


Abstract
We show here that under physiologically reasonable conditions, CGG repeats in RNA readily form hairpins. In contrast to its DNA counterpart that forms a complex mixture of hairpins and tetraplexes, r(CGG)22 forms a single stable hairpin with no evidence for any other folded structure even at low pH. RNA with the sequence (CGG)9AGG (CGG)12AGG(CGG)97, found in a fragile X syndrome pre-mutation allele, forms a number of different hairpins. The most prominent hairpin forms in the 3′ part of the repeat and involves the 97 uninterrupted CGG repeats. In contrast to the CUG-RNA hairpins formed by myotonic dystrophy type 1 repeats, we found no evidence that CGG-RNA hairpins activate PKR, the interferon-inducible protein kinase that is activated by a wide range of double-stranded RNAs. However, we do show that the CGG-RNA is digested, albeit inefficiently, by the human Dicer enzyme, a step central to the RNA interference effect on gene expression. These data provide clues to the basis of the toxic effect of CGG-RNA that is thought to occur in fragile X pre-mutation carriers. In addition, RNA hairpins may also account for the stalling of the 40S ribosomal subunit that is thought to contribute to the translation deficit in fragile X pre-mutation and full mutation alleles.
INTRODUCTION
Fragile X mental retardation syndrome (FXS) is caused by the expansion of a CGG·CCG repeat tract in the 5′-untranslated region (5′ UTR) of the FMR1 gene (1,2). This expansion occurs from a pre-mutation allele containing 55–200 repeats to one that has a much larger number of repeats. In addition to the risk of having a child with a full mutation allele, female carriers of pre-mutation alleles have elevated levels of premature ovarian failure (3,4). A high incidence of cerebellar degeneration is also seen in both female and male carriers (5). These symptoms are thought to reflect some property of the transcript from pre-mutation alleles rather than insufficiency of the protein product of the FMR1 gene.
A number of other repeat expansion diseases are known where the repeat-containing RNA has been implicated in disease pathology. The best characterized of these is myotonic dystrophy (DM) type 1 which is caused by expansion of a CTG·CAG tract in the 3′ UTR of the myotonic dystrophy protein kinase gene, DMPK [for recent review see Ranum and Day (6)]. Both CTG repeats and, more recently, their RNA counterparts the CUG repeats, have been shown to form hairpins (7,8). DM RNA hairpins have been suggested to cause pathology in a variety of ways including via activation of enzymes such as the interferon-inducible protein kinase PKR that leads to inhibition of translation and ultimately to apoptosis (9).
While the DNA structure of long FXS pre-mutation alleles has not been described, DNAs with CGG repeat numbers in the normal range are known to form a complex mixture of folded structures including hairpins (10,11) and tetraplexes (12–16). We reasoned that RNA with a large number of CGG repeats might form a similar array of structures that, as is the case in DM1, may have consequences for disease pathology. We describe here our analysis of the structures formed by RNA corresponding to a normal allele, as well as a long FXS pre-mutation allele with an AGG interspersion pattern commonly seen in humans. We also describe the effect of these structures on PKR and Dicer, two enzymes that can have significant effects on gene expression and that are sensitive to RNA with double-stranded character. We discuss the implications of our findings for FXS and pre-mutation carrier symptoms.
MATERIALS AND METHODS
Cell lines and plasmids
The HEK293-derived cell lines expressing 0, 22 and 176 transcribed but untranslated CGG·CCG repeats from the same doxycycline-regulatable promoter in the same chromosomal locus will be described in detail elsewhere (V.Handa, D.Goldwater, T.Saha and K.Usdin, in preparation). Briefly, the cell lines were generated using the Flp-In T-Rex system (Invitrogen, Carlsbad, CA) in which the repeats were inserted downstream of a CMV–TetO hybrid promoter in a plasmid containing an FRT site [pcDNA5/FRT/TO (Invitrogen)]. Flp recombinase-mediated plasmid integration was carried out by co-transfection of the repeat-containing plasmid together with a non-replicating plasmid encoding the Flp recombinase into a cell line containing an integrated FRT site as recommended by the supplier. The PKR expression plasmid, pEGST·PKR/λPP, which expresses both a GST·PKR fusion protein and the bacteriophage λ protein phosphatase was a gift of Dr Takayasu Date [Kanazawa Medical University, Japan (17)]. Clones containing 22 repeats of either CGG·CCG, CAG·CTG or ATT·TTA were generated as previously described using the plasmid pREX+ which contains a T7 promoter sequence upstream of the repeats (18). To reduce the amount of non-repeat sequence that would be generated by transcription from these templates, we deleted the region between the BglII and SpeI sites in the vector. A T7 RNA polymerase template containing the sequence (CGG)9AGG(CGG)12AGG(CGG)97 was generated by PCR amplification of a large human pre-mutation allele that we had previously inserted into transgenic mice (19). The forward primer used was T7CGG-F (5′-GCATGAATTCTAATACGACTCACTATAGGGAGGGCGTGCGGCAGCG-3′), which contains a T7 promoter, and the reverse primer T7CGG-R (5′-TTTTggatccGCTGCGGG CGCTCGAGGCCCAG-3′). The PCR fragment was digested with BamHI and cloned into SmaI- and BglII-digested pGL3-Basic (Promega, Madison, WI).
