidylinositol-3-kinase-Akt pathway. Cardiac microvascular endothelial cells express neuregulin, and recombinant neuregulin-1β protects cardiomyocytes against anthracycline-, β-adrenergic receptor-, and H2O2-induced cardiomyocyte death (56-58). H2O2 induces neuregulin-1β release from endothelial cells in a concentration-dependent manner and conditioned medium from the cells activates erbB4 signaling in cardiomyocytes via paracrine mechanisms (56). Cocultured cardiac endothelial cells protect H2O2-induced cardiomyocyte apoptosis through neuregulin-erbB4 signaling.
Cardiomyocytes are also protected from apoptosis by endothelial cells in three-dimensional culture (59). In this setting, there is no blood flow to consider, and the effects of endothelial cells are thus due to secreted factors or to cell-cell contact. Preliminary data from our laboratory suggest that endothelial cells, when in contact with cardiomyocytes, secrete PDGF-B, which protects cardiomyocytes through phosphatidylinositol-3-kinase-Akt signaling (P.C.H. Hsieh, unpublished data). Interestingly, PDGF mediates cardiac microvascular endothelial cell hemostatic and angiogenic activity (60), and injection of PDGF may decrease the extent of myocardial infarction after coronary occlusion in young, but not in old, rat hearts (61, 62).
In addition to neuregulin and PDGF-B, another candidate endothelial protein mediating cardiomyocyte survival is angiopoietin-1. As described above, mice with genetic deletion of angiopoietin-1 die during development because of cardiac defects (26). These defects are thought to be due to impairment in endothelial functions such as vascular maturation and endocardial formation. Angiopoietin-1 not only promotes endothelial cell survival but can also improve cell survival in skeletal myocytes and cardiomyocytes (63). Furthermore, adenoviral overexpression of angiopoetin-1 reduces infarct size in experimental myocardial infarction (64).
Endothelial Cells Guide Cardiomyocyte Organization
During vasculogenesis, endothelial cells recruit mural cells to the outside of the growing tube, where they adhere and differentiate to form the media of the new vessel. Endothelial cells can also recruit cardiomyocytes in a similar fashion. When cultured in a three-dimensional scaffold, endothelial cells form typical vascular networks with capillary-like tubes, whereas primary cardiomyocytes form small islands of cells that die. When endothelial cells and cardiomyocytes are cultured together, however, the endothelial cells form tube-like structures and the cardiomyocytes position themselves on the outside of these tubes (59). These data suggest that an endothelial factor directs the assembly of cardiomyocytes on the capillary-like endothelial tube. Furthermore, endothelial cells promote the synthesis of Connexin43, a principal gap junction protein of cardiomyocytes. In the presence of endothelial cells, cardiomyocyte contraction is more coordinated, suggesting that the physiological coupling of cardiomyocytes is, in part, endothelial dependent. These data raise the intriguing hypothesis that achieving proper capillarycardiomyocyte architecture in cardiac repair may provide electrical stability in addition to mechanical functional improvement.
Endothelial Cells Transdifferentiatate into Cardiomyocytes
Whereas endothelial cells may direct differentiation, survival, and organization of cardiomyocytes, some endothelial progenitor cells may have the capacity to transdifferentiate into cardiomyocytes. Condorelli et al. (65) reported that various types of endothelial cells are capable of expressing putative cardiomyocyte markers when cultured with cardiomyocytes. Surprisingly, this transdifferentiation may occur in both adult committed endothelial cells from different tissues as well as in putative endothelial progenitor cells (65-67). Transdifferentiation may require direct cell-cell contact between the endothelial progenitor cell and the cardiomyocyte (66, 68). Because in vivo studies suggest that transdifferentiation into cardiomyocytes is very rare (53), many experimental studies may show transdifferentiation because of cell fusion, which can explain the apparent phenotypic change. Indeed, injection of bone marrow-derived cells into the myocardium can cause fusion with cardiomyocytes without transdifferentiation into cardiomyocytes (69). Interestingly, fused cardiomyocytes express cardiac troponin-I and intercalate with neighboring cardiomyocytes with mature gap junction formation, implying a true potential mechanism for cardiac regeneration. Moreover, a recent study showed that endothelial and endothelial progenitor cells can fuse with cardiomyocytes in vitro and in vivo (70). The endothelial-cardiomyocyte fused cells express predominantly cardiomyocyte markers and, surprisingly, also express Ki67, phosphohistone H3, and cyclinB1 and reenter the G2/M cell cycle. Although direct cardiomyocyte division was not observed after fusion, this study suggests that augmented cell fusion with endothelial cells may rescue injured cardiomyocytes.
Clinical Approaches Using Endothelial Cells for Cardiac Regeneration
To date, most studies using endothelial cells have aimed to enhance the endogenous postinfarct angiogenic response with growth factors or to improve angiogenesis with endothelial progenitor cell therapy. It is worth noting that injured myocardium is itself angiogenic initially and that native cardiomyocytes respond to hypoxia and other stress signals with the release of angiogenic factors, including VEGF (71). In humans, angiogenic factors are increased in the plasma and the myocardium after acute myocardial ischemia and infarction (72, 73). Thus, with therapeutic angiogenesis it generally is assumed that increasing angiogenic stimuli beyond the normal response to injury is beneficial. This is not always a good assumption, however, because excessive delivery of angiogenic factors can lead to aberrant vasculature and hemangiomas (74).
Ex vivo-expanded endothelial progenitor cells have shown promise in animal studies, improving cardiac function following myocardial infarction (51, 75, 76). This has led to several clinical trials including the TOPCARE-AMI (77) and MAGIC (78) trials. These studies used either endothelial progenitors or peripheral blood mononuclear cells for repair of the damaged myocardium (TOPCARE-AMI) or the progenitor-mobilizing factor granulocyte colony-stimulating factor (in MAGIC). The use of granulocyte colony-stimulating factor was associated with early restenosis within stented coronary arteries, suggesting that systemic mobilization of endothelial precursors may have adverse effects. It is far too early to draw conclusions from myocardial angiogenesis and endothelial cell therapy trials. It is possible that early attempts to overdrive angiogenesis will fail, whereas carefully timed delivery of angiogenic factors or endothelial precursors can successfully protect cardiomyocytes or even promote regeneration and functional integration of new cardiomyocytes. Future studies that define precisely how endothelial cells protect and stimulate cardiomyocytes may allow simple and practical ways to prevent heart failure.
CONCLUSIONS
Studies to date indicate that cardiac regeneration in mammals may be feasible, but the response is inadequate to preserve myocardial function after a substantial injury. Thus, understanding how normal myocardial structure can be regenerated in adult hearts is essential. It is clear that endothelial cells play a role in cardiac morphogenesis and most likely also in survival and function of mature cardiomyocytes. Initial attempts to promote angiogenesis in myocardium were based on the premise that persistent ischemia could be alleviated. However, it is also possible that endothelial-cardiomyocyte interactions are essential in normal cardiomyocyte function and for protection from injury. Understanding the molecular and cellular mechanisms controlling these cell-cell interactions will not only enhance our understanding of the establishment of vascular network in the heart but also allow the development of new targeted therapies for cardiac regeneration by improving cardiomyocyte survival and maturation.
LITERATURE CITED
- 1.Cleaver O, Melton DA. Endothelial signaling during development. Nat. Med. 2003;9:661–68. doi: 10.1038/nm0603-661. [DOI] [PubMed] [Google Scholar]
- 2.Brutsaert DL. Cardiac endothelialmyocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 2003;83:59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 3.Korecky B, Hai CM, Rakusan K. Functional capillary density in normal and transplanted rat hearts. Can. J. Physiol. Pharmacol. 1982;60:23–32. doi: 10.1139/y82-003. [DOI] [PubMed] [Google Scholar]
- 4.Lough J, Sugi Y. Endoderm and heart development. Dev. Dyn. 2000;217:327–42. doi: 10.1002/(SICI)1097-0177(200004)217:4<327::AID-DVDY1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 5.Harvey RP. Patterning the vertebrate heart. Nat. Rev. Genet. 2002;3:544–56. doi: 10.1038/nrg843. [DOI] [PubMed] [Google Scholar]
- 6.Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol. Rev. 2003;83:1223–67. doi: 10.1152/physrev.00006.2003. [DOI] [PubMed] [Google Scholar]
- 7.Linask KK, Lash JW. Early heart development: dynamics of endocardial cell sorting suggests a common origin with cardiomyocytes. Dev. Dyn. 1993;196:62–69. doi: 10.1002/aja.1001960108. [DOI] [PubMed] [Google Scholar]
- 8.Marchionni MA. Cell-cell signalling: neu tack on neuregulin. Nature. 1995;378:334–35. doi: 10.1038/378334a0. [DOI] [PubMed] [Google Scholar]
- 9.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–90. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 10.Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, et al. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–94. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- 11.Lee KF, Simon H, Chen H, Bates B, Hung MC, et al. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378:394–98. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- 12.Zhao YY, Sawyer DR, Baliga RR, Opel DJ, Han X, et al. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J. Biol. Chem. 1998;273:10261–69. doi: 10.1074/jbc.273.17.10261. [DOI] [PubMed] [Google Scholar]
- 13.Lin AE, Birch PH, Korf BR, Tenconi R, Niimura M, et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am. J. Med. Genet. 2000;95:108–17. doi: 10.1002/1096-8628(20001113)95:2<108::aid-ajmg4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 14.Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, et al. Nf1 has an essential role in endothelial cells. Nat. Genet. 2003;33:75–79. doi: 10.1038/ng1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoch RV, Soriano P. Roles of PDGF in animal development. Development. 2003;130:4769–84. doi: 10.1242/dev.00721. [DOI] [PubMed] [Google Scholar]
- 16.Bjarnegard M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, et al. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131:1847–57. doi: 10.1242/dev.01080. [DOI] [PubMed] [Google Scholar]
- 17.Haigh JJ, Gerber HP, Ferrara N, Wagner EF. Conditional inactivation of VEGF-A in areas of collagen2a1 expression results in embryonic lethality in the heterozygous state. Development. 2000;127:1445–53. doi: 10.1242/dev.127.7.1445. [DOI] [PubMed] [Google Scholar]
- 18.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 19.Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, et al. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc. Natl. Acad. Sci. USA. 2001;98:5780–85. doi: 10.1073/pnas.091415198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–46. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- 21.Dor Y, Camenisch TD, Itin A, Fishman GI, McDonald JA, et al. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 2001;128:1531–38. doi: 10.1242/dev.128.9.1531. [DOI] [PubMed] [Google Scholar]
- 22.Dor Y, Klewer SE, McDonald JA, Keshet E, Camenisch TD. VEGF modulates early heart valve formation. Anat. Rec. 2003;271:202–8. doi: 10.1002/ar.a.10026. [DOI] [PubMed] [Google Scholar]
- 23.Lambrechts D, Carmeliet P. Sculpting heart valves with NFATc and VEGF. Cell. 2004;118:532–34. doi: 10.1016/j.cell.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 24.Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, et al. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–63. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 26.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–80. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 27.Puri MC, Partanen J, Rossant J, Bernstein A. Interaction of the TEK and TIE receptor tyrosine kinases during cardiovascular development. Development. 1999;126:4569–80. doi: 10.1242/dev.126.20.4569. [DOI] [PubMed] [Google Scholar]
- 28.Ward NL, Van Slyke P, Sturk C, Cruz M, Dumont DJ. Angiopoietin 1 expression levels in the myocardium direct coronary vessel development. Dev. Dyn. 2004;229:500–9. doi: 10.1002/dvdy.10479. [DOI] [PubMed] [Google Scholar]
- 29.Godecke A, Heinicke T, Kamkin A, Kiseleva I, Strasser RH, et al. Inotropic response to β-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. J. Physiol. 2001;532:195–204. doi: 10.1111/j.1469-7793.2001.0195g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Champion HC, Georgakopoulos D, Takimoto E, Isoda T, Wang Y, et al. Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circ. Res. 2004;94:657–63. doi: 10.1161/01.RES.0000119323.79644.20. [DOI] [PubMed] [Google Scholar]
- 31.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–39. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 32.Ozcelik C, Erdmann B, Pilz B, Wettschureck N, Britsch S, et al. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA. 2002;99:8880–85. doi: 10.1073/pnas.122249299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 2002;8:459–65. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 34.Lemmens K, Fransen P, Sys SU, Brutsaert DL, DeKeulenaer GW. Neuregulin-1 induces a negative inotropic effect in cardiac muscle: role of nitric oxide synthase. Circulation. 2004;109:324–26. doi: 10.1161/01.CIR.0000114521.88547.5E. [DOI] [PubMed] [Google Scholar]
- 35.Rich S, McLaughlin VV. Endothelin receptor blockers in cardiovascular disease. Circulation. 2003;108:2184–90. doi: 10.1161/01.CIR.0000094397.19932.78. [DOI] [PubMed] [Google Scholar]
- 36.Zolk O, Quattek J, Sitzler G, Schrader T, Nickenig G, et al. Expression of endothelin-1, endothelin-converting enzyme, and endothelin receptors in chronic heart failure. Circulation. 1999;99:2118–23. doi: 10.1161/01.cir.99.16.2118. [DOI] [PubMed] [Google Scholar]
- 37.Luscher TF, Enseleit F, Pacher R, Mitrovic V, Schulze MR, et al. Hemodynamic and neurohumoral effects of selective endothelin A (ET(A)) receptor blockade in chronic heart failure: the Heart Failure ET(A) Receptor Blockade Trial (HEAT) Circulation. 2002;106:2666–72. doi: 10.1161/01.cir.0000038497.80095.e1. [DOI] [PubMed] [Google Scholar]
- 38.O’Connor CM, Gattis WA, Adams KF, Jr, Hasselblad V, Chandler B, et al. Tezosentan in patients with acute heart failure and acute coronary syndromes: results of the Randomized Intravenous TeZosentan Study (RITZ-4) J. Am. Coll. Cardiol. 2003;41:1452–55. doi: 10.1016/s0735-1097(03)00194-3. [DOI] [PubMed] [Google Scholar]
- 39.Packer M, McMurray J, Massie BM, Caspi A, Charlon V, et al. Clinical effects of endothelin receptor antagonism with bosentan in patients with severe chronic heart failure: results of a pilot study. J. Card. Fail. 2005;11:12–20. doi: 10.1016/j.cardfail.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Becker RO, Chapin S, Sherry R. Regeneration of the ventricular myocardium in amphibians. Nature. 1974;248:145–47. doi: 10.1038/248145a0. [DOI] [PubMed] [Google Scholar]
- 41.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–90. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 42.Anversa P. Myocyte death in the pathological heart. Circ. Res. 2000;86:121–24. doi: 10.1161/01.res.86.2.121. [DOI] [PubMed] [Google Scholar]
- 43.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 44.Engel FB, Schebesta M, Duong MT, Lu G, Ren SX, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–87. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deb A, Wang S, Skelding KA, Miller D, Simper D, et al. Bone marrow-derived cardiomyocytes are present in adult human heart: A study of gendermismatched bone marrow transplantation patients. Circulation. 2003;107:1247–49. doi: 10.1161/01.cir.0000061910.39145.f0. [DOI] [PubMed] [Google Scholar]
- 46.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. USA. 2003;100:12313–18. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laugwitz KL, Moretti A, Lam J, Gruber P, Chen YH, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–53. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenblatt-Velin N, Lepore MG, Cartoni C, Beermann F, Pedrazzini T. FGF-2 controls the differentiation of resident cardiac precursors into functional cardiomyocytes. J. Clin. Invest. 2005;115:1724–33. doi: 10.1172/JCI23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reffelmann T, Kloner RA. Cellular cardiomyoplasty: cardiomyocytes, skeletal myoblasts, or stem cells for regenerating myocardium and treatment of heart failure? Cardiovasc. Res. 2003;58:358–68. doi: 10.1016/s0008-6363(02)00739-3. [DOI] [PubMed] [Google Scholar]
- 50.Zimmermann WH, Schneiderbanger K, Schubert P, Didie M, Munzel F, et al. Tissue engineering of a differentiated cardiac muscle construct. Circ. Res. 2002;90:223–30. doi: 10.1161/hh0202.103644. [DOI] [PubMed] [Google Scholar]
- 51.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, et al. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 2001;7:430–36. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 52.Itescu S, Kocher AA, Schuster MD. Myocardial neovascularization by adult bone marrow-derived angioblasts: strategies for improvement of cardiomyocyte function. Heart Fail. Rev. 2003;8:253–58. doi: 10.1023/a:1024721717926. [DOI] [PubMed] [Google Scholar]
- 53.Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004;428:664–68. doi: 10.1038/nature02446. [DOI] [PubMed] [Google Scholar]
- 54.Rehman J, Li JL, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. 2003;107:1164–69. doi: 10.1161/01.cir.0000058702.69484.a0. [DOI] [PubMed] [Google Scholar]
- 55.Annex BH, Simons M. Growth factor-induced therapeutic angiogenesis in the heart: protein therapy. Cardiovasc. Res. 2005;65:649–55. doi: 10.1016/j.cardiores.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 56.Kuramochi Y, Cote GM, Guo X, Lebrasseur NK, Cui L, et al. Cardiac endothelial cells regulate reactive oxygen species-induced cardiomyocyte apoptosis through neuregulin-1β/erbB4 signaling. J. Biol. Chem. 2004;279:51141–47. doi: 10.1074/jbc.M408662200. [DOI] [PubMed] [Google Scholar]
- 57.Fukazawa R, Miller TA, Kuramochi Y, Frantz S, Kim YD, et al. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J. Mol. Cell. Cardiol. 2003;35:1473–79. doi: 10.1016/j.yjmcc.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 58.Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, et al. β-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ. Res. 2003;92:136–38. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- 59.Narmoneva D, Davis ME, Vukmirovich R, Kamm RD, Lee RT. Endothelial cells promote cardiac myocyte survival and spatial reorganization: implications for cardiac regeneration. Circulation. 2004;110:962–68. doi: 10.1161/01.CIR.0000140667.37070.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edelberg JM, Aird WC, Wu W, Rayburn H, Mamuya WS, et al. PDGF mediates cardiac microvascular communication. J. Clin. Invest. 1998;102:837–43. doi: 10.1172/JCI3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edelberg JM, Lee SH, Kaur M, Tang L, Feirt NM, et al. Platelet-derived growth factor-AB limits the extent of myocardial infarction in a rat model: feasibility of restoring impaired angiogenic capacity in the aging heart. Circulation. 2002;105:608–13. doi: 10.1161/hc0502.103672. [DOI] [PubMed] [Google Scholar]
- 62.Xaymardan M, Zheng JG, Duignan I, Chin A, Holm JM, et al. Senescent impairment in synergistic cytokine pathways that provide rapid cardioprotection in the rat heart. J. Exp. Med. 2004;199:797–804. doi: 10.1084/jem.20031639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dallabrida SM, Ismail N, Oberle JR, Himes BE, Rupnick MA. Angiopoietin-1 promotes cardiac and skeletal myocyte survival through integrins. Circ. Res. 2005;96:e8–24. doi: 10.1161/01.RES.0000158285.57191.60. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi K, Ito Y, Morikawa M, Kobune M, Huang J, et al. Adenoviraldelivered angiopoietin-1 reduces the infarction and attenuates the progression of cardiac dysfunction in the rat model of acute myocardial infarction. Mol. Ther. 2003;8:584–92. doi: 10.1016/s1525-0016(03)00230-2. [DOI] [PubMed] [Google Scholar]
- 65.Condorelli G, Borello U, De Angelis L, Latronico M, Sirabella D, et al. Cardiomyocytes induce endothelial cells to trans-differentiate into cardiac muscle: implications for myocardium regeneration. Proc. Natl. Acad. Sci. USA. 2001;98:10733–38. doi: 10.1073/pnas.191217898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Badorff C, Brandes RP, Popp R, Rupp S, Urbich C, et al. Transdifferentiation of blood-derived human adult endothelial progenitor cells into functionally active cardiomyocytes. Circulation. 2003;107:1024–32. doi: 10.1161/01.cir.0000051460.85800.bb. [DOI] [PubMed] [Google Scholar]
- 67.Yeh ET, Zhang S, Wu HD, Korbling M, Willerson JT, et al. Transdifferentiation of human peripheral blood CD34+-enriched cell population into cardiomyocytes, endothelial cells, and smooth muscle cells in vivo. Circulation. 2003;108:2070–73. doi: 10.1161/01.CIR.0000099501.52718.70. [DOI] [PubMed] [Google Scholar]
- 68.Koyanagi M, Brandes RP, Haendeler J, Zeiher AM, Dimmeler S. Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: a novel mechanism for cell fate changes? Circ. Res. 2005;96:1039–41. doi: 10.1161/01.RES.0000168650.23479.0c. [DOI] [PubMed] [Google Scholar]
- 69.Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, et al. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–73. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- 70.Matsuura K, Wada H, Nagai T, Iijima Y, Minamino T, et al. Cardiomyocytes fuse with surrounding noncardiomyocytes and reenter the cell cycle. J. Cell Biol. 2004;167:351–63. doi: 10.1083/jcb.200312111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yue X, Tomanek RJ. Stimulation of coronary vasculogenesis/angiogenesis by hypoxia in cultured embryonic hearts. Dev. Dyn. 1999;216:28–36. doi: 10.1002/(SICI)1097-0177(199909)216:1<28::AID-DVDY5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 72.Lee KW, Lip GY, Blann AD. Plasma angiopoietin-1, angiopoietin-2, angiopoietin receptor tie-2, and vascular endothelial growth factor levels in acute coronary syndromes. Circulation. 2004;110:2355–60. doi: 10.1161/01.CIR.0000138112.90641.7F. [DOI] [PubMed] [Google Scholar]
- 73.Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, et al. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N. Engl. J. Med. 2000;342:626–33. doi: 10.1056/NEJM200003023420904. [DOI] [PubMed] [Google Scholar]
- 74.Lee RJ, Springer ML, Blanco-Bose WE, Shaw R, Ursell PC, et al. VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation. 2000;102:898–901. doi: 10.1161/01.cir.102.8.898. [DOI] [PubMed] [Google Scholar]
- 75.Kawamoto A, Gwon HC, Iwaguro H, Yamaguchi JI, Uchida S, et al. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation. 2001;103:634–37. doi: 10.1161/01.cir.103.5.634. [DOI] [PubMed] [Google Scholar]
- 76.Kawamoto A, Tkebuchava T, Yamaguchi J, Nishimura H, Yoon YS, et al. Intramyocardial transplantation of autologous endothelial progenitor cells for therapeutic neovascularization of myocardial ischemia. Circulation. 2003;107:461–68. doi: 10.1161/01.cir.0000046450.89986.50. [DOI] [PubMed] [Google Scholar]
- 77.Assmus B, Schachinger V, Teupe C, Britten M, Lehmann R, et al. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE-AMI) Circulation. 2002;106:3009–17. doi: 10.1161/01.cir.0000043246.74879.cd. [DOI] [PubMed] [Google Scholar]
- 78.Kang HJ, Kim HS, Zhang SY, Park KW, Cho HJ, et al. Effects of intracoronary infusion of peripheral blood stemcells mobilised with granulocyte-colony stimulating factor on left ventricular systolic function and restenosis after coronary stenting in myocardial infarction: the MAGIC cell randomised clinical trial. Lancet. 2004;363:751–56. doi: 10.1016/S0140-6736(04)15689-4. [DOI] [PubMed] [Google Scholar]