

Abstract
Mon1 and Ccz1 were identified from a gene deletion library as mutants defective in the vacuolar import of aminopeptidase I (Ape1) via the cytoplasm to vacuole targeting (Cvt) pathway. The mon1Δ and ccz1Δ strains also displayed defects in autophagy and pexophagy, degradative pathways that share protein machinery and mechanistic features with the biosynthetic Cvt pathway. Further analyses indicated that Mon1, like Ccz1, was required in nearly all membrane-trafficking pathways where the vacuole represented the terminal acceptor compartment. Accordingly, both deletion strains had kinetic defects in the biosynthetic delivery of resident vacuolar hydrolases through the CPY, ALP, and MVB pathways. Biochemical and microscopy studies suggested that Mon1 and Ccz1 functioned after transport vesicle formation but before (or at) the fusion step with the vacuole. Thus, ccz1Δ and mon1Δ are the first mutants identified in screens for the Cvt and Apg pathways that accumulate precursor Ape1 within completed cytosolic vesicles. Subcellular fractionation and co-immunoprecipitation experiments confirm that Mon1 and Ccz1 physically interact as a stable protein complex termed the Ccz1-Mon1 complex. Microscopy of Ccz1 and Mon1 tagged with a fluorescent marker indicated that the Ccz1-Mon1 complex peripherally associated with a perivacuolar compartment and may attach to the vacuole membrane in agreement with their proposed function in fusion.
Compartmentalization allows eukaryotic cells to regulate intracellular functions by separating competing reactions and localizing enzymes and substrates at specific locations within the cell. Efficient compartmentalization necessitates dynamic protein trafficking processes by which cells are able to establish and maintain the identity and function of each organelle. The vacuole (lysosome) of the yeast Saccharomyces cerevisiae plays a central role in the turnover of cytoplasmic organelles, degradation of intracellular/extracellular components, and maintenance of cellular physiology (1). To carry out these functions, the vacuole maintains a variety of degradative enzymes. Both resident hydrolases and their substrates arrive at this destination through a variety of sorting pathways. The main routes by which vacuolar hydrolases are delivered to this organelle are the carboxypeptidase Y (CPY),1 alkaline phosphatase (ALP), and multivesicular body (MVB) pathways, which involve transit through a portion of the secretory pathway, and the cytoplasm to vacuole targeting (Cvt) pathway by which the cargo molecules are packaged as cytosolic membrane-bound intermediates (2, 3). Resident proteins are also transmitted by inheritance from mother cell vacuoles to daughter cells during cell division (4). Substrates enter the vacuole through endocytosis, autophagy and the vacuole import and degradation pathway (reviewed in Ref. 5). One common feature in all of these processes is membrane fusion. The membrane fusion mechanism acts to ensure specificity for the directed movement of proteins while also maintaining the distinct composition of each organelle within the highly compartmentalized eukaryotic cell.
The cytoplasm to vacuole targeting pathway that is used to deliver the soluble hydrolase aminopeptidase I (Ape1) to the vacuole has been under investigation (for reviews see Refs. 2, 5, and 6). Under vegetative conditions, precursor Ape1 (prApe1) is assembled into a large Cvt complex composed in part of multiple prApe1 dodecamers in the cytosol that becomes enwrapped within a double-membrane Cvt vesicle (7). Upon completion, the cytosolic Cvt vesicle targets to the vacuole. The outer membrane of the Cvt vesicle fuses with the vacuole membrane and the intact inner vesicle (Cvt body) passes into the vacuole lumen (8). The Cvt body is ultimately broken down by resident vacuolar hydrolases, resulting in the release and maturation of prApe1. Precursor Ape1 is transported to the vacuole by another pathway, termed autophagy (Apg), under starvation conditions (2, 9). In the Apg pathway, portions of cytoplasm are sequestered within relatively larger double membrane vesicles (autophagosomes) that are also targeted to the vacuole (7). Although Apg is a degradative process, mutants defective in autophagy, apg/aut, overlap with cvt mutants (10). Morphological and biochemical analyses further indicate that the Cvt and Apg pathways use analogous mechanisms (2, 5, 9).
To gain additional insight into the Cvt/Apg pathways, we screened a gene deletion library for mutants that are defective in prApe1 maturation. We found two mutants that are required for Cvt/Apg import that had not been previously implicated in these pathways. The product of one of these genes, Ccz1, has been suggested to be involved in multiple trafficking pathways to the vacuole (11). Overexpression of the Rab protein Ypt7 rescues the sensitivity to calcium, caffeine, and zinc observed with the ccz1Δ strain. The Ypt7K127E mutant has been identified as a specific mutation that suppresses the ccz1Δ phenotype (12). Co-immunoprecipitation data further support the physical interaction between Ccz1 and Ypt7 (12). The mon1Δ strain is sensitive to monensin and brefeldin A (13), but is otherwise uncharacterized. In this study, we show that strains lacking either of these two proteins have similar phenotypes. Both Mon1 and Ccz1 are required not only for the Cvt/Apg pathways but also other vacuole biogenesis processes including the sorting of newly synthesized vacuolar proteins through the CPY, ALP, and MVB pathways and endocytosis. Biochemical and morphological evidence further indicate that the Cvt/Apg pathways are blocked at a stage after the formation of the sequestering vesicles but prior to their fusion with the vacuole. These studies also suggest that Ccz1 and Mon1 co-localize to a unique membrane and that they physically interact. Finally, we demonstrate the in vivo localization of these two proteins to a perivacuolar compartment and the vacuole membrane, a site consistent with their proposed role in fusion.
Experimental Procedures
Strains, Media, and Growth Conditions
The yeast strains used in this study are listed in Table I. Synthetic minimal medium (SMD) contained 0.67% yeast nitrogen base without amino acids, 2% glucose, and auxotrophic amino acids and vitamins as needed. Nitrogen starvation medium (SD-N) contained 0.17% yeast nitrogen base without amino acids and ammonium sulfate and 2% glucose. YPD medium contained 1% yeast extract, 2% peptone, and 2% glucose. S. cerevisiae strains were grown at 30 °C. Yeast cells used for this study were grown in the appropriate SMD medium to mid-log (OD600 of 0.6).
TABLE I. Yeast strains used in this study.
| Strain | Genotype | Reference |
|---|---|---|
| SEY6210 | MATα leu2–3,112 ura3–52 his3–Δ200 trp1–Δ901 lys2-801 suc2–Δ9 GAL | (48) |
| CWY3 | SEY6210 ccz1Δ∷HIS5 | This study |
| JSY1 | SEY6210 mon1Δ∷HIS5 | This study |
| BY4742 | MATα his3Δ leu2Δ lys2Δ ura3Δ | ResGen |
| ccz1Δ | BY4742 ccz1Δ∷KanMX | ResGen |
| mon1Δ | BY4742 mon1Δ∷KanMX | ResGen |
| D3Y102 | SEY6210 vac8Δ | (31) |
| NNY20 | MATa ura3 trp1 leu2 apg1Δ∷LEU2 | (31) |
| vps5 | SEY6210 vps5 | (48) |
| CWY40 | SEY6210 vam3Δ∷TRP1 | This study |
| WSY99 | SEY6210 ypt7Δ∷HIS3 | (49) |
| VDY101 | SEY6210 apg7Δ∷LEU2 | (37) |
| PSY35 | SEY6210 MON1-HA∷TRP1 | This study |
| PSY36 | SEY6210 CCZ1-HA∷TRP1 | This study |
| PSY42 | SEY6210 CCZ1-YFP∷HIS3 | This study |
| PSY44 | SEY6210 CCZ1-CFP∷KanMX | This study |
| PSY45 | SEY6210 CCZ1-CFP∷KanMX pCUP1-YFP-MON1∷URA3 | This study |
| PSY46 | SEY6210 CCZ1-GFP∷HIS3 | This study |
| PSY47 | SEY6210 MON1-GFP∷HIS3 | This study |
| TVY1 | SEY6210 pep4Δ∷LEU2 | (50) |
Reagents and Antisera/Antibodies
Reagents for growth medium were from Difco Laboratories (Detroit, MI). DNA restriction enzymes, T4 DNA ligase and calf intestinal alkaline phosphatase were obtained from New England Biolabs, Inc. (Beverly, MA). Tran[35S] label was obtained from ICN (Costa Mesa, CA). Oxalyticase was from Enzogenetics (Corvallis, OR). OptiPrep™ was from Accurate Chemical and Scientific Corp. (Westbury, NY). Complete™ EDTA-free protease inhibitor was obtained from Roche Molecular Biochemicals. The pME3 vector containing the Schizosaccharomyces pombe HIS5 auxotrophic marker was a gift from Dr. Neta Dean (State University of New York, Stony Brook, NY). The pFA6a knockout and tagging vectors containing TRP1, HIS3, or KanMX markers were generous gifts from Dr. Mark Longtine (Oklahoma State University) (14). The CFP (pDH3) and YFP (pDH5) plasmids were from the Yeast Resource Center (University of Washington). FM 4-64 dye was obtained from Molecular Probes (Eugene, OR). All other reagents were from Sigma-Aldrich. Antisera against Ape1 (15), Prc1 (16), and Pep4 (16) have been described. Antisera against Pgk1, Ypt7, and Anp1 were provided by Dr. Jeremy Thorner (University of California, Berkeley, CA), Dr. William Wickner (Dartmouth Medical School, Hanover, NH), and Dr. Sean Munro (MRC Laboratory of Molecular Biology, Cambridge, UK), respectively. Antibodies against Pho8, Dpm1, and Pep12 were obtained from Molecular Probes, and the anti-HA antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). To prepare antiserum against Mon1, the NH2 terminus of the Mon1 ORF (1–585 bp) was PCR-amplified and fused to the COOH terminus of the maltose-binding protein. The resulting plasmid was transformed into E. coli strain BL21. Fusion protein purification and antiserum generation were as described (17).
Screening the Haploid Gene Deletion Library
A MATα haploid gene deletion library was obtained from ResGen/Invitrogen Corporation (Huntsville, AL). The mutants provided from the company were inoculated on YPD plates and incubated at 30 °C for 12–24 h. The cells on YPD plates were collect and resuspended in 50 μl of MURB (50 mm NaPO4, 25 mm MES, pH 7.0, 1% SDS, 3 m urea, 0.5% β-mercaptoethanol, 1 mm NaN3, and 0.05% bromphenol blue) and converted into crude cell extracts by glass bead lysis and boiling. The extracts were subjected to immunoblot analysis using anti-Ape1 antisera.
Disruption, Epitope Tagging, and Gene Cloning
The chromosomal MON1 and CCZ1 loci were deleted by a PCR-based, one-step procedure (18). In brief, the corresponding auxotrophic marker was amplified from the pME3 or pFA6a knockout plasmids by PCR using oligonucleotides that contained sequences outside of the marker, flanked by sequences that encode regions at the beginning and end of the corresponding ORFs. PCR products were used to transform yeast strain SEY6210. Putative knockout strains were checked by Western blot for the Ape1 phenotype. Similar strategies were applied for the chromosomal HA and fluorescent protein tagging. To clone the MON1 and CCZ1 genes, both ORFs and their upstream/downstream sequences were PCR-amplified using genomic DNA as template. The resulting PCR products for MON1 include 360 bp before the sequence encoding the start codon and 405 bp after the stop codon. The fragments were digested with SacI and SmaI and inserted into the SacI and SmaI site of the pRS416/426 vector to generate plasmids pMON1(416/426). The PCR products for the cloning of CCZ1 contain ∼300-bp upstream and 700-bp downstream of the CCZ1 ORF. The PCR products were digested with KpnI to generate pCCZ1(416/426). To construct COOH-terminal HA epitope-tagged Ccz1, the CCZ1 ORF was PCR-amplified using pCCZ1(416) as a template. The resulting PCR product was digested and inserted into pRS416HA and pRS426HA that contains a 3×HA epitope (19). To construct an NH2-terminal YFP fusion to Mon1, the MON1 ORF was PCR-amplified using pMON1 (416) as a template. The resulting PCR products were inserted into pCuYFP (306) to generate pCuYFP-MON1 (306). The construct was linearized with KpnI and transformed into strain PSY44 to replace endogenous MON1 with pCuYFP-MON1 (strain PSY45). The plasmids pCvt19-CFP(414) (20), pSte3-GFP(316) (21), pCuGFP-Aut7 (416) (22), pGFP-Pho8(426) (23), and pSna3-GFP(416) (24) were described previously. All oligonucleotide sequences and additional details of the plasmid constructions will be provided upon request.
Immunoblot Analysis, Pulse/Chase Labeling, and Immunoprecipitation
Immunoblot analysis was carried out essentially as described previously (25). For kinetic analysis of Prc1, yeast cells were grown to an OD600 of 1.0 and converted into spheroplasts. The spheroplasts from 20 OD600 units of cells were resuspended in 300 μl of SMD medium containing 1.3 m sorbitol, and labeled with 20 μCi of Tran[35S] label for 5 min, followed by a chase reaction in SMD containing 1.3 m sorbitol, 0.2% yeast extract, 4 mm methionine, and 2 mm cysteine at a final density of 2.0 OD600/ml. Samples were removed at the indicated time points and 1 mm NaN3 was added to stop the reaction. The samples were subjected to a 5,000 × g centrifugation for 3 min. The resulting supernatant and pellet fractions were separately precipitated with 10% trichloroacetic acid. Trichloroacetic acid precipitates were resuspended in MURB buffer and subjected to immunoprecipitation as described previously (25). For kinetic analyses of Ape1, Pep4, and Ste3, yeast cells were grown to an OD600 of 1.0 in SMD medium. Cells (20 OD600 units) were resuspended in 300 μl of SMD medium and labeled with 20 μCi of Tran[35S] label for 5–10 min, followed by a chase reaction as above at a final density of 20 OD600/ml. Samples were removed at the indicated time points and precipitated with 10% trichloroacetic acid. Crude extracts were prepared by glass bead lysis and subjected to immunoprecipitation as described previously (25).
Analyses of the Cvt Pathway and Autophagy
Cell viability and starvation curves and peroxisome degradation rates were determined as described previously (17). The membrane flotation assay was performed essentially by the method described previously (26) with minor modifications. Spheroplasts derived from the mon1Δ strain were resuspended in PS200 lysis buffer (20 mm PIPES, pH 6.8, 200 mm sorbitol) containing 5 mm MgCl2 at a spheroplast density of 20 OD600/ml. The lysate was centrifuged at 13,000 × g for 5 min at 4 °C. The pellet fractions from 10 OD600 units of cells were resuspended in 100 μl of 15% Ficoll-400 (w/v) in lysis buffer with or without the addition of 0.2% Triton X-100. The resuspended pellet fractions were overlaid with 1 ml of 13% Ficoll-400 in lysis buffer and then overlaid with 200 μl of 2% Ficoll-400 in lysis buffer. The resulting step gradient was subjected to centrifugation at 13,000 × g for 10 min at 4 °C. The top 500 μl was designated as the float fraction (F), the remaining solution was considered as the nonfloat fraction (NF), and the gradient pellet was designated as the pellet fraction (P). The three fractions were trichloroacetic acid-precipitated, washed twice with acetone, and analyzed by immunoblot. The protease protection assay was performed as described previously (17). In brief, log-phase cultures were subjected to osmotic lysis in PS200 containing 5 mm MgCl2. The lysates were centrifuged at 13,000 × g for 10 min, and the pellet fractions (P13) were resuspended in lysis buffer in the presence or absence of 50 μg/ml proteinase K and/or 0.2% Triton X-100. Reactions were carried out on ice for 30 min followed by trichloroacetic acid precipitation and immunoblot analysis.
Subcellular Fractionation and OptiPrep™ Density Gradient Analysis
Mon1-HA cells expressing pCcz1-HA(416) were grown to mid-log phase (OD600 = 0.6) in SMD medium. The cells were converted into spheroplasts and resuspended in PS200 lysis buffer containing 5 mm MgCl2 and the Complete™ EDTA-free protease inhibitor mixture at a density of 20 OD600/ml. After a preclearing spin (500 × g, for 5 min, at 4 °C), the total lysate was subjected to low-speed centrifugation (13,000 × g for 10 min), resulting in the supernatant (S13) and pellet (P13) fractions. The S13 fraction was subjected to high speed centrifugation (100,000 × g for 30 min at 4 °C) to generate the supernatant (S100) and pellet (P100) fractions. The resulting fractions were subjected to immunoblot analysis. To examine membrane binding of Ccz1-HA and Mon1-HA the membrane fractions from lysed spheroplasts were treated with 1 m KCl, 0.1 m Na2CO3 (pH 10.5), 3 m urea, or 1% Triton X-100 as described previously (17). OptiPrep™ density gradient analysis was performed using a modification of a previously described procedure (17). In brief, a Mon1-HA strain expressing Ccz1-HA was grown to mid-log phase (OD600 = 0.6) and converted into spheroplasts. The spheroplasts were subjected to osmotic lysis in PS200 containing 1 mm EDTA, 1 mm MgCl2, and a protease inhibitor mixture (Complete™ EDTA-free protease inhibitor tablets, 1 μg/ml leupeptin and 1 μg/ml pepstatin A). The lysate was subjected to very low speed centrifugation (800 × g for 5 min) to remove the remaining intact spheroplasts. After this preclearing step, the crude lysate from 35 OD600 units of cells was centrifuged at 100,000 × g for 20 min at 4 °C. The resulting total membrane fraction was resuspended in 200 μl of lysis buffer and then applied on top of a density gradient (12 ml, linear) consisting of 10–55% OptiPrep™ in PS200 lysis buffer containing 1 mm EDTA, 1 mm MgCl2, 1 mm dithiothreitol, and a protease inhibitor mixture. The gradients were subjected to centrifugation at 100,000 × g for 12 h at 4 °C in a Sorvall Th-641 rotor. Samples were collected from the top of the gradients into 14 fractions. The fractions were trichloroacetic acid-precipitated and washed twice with acetone followed by immunoblot analyses.
Native Immunoprecipitation
The protocol for co-immunoprecipitation with Ccz1-HA was modified from a previously described procedure (12). In brief, 10 OD600 units of log-phase cells were lysed with glass beads in lysis buffer (50 mm HEPES, pH 7.4, 150 mm KCl, 1 mm EDTA, 0.5% Triton X-100) with the addition of protease inhibitor mixture and 1 mm phenylmethylsulfonyl fluoride. After a 10-min solubilization on ice, total cell lysates were centrifuged at 13,000 × g for 15 min at 4 °C. To the resulting supernatant, 10 μl of anti-HA antiserum was added followed by incubation with protein A-Sepharose at 4 °C overnight. Sepharose beads were washed with lysis buffer a total of eight times. Bound proteins were eluted in MURB followed by SDS-PAGE and Western blot analysis.
Microscopy
All strains used for microscopy were grown in SMD medium to mid-log phase. In vivo FM 4–64 staining was performed as described previously (27). Microscopy analysis was performed using a Nikon E-800 fluorescent microscope (Mager Scientific Inc., Dexter, MI). Images were captured by an ORCA II CCD camera (Hamamatsu Corp., Bridgewater, NJ) using Openlab 3 software (Improvision, Inc., Lexington, MA).
Results
Mon1 and Ccz1 Are Required for the Cvt, Autophagy, and Pexophagy Pathways
Although various cvt, apg, and aut mutants defective in the Cvt and Apg pathways have been isolated and analyzed (reviewed in Refs. 5 and 28), many questions concerning these pathways remain to be answered. We are interested in the molecular mechanism governing the dynamic aspects of the Cvt and Apg pathways. We reasoned that the identification of additional mutants would provide further insight into the protein machinery of these processes. Accordingly, we screened a haploid gene deletion library based on the accumulation of prApe1, a cargo protein that is delivered to the vacuole through the Cvt/Apg pathways. Among the new mutants identified, mon1Δ and ccz1Δ showed a complete block in prApe1 maturation. Although mon1Δ has not been previously reported as having a role in the Cvt pathway, complementation analyses indicate that CCZ1 is allelic with CVT16, a previously uncharacterized CVT gene (10). The ccz1Δ mutant was originally identified due to its sensitivity to caffeine, calcium, and zinc (29). It has also been shown that the strain displays a severe vacuole protein-sorting defect. Immunofluorescent data suggest that Ccz1 localizes to the endosomal compartment, and it has been suggested to act in concert with the Rab protein Ypt7 (11, 12). There has not been a published report describing Mon1 function. The MON1 gene, YGL124c, encodes a 644-amino acid protein with a predicted molecular mass of 73.5 kDa. A data base search indicates that Mon1 does not have homology with other proteins of S. cerevisiae. However, possible homologues having 24–37% identity with Mon1 exist in S. pombe, Caenorhabditis elegans, and Drosophila melanogaster. Ccz1 has no significant homologues.
When wild type cells are grown under nutrient-rich conditions, the majority of Ape1 is present as the 50-kDa mature form (Fig. 1A), although a small fraction is present as the 61-kDa precursor. In contrast, both the mon1Δ and ccz1Δ strains accumulated only the precursor form of Ape1. The defect in prApe1 processing in these mutants was rescued by expressing either single or multicopy versions of the corresponding genes on plasmids, confirming the essential roles of Mon1 and Ccz1 for the Cvt pathway (Fig. 1A). Precursor Ape1 is delivered to the vacuole through autophagy under starvation conditions. We utilized a starvation-sensitivity analysis to determine whether the mon1Δ and ccz1Δ strains were able to carry out autophagy. Wild type cells, or mutants specific to the Cvt pathway, are starvation-resistant while mutants defective for autophagy lose viability in the absence of nitrogen (17). As shown in Fig. 1B, the wild type strain was resistant to starvation over the time course examined. In contrast, mon1Δ and ccz1Δ strains, similar to the apg1Δ mutant, displayed a rapid loss of viability in SD-N medium. Viability in the mon1Δ strain was restored when these cells expressed Mon1 from a CEN-based plasmid.
FIG. 1. The ccz1Δ and mon1Δ strains are defective in the Cvt, autophagy, and pexophagy pathways.
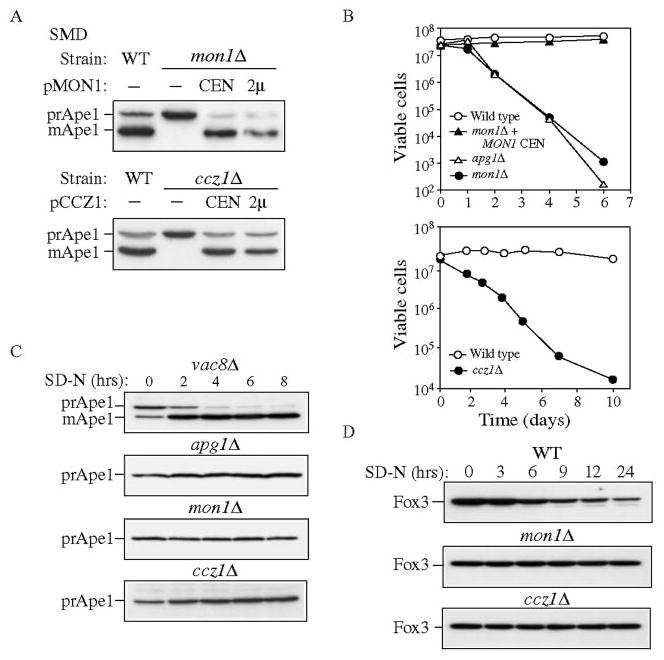
A, cloning and characterization of CCZ1 and MON1. Wild type (WT, SEY6210), ccz1Δ (CWY3), and mon1Δ (JSY1) strains and the knockout strains expressing the respective single copy (CEN) or multicopy (2μ) plasmids were grown in SMD medium and analyzed by immunoblot against Ape1. B, mon1Δ and ccz1Δ strain are sensitive to nitrogen-starvation conditions. The wild type, apg1Δ, and mon1Δ strains and the mon1Δ strain harboring pMON1(416) or the wild type and ccz1Δ strains were grown to mid-log phase in SMD medium and shifted to SD-N medium. At the indicated time, aliquots were removed and spread onto YPD plates in triplicate. The number of viable colonies was counted after 2 days incubation at 30 °C. C, mon1Δ and ccz1Δ mutants do not bypass the prApe1 accumulation defect when autophagy is induced. The vac8Δ (D3Y102), apg1Δ (NNY20), ccz1Δ, and mon1Δ strains were grown to mid-log phase in SMD and shifted to SD-N medium. At the indicated time, aliquots were removed and subjected to immunoblot against Ape1. D, mon1Δ and ccz1Δ strains are defective for pexophagy. The wild type, ccz1Δ and mon1Δ strains in the BY4742 background were grown in YPD to mid-log phase, transferred to oleic acid medium to induce peroxisome production and shifted to SD-N. Aliquots were removed at the indicated times and analyzed by Western blot with antiserum to Fox3.
Starvation-sensitivity indicates that autophagy is not fully functional in the mon1Δ and ccz1Δ strains. Recently, however, we have demonstrated that some mutants that are autophagy defective by this criterion are still able to induce the formation of autophagosomes under starvation conditions. For example, the aut7Δ strain is starvation-sensitive but is able to induce the formation of small, abnormal autophagosomes in SD-N (30). In addition, some components of the Cvt and Apg pathways are only essential for one of these two pathways. For example, Vac8 and Cvt9 are only required for the Cvt pathway whereas Apg17 appears to function only in autophagy (26, 31, 32). Accordingly, these types of mutants are able to mature prApe1 under starvation conditions. We extended our analysis of autophagy by examining the role of Ccz1 and Mon1 in prApe1 import under nutrient-deprivation conditions. Strains were grown in SMD to mid-log phase, shifted to medium lacking nitrogen (SD-N), and the time course of prApe1 processing was examined by Western blot (Fig. 1C). As expected, the vac8Δ strain showed a reversal of the prApe1 accumulation defect after cells were shifted to SD-N. In contrast, the apg1Δ mutant that is defective for both the Cvt and Apg pathways was unable to process prApe1 due to its defect in autophagosome formation. Similar to the apg1Δ strain, the mon1Δ and ccz1Δ strains retained the precursor form of Ape1 in starvation conditions, suggesting that these two proteins are absolutely required for autophagy. The block in prApe1 maturation in SD-N was consistent with the starvation sensitivity phenotype. Thus, we conclude that Mon1 and Ccz1 are required for both the Cvt and autophagy pathways.
We have reported previously that the peroxisome degradation pathway, pexophagy, uses similar molecular components as the Cvt and autophagy pathways (33). To investigate if Mon1 and Ccz1 are also required for pexophagy, we induced the expression of peroxisomes by growing cells in oleic acid in the wild type, mon1Δ, and ccz1Δ strains, and then monitored the degradation of Fox3 after cells were shifted to glucose. Crude cell extracts were collected at the times indicated and examined by Western blot. In wild type cells, Fox3 levels decreased in SD-N, reflecting peroxisome degradation (Fig. 1D). In contrast, both the mon1Δ and ccz1Δ strains maintained Fox3 at the initial level indicating a defect in peroxisome degradation. Therefore, we conclude that both Mon1 and Ccz1 are part of the mechanism shared by the Cvt, autophagy, and pexophagy pathways.
Ccz1 and Mon1 Are Required for Multiple Vacuole Delivery Pathways
It has been reported that the ccz1Δ strain exhibits a severe vacuolar hydrolase sorting defect as well as a fragmented vacuole phenotype (11). To gain a better understanding of vacuole protein delivery in the mon1Δ strain, we examined different cargo proteins that are targeted to the vacuole by various mechanisms. Carboxypeptidase Y, Prc1, is transported to the vacuole through the CPY pathway, a transport itinerary that includes the ER, Golgi complex, and endosome. In the wild type strain, Prc1 is matured (mPrc1) with a half-time of 5–10 min. Approximately 5% of Prc1 is secreted from the cell under standard conditions used for this type of analysis (Fig. 2A). In contrast, in typical vps mutants such as vps5, Prc1 remains as precursor, and the majority is secreted into the extracellular fraction as the p2 (Golgi-modified precursor) form. In the mon1Δ strain, a small amount of mPrc1 (∼5%) was found in the intracellular fraction. However, the majority of the protein was found in the p2 form even after 30 min of chase, and approximately half was missorted to the extracellular fraction (Fig. 2A). Similar results were seen with Pep4 (data not shown). The Prc1-processing defect in the ccz1Δ strain has already been published (11). Consistent with the published data, the ccz1Δ strain showed a Prc1 sorting defect by pulse/chase analysis but accumulated a substantial amount of mPrc1 under steady-state conditions (data not shown). The steady-state accumulation of mPrc1 probably reflects a block in exit from a pre-vacuolar compartment that has attained protease-processing capacity (34).
FIG. 2. Multiple vacuole transport pathways are blocked in the ccz1Δ and mon1Δ strains.
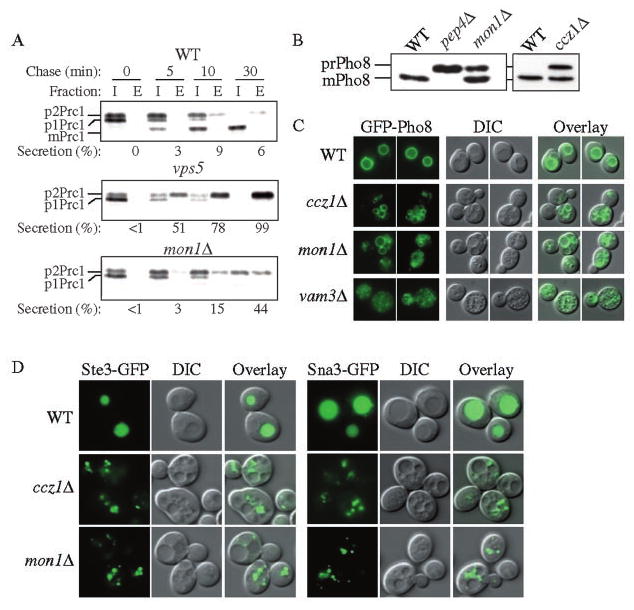
A, the mon1Δ strain missorts Prc1 into the extracellular fraction. The wild type (WT, SEY6210), vps5, and mon1Δ (JSY1) strains were grown to mid-log phase and converted into spheroplasts. The spheroplasts were labeled for 5 min and subjected to a non-radioactive chase for the time indicated at 30 °C. Samples were separated into intracellular (I) and extracellular (E) fractions, immunoprecipitated with antiserum to Prc1, and separated by SDS-PAGE. B, mon1Δ and ccz1Δ strains accumulate precursor Pho8. The wild type, pep4Δ (TVY1), ccz1Δ (CWY3), and mon1