RNA synthesis
Synthesis of RNA with 22 repeats was carried out using plasmid templates. These templates were linearized with a restriction enzyme with a recognition site immediately downstream of the repeats. The generation of a long pre-mutation FXS allele has been previously described (19). This allele contains the following sequence (CGG)9AGG (CGG)12AGG(CGG)97. Templates containing this allele were prepared by PCR from a plasmid in which the repeats were stable, using a primer pair one of which had a T7 promoter site. The templates were then phenol extracted, ethanol precipitated and resuspended in TE buffer. Transcription was carried out using the Megashortscript Kit (Ambion, Austin, TX). For T1 and S1 mapping, 20 µCi of [γ-32P]GTP (ICN, Irvine, CA) was added to each reaction, and the mixture incubated at 37°C for 2 h. For the Dicer reaction, transcription was carried out using a mixture of all four [α-32P]NTPs (ICN). The reactions were terminated by addition of formamide-EDTA solution (96% formamide, 2 mM EDTA) and the RNA precipitated with 3 vol of ethanol. The RNA was then resuspended in gel loading buffer (formamide-EDTA solution, 0.025% bromophenol blue, 0.025% xylene cyanol) and subjected to electrophoresis on a polyacrylamide gel containing 7 M urea. The full-length RNA was excised from the gel, extracted by incubation in formamide-EDTA solution overnight at 37°C, extracted with phenol:chloroform:isoamylalcohol and then ethanol precipitated.
Enzymatic treatment of RNA
RNA samples were resuspended in TE buffer and heated to 95°C for 3 min. For reactions carried out in the presence of K+, KCl was added to a final concentration of 40 mM before cooling. After digestion by S1, T1 or Dicer, the samples were phenol extracted, ethanol precipitated and dissolved in gel loading buffer for electrophoresis on a polyacrylamide gel containing 7 M urea.
S1. Digestion was carried out in a 100 µl reaction containing S1 nuclease (Amersham Biosciences Corp., Piscataway, NJ), 30 mM NaOAc pH 4.6, 50 mM NaCl, 1 mM ZnCl2, 5% glycerol and 8 mM Escherichia coli tRNA. The reactions were incubated at 20°C for 20 min.
T1. Digestion was carried out with T1 nuclease (Ambion) in a 100 µl reaction containing either T1 nuclease buffer (T1B) (10 mM Tris–HCl pH 7.5, 40 mM NaCl, 1 mM MgCl2 and 8 mM E.coli tRNA), or the buffer used to carry out the Dicer digestion (see below). Reactions were carried out for 15 min at 37°C.
Dicer. Digestion with recombinant human Dicer (Gene Therapy Systems, San Diego, CA) was carried out in accordance with the manufacturer’s instructions using a buffer containing 250 mM NaCl, 30 mM HEPES pH 8.0, 0.05 mM EDTA, 2.5 mM MgCl2. ATP was added to a final concentration of 1 mM and digestion was carried out for 17 h at 37°C. The same mass of RNA was used for each reaction. Samples containing the same amount of radiolabeled product were loaded on each gel. Gel images were obtained using Fujifilm type BAS-III phosphoimager screens and a Fujifilm BAS 15000 reader. Analysis and quantification was performed with Sciencelab Image Gauge 3.0 (Mac) software. The relative amount of Dicer product produced from the CGG-RNA substrate was determined both by comparing the amount of this product relative to the amount of full-length material and by comparison with the amount of reaction product produced using the bona fide double-stranded RNA substrate.
Computer analysis
The (CGG)9AGG(CGG)12AGG(CGG)97 RNA was analyzed in silico using the Mfold web server (20) using either the default settings or the constraints on unpaired bases obtained from the T1 and S1 digestions.
Purification of human recombinant PKR
Escherichia coli BL21(DE3) pLysS freshly transformed with pEGST·PKR/λPP was grown in 2× YT with 100 µg/ml ampicillin at 30°C to an OD600 of 0.8. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM. After induction, cultures were continued at the same temperature for another 4 h. The bacterial cells were harvested by centrifugation and resuspended in sonication buffer [50 mM Tris–HCl pH 7.5, 50 mM KCl, 1 mM phenylmethylsulfonylfluoride (PMSF), 5 mM dithiothreitol (DTT), 20% (v/v) glycerol]. Cells were broken open by sonication on ice and Triton X-100 was added to a final concentration of 1% (v/v). Cell debris was removed by centrifugation at 4°C for 30 min at 15 000 r.p.m.. The supernatant was added to GST–agarose beads (Pierce, Rockford, IL), and gently rocked at 4°C for 1 h before centrifugation at 2500 r.p.m. for 5 min. The supernatant was removed and the resin resuspended in sonication buffer. Resin-bound GST fusion protein was transferred to a spin column (Pierce) and washed three times with sonication buffer. The GST fusion products were eluted by addition of 0.5 ml of elution buffer [50 mM Tris–HCl pH 7.5, 50 mM KCl, 5 mM DTT, 20% (v/v) glycerol] containing 2.5, 5 and 10 mM glutathione. The eluted affinity-purified GST fusion proteins were assayed by western blot using antibody against GST (Pierce).
In vitro protein kinase assay
A 10 µl mixture containing 50 mM Tris–HCl pH 7.5, 2 mM MgCl2, 50 mM KCl, 20 µM ATP, 25 µCi of [γ-32P]ATP and partially purified GST–PKR (2.5 mM glutathione fraction containing ∼2 µg of total protein) was incubated with the indicated concentrations of poly(I)·poly(C), (AUU)88 RNA, (CGG)22 RNA or (CGG)9AGG(CGG)12AGG(CGG)97 RNA for 30 min at 23°C. A 1:100 dilution of the phosphatase inhibitor cocktails I and II (Sigma, St Louis, MO) were added to reduce the activity of any co-purifying phosphatase. Reactions were resolved on 10% polyacrylamide–SDS gels, fixed, dried and subjected to autoradiography. Images were obtained using Fujifilm type BAS-III phosphoimager screens and a Fujifilm BAS 15000 reader. Analysis and quantification were performed with Sciencelab Image Gauge 3.0 (Mac) software.
Lipofectin-mediated delivery of RNA
A 36 µl aliquot of lipofectin (2 mg/ml) (Invitrogen) was added to serum and antibiotic-free Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) and mixed well. An 18 µg aliquot of RNA was added to the lipofectin–DMEM mixture to a final volume of 1.5 ml and incubated at room temperature for 10 min; 4.5 ml of DMEM was then added. HeLa cells grown in 12-well dishes to a confluency of 70–80% were washed twice with DMEM that was serum and antibiotic free. A 750 µl aliquot of the RNA–lipofectin mixture was then added to each of eight wells. After 6 h, the medium was changed, and incubations were carried out for the indicated length of time.
Analysis of PKR activation in vivo
Cell extracts were made using standard procedures. A 40 µg aliquot of protein was subjected to SDS–PAGE, and transferred to a nitrocellulose membrane (MSI, Westboro, MA). The membrane was blocked overnight in Tris-buffered saline (TBS) containing 5% skim milk at 4°C and then incubated at room temperature for 3 h with a rabbit anti-eIF2α antibody specific for the phosphorylated form of the protein (Biosource, Camarillo, CA) diluted 1:1000. The membrane was washed in TBS with 0.05% Tween-20 and then incubated with anti-rabbit immunoglobulin conjugated to horseradish peroxidase (Upstate Biotechnology, Charlottesville, VA). An ECL kit was used for signal detection as per the manufacturer’s instructions (Amersham Biosciences).
RESULTS
CGG-RNA forms hairpins
RNA containing 22 CGG repeats produced a very simple pattern of T1 and S1 cleavage, with a single region of cleavage located in the middle of the repeat tract (Fig. 1). In contrast, RNA containing the sequence (CGG)9AGG (CGG)12AG G(CGG)97 produced a complex mixture of bands when treated with T1 nuclease at neutral pH or S1 nuclease at low pH (Fig. 1). The pattern of cleavage of (CGG)22 is consistent with formation of a single hairpin, while the pattern of cleavage of the longer repeat suggests that more than one structure is formed. All reactions using the longer RNA produced somewhat similar amounts of cleavage products ∼265, ∼232, ∼85 and ∼47 bases long. Products of ∼213 and ∼200 nucleotides were also seen in all reactions but the amount of this material differed depending on the reaction conditions. In addition to these products, a fragment of ∼158 bases is seen in S1 reactions and to a much lesser degree in T1 reactions carried out in the presence of K+ (Fig. 1).
Figure 1.

Analysis of CGG-RNA structure. S1 and T1 nuclease digestion of CGG-RNA. S1 and T1 treatment of RNA containing (CGG)22 and (CGG)9AGG(CGG)12AGG(CGG)97 [referred to as (CGG)120]. The sizes of the S1 and T1 cleavage products were determined by comparison with both a pBR322 MspI digest and the background of T1 cleavage products resulting from a small amount of unstructured RNA in the reaction. T1B, S1B and DB refer to reactions carried out in the T1 nuclease, S1 nuclease and Dicer buffers as described in Materials and Methods.
T1 reactions carried out in the presence of K+ produce significant amounts of the ∼47, ∼85 and ∼232 cleavage products and less of those fragments resulting from cleavage at position ∼265. One of the most stable structures generated by computer analysis of the repeat region using the Mulfold algorithm of Zuker (20) without any base pairing constraints was one containing three hairpins with loops at ∼47, ∼85 and ∼232 that had a ΔG of –267.3 [Fig. 2(ii)]. Applying the constraint that bases 264–266 had to be unpaired resulted in the generation of a variety of structures, the most stable of which is the structure shown in Figure 2(iii). This structure which is comprised of two hairpins with loops at ∼85 and ∼265 bases has a ΔG of –263.9. Alternative structures containing bases ∼213 or ∼200 in the loops as shown in Fig. 2(iv) and (v) had ΔGs of –248.4 and –263.5, respectively. These data suggest that the most likely conformation for the (CGG)9AGG(CGG)12AGG(CGG)97 RNA is the one that is comprised of three smaller hairpins shown in Figure 2(ii). The stability of the largest of these three hairpins is –226.7, which is significantly lower than the ΔG for the hairpin containing all the bases in the repeat tract (–263.5), and the ΔG for a single hairpin formed by the uninterrupted repeat (–279.5). These data illustrate the different ways that the AGG interruptions may reduce the effect of long CGG tracts in RNA: they not only prevent the formation of a more stable single hairpin lacking any mismatches, but they also favor the formation of a series of smaller and less stable hairpins, over a single more stable hairpin with mismatches. These smaller hairpins may provide less of an impediment to translocation of the 40S ribosomal subunit.
Figure 2.

Diagrammatic representation of the sequence of the (CGG)9AGG(CGG)12AGG(CGG)97 RNA and the structures consistent with the S1 and T1 cleavage products. The repeat region is shown in black and the flanking sequence in gray. The arrows indicate the regions of nuclease cleavage. The tetrads and base pairs shown are for illustrative purposes only and are not meant to represent the actual number of hydrogen bonds formed.
The ∼158 base fragments seen most clearly in the S1 reaction could be the result of cleavage of the first of the three loops of a tetraplex formed by the 97 uninterrupted CGG repeats at the 3′ end of the repeat tract as illustrated in Figure 2(vi). The central loop of this tetraplex would co-migrate with the products of cleavage of the hairpin formed by the same region. While no distinct product of ∼297 bases could be seen for what would be cleavage of the third loop of the tetraplex, support for tetraplex formation is bolstered by the fact that the appearance of the ∼155 base fragment is favored by low pH and the presence of K+, both factors known to stabilize the CGG-DNA tetraplex.
CGG-RNA does not activate PKR
We assessed the ability of CGG-RNA to activate partially purified human PKR expressed in bacteria. Since bacterially produced PKR is normally fully activated, we made use of a construct containing both the human PKR and the λ phosphatase genes. We incubated PKR in the presence of [γ-32P]ATP and in vitro transcribed RNA containing either 22 CGG repeats, corresponding to a normal allele, or 120 repeats, corresponding to the repeats in a pre-mutation allele. The amount of PKR autophosphorylation is shown in Figure 3A. While a significant increase in the extent of phosphorylation was seen with increasing amounts of the synthetic double-stranded RNA polymer polyI:polyC, no such increase could be seen with either (CGG)22 or (CGG)9AGG (CGG)12AG G(CGG)97.
Figure 3.

Effect of CGG-RNA on PKR activation. (A) Phosphorylation of partially purified human PKR in vitro in response to added RNAs. The data are expressed as the amount of phosphorylated PKR relative to the endogenous levels of phosphorylated material present in the absence of added RNA. The level of PKR phosphorylation in response to added RNA for poly(I):poly(C) is indicated by triangles, (AUU)88 by crosses, (CGG)22 by diamonds and (CGG)9AGG(CGG)12AGG(CGG)97 by squares. (B) The amount of phosphorylated eIF2α in cells transfected with in vitro synthesized RNA. The data are expressed as the amount of phosphorylated eIF2α at the indicated time points relative to the amount before addition of the RNA. The gray bars indicate the amount of phosphorylated eIF2α in cells transfected with poly(I):poly(C) and the black bars the amount detected in cells transfected with the same amount of (CGG)9AGG(CGG)12AG G(CGG)97 RNA. (C) The amount of phosphorylated eIF2α in cells expressing an otherwise identical transcript containing either 0 (FRT0) or 176 (FRT176) CGG repeats. A small increase in the amount of phosphorylated eIF2α is seen in both cell lines at 48 h but, at least in the case of the FRT176 cell line, this does not seem to be related to expression of the CGG-containing RNA since more phosphorylated product is seen in the absence of dox than in its presence.
We also tested the ability of these RNAs to activate PKR in vivo by measuring the amount of phosphorylation of a downstream target of activated PKR, eIF2α, in HeLa cells transfected with in vitro synthesized (CGG)9AGG (CGG)12AGG(CGG)97 RNA or in otherwise isogenic cell lines that express either 0 or 176 CGG repeats under the control of a doxycycline-regulatable CMV promoter. While an increase in the amount of phosphorylated eIF2α was seen when poly(I):poly(C) was introduced into cells, no increase in the amount of phosphorylated eIF2α was seen with the exogenously produced CGG-containing RNA (Fig. 3B). No increase in the amount of phosphorylated eIF2α was seen with the endogenous source of CGG-RNA either (Fig. 3C).
CGG-RNA is a substrate for human recombinant Dicer in vitro
Digestion of a ‘single-stranded’ RNA generated by T7 transcription of one strand of the luciferase gene showed no evidence of cleavage by purified recombinant human Dicer. In contrast, digestion of a bona fide double-stranded RNA produced by transcription from the same template transcribed in both directions yielded significant amounts of a 22 bp fragment (Fig. 4). Treatment of (CGG)9AGG (CGG)12AGG(CGG)97 RNA produced a product of the same size, indicating that the RNA is a substrate for Dicer. However, the yield of this product was only ∼10% that obtained for the bona fide double-stranded RNA substrate. Its relatively inefficient conversion into small interfering (siRNA) could be due in part to the fact that the constituent hairpins contain a large number of G·G mismatches.
Figure 4.
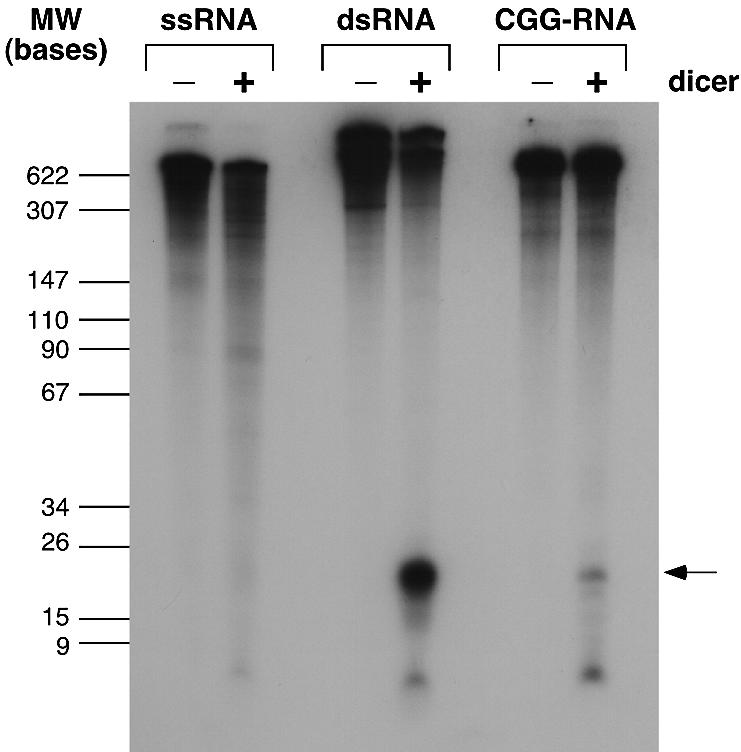
Dicer digestion of CGG-RNA. Bona fide single- and double-stranded RNAs were digested alongside (CGG)9AGG(CGG)12AGG(CGG)97 RNA with human recombinant Dicer enzyme as described in Materials and Methods. The relative amount of ∼22 bp Dicer product produced was calculated by comparing the amount of this product with the amount of undigested material remaining.
DISCUSSION
We have shown that the predominant structures formed by CGG repeat RNA under physiologically reasonable conditions are hairpins. Formation of these structures may explain the stalling of the 40S ribosomal subunits that is seen on FMR1 alleles with long CGG repeat tracts. This stalling is believed to contribute to FXS disease pathology by reducing the translation efficiency of these transcripts.
In addition, since double-stranded RNA potentially has a number of negative consequences for a cell, it is possible that hairpin formation also contributes to the pathology seen in FXS ‘pre-mutation’ carriers. However, in contrast to what has been demonstrated for the DM1 repeats, we found no evidence that CGG-RNA can activate PKR either in vitro or in vivo. It is known that not all RNAs with duplex character activate PKR, and some RNAs such as the human immunodeficiency virus TAR RNA (21) and adenovirus VA I RNA (22) actually inhibit PKR activation. One common feature of these RNAs seems to be the presence of numerous unpaired or non-Watson–Crick base pairs (23). CGG-RNA with its likely 2:1 ratio of Watson–Crick C·G and Hoogsteen G·G base pairs does fit this description. The absence of detectable PKR activation by pre-mutation length CGG repeats suggests that any potential toxicity of CGG-RNA lies elsewhere.
The fact that pre-mutation length CGG-RNA is a substrate for Dicer suggests one possible way that long CGG tracts in RNA could be deleterious, i.e. via translational suppression or RNAi-mediated gene silencing. Since these modes of silencing are homology based, it is possible that overexpression of CGG-RNA in ‘pre-mutation’ carriers could affect the expression of other CGG·CCG-containing genes. This in turn could have pleiotropic effects on development and cell viability. However, other potential mechanisms of RNA toxicity are also possible. By analogy with one hypothesis for the pathogenesis of DM1, it may be that the CGG-RNA sequesters one or more CGG-RNA-binding proteins, leading to defects in the pathways in which these proteins act [for recent review see Ranum and Day (6)]. This possibility is currently under investigation.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to thank the other members of the Usdin laboratory, namely Eriko Greene, Lata Mahishi and Daman Kumari, for many helpful suggestions and the occasional buffer.
REFERENCES
- 1.Fu Y.H., Kuhl,D.P., Pizzuti,A., Pieretti,M., Sutcliffe,J.S., Richards,S., Verkerk,A.J., Holden,J.J., Fenwick,R.G.,Jr, Warren,S.T. et al. (1991) Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell, 67, 1047–1058. [DOI] [PubMed] [Google Scholar]
- 2.Verkerk A.J., Pieretti,M., Sutcliffe,J.S., Fu,Y.H., Kuhl,D.P., Pizzuti,A., Reiner,O., Richards,S., Victoria,M.F., Zhang,F.P. et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell, 65, 905–914. [DOI] [PubMed] [Google Scholar]
- 3.Allingham-Hawkins D.J., Babul-Hirji,R., Chitayat,D., Holden,J.J., Yang,K.T., Lee,C., Hudson,R., Gorwill,H., Nolin,S.L., Glicksman,A. et al. (1999) Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study—preliminary data. Am. J. Med. Genet., 83, 322–325. [PMC free article] [PubMed] [Google Scholar]
- 4.Sherman S.L. (2000) Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet., 97, 189–194. [DOI] [PubMed] [Google Scholar]
- 5.Greco C.M., Hagerman,R.J., Tassone,F., Chudley,A.E., Del Bigio,M.R., Jacquemont,S., Leehey,M. and Hagerman,P.J. (2002) Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain, 125, 1760–1771. [DOI] [PubMed] [Google Scholar]
- 6.Ranum L.P. and Day,J.W. (2002) Dominantly inherited, non-coding microsatellite expansion disorders. Curr. Opin. Genet. Dev., 12, 266–271. [DOI] [PubMed] [Google Scholar]
- 7.Napierala M. and Krzyosiak,W.J. (1997) CUG repeats present in myotonin kinase RNA form metastable ‘slippery’ hairpins. J. Biol. Chem., 272, 31079–31085. [DOI] [PubMed] [Google Scholar]
- 8.Koch K.S. and Leffert,H.L. (1998) Giant hairpins formed by CUG repeats in myotonic dystrophy messenger RNAs might sterically block RNA export through nuclear pores. J. Theor. Biol., 192, 505–514. [DOI] [PubMed] [Google Scholar]
- 9.Tian B., White,R.J., Xia,T., Welle,S., Turner,D.H., Mathews,M.B. and Thornton,C.A. (2000) Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA, 6, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nadel Y., Weisman-Shomer,P. and Fry,M. (1995) The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J. Biol. Chem., 270, 28970–28977. [DOI] [PubMed] [Google Scholar]
- 11.Mitas M., Yu,A., Dill,J. and Haworth,I.S. (1995) The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn·Ganti base pairs. Biochemistry, 34, 12803–12811. [DOI] [PubMed] [Google Scholar]
- 12.Kettani A., Kumar,R.A. and Patel,D.J. (1995) Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol., 254, 638–656. [DOI] [PubMed] [Google Scholar]
- 13.Fry M. and Loeb,L.A. (1994) The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl Acad. Sci. USA, 91, 4950–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Usdin K. and Woodford,K.J. (1995) CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res., 23, 4202–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Usdin K. (1998) NGG-triplet repeats form similar intrastrand structures: implications for the triplet expansion diseases. Nucleic Acids Res., 26, 4078–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel P.K., Bhavesh,N.S. and Hosur,R.V. (2000) Cation-dependent conformational switches in d-TGGCGGC containing two triplet repeats of fragile X syndrome: NMR observations. Biochem. Biophys. Res. Commun., 278, 833–838. [DOI] [PubMed] [Google Scholar]
- 17.Matsui T., Tanihara,K. and Date,T. (2001) Expression of unphosphorylated form of human double-stranded RNA-activated protein kinase in Escherichia coli. Biochem. Biophys. Res. Commun., 284, 798–807. [DOI] [PubMed] [Google Scholar]
- 18.Grabczyk E. and Usdin,K. (1999) Generation of microgram quantities of trinucleotide repeat tracts of defined length, interspersion pattern and orientation. Anal. Biochem., 267, 241–243. [DOI] [PubMed] [Google Scholar]
- 19.Lavedan C., Grabczyk,E., Usdin,K. and Nussbaum,R.L. (1998) Long uninterrupted CGG repeats within the first exon of the human FMR1 gene are not intrinsically unstable in transgenic mice. Genomics, 50, 229–240. [DOI] [PubMed] [Google Scholar]
- 20.Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res., 31, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunnery S., Rice,A.P., Robertson,H.D. and Mathews,M.B. (1990) Tat-responsive region RNA of human immunodeficiency virus 1 can prevent activation of the double-stranded-RNA-activated protein kinase. Proc. Natl Acad. Sci. USA, 87, 8687–8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathews M.B. and Shenk,T. (1991) Adenovirus virus-associated RNA and translation control. J. Virol., 65, 5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bevilacqua P.C., George,C.X., Samuel,C.E. and Cech,T.R. (1998) Binding of the protein kinase PKR to RNAs with secondary structure defects: role of the tandem A-G mismatch and noncontiguous helixes. Biochemistry, 37, 6303–6316. [DOI] [PubMed] [Google Scholar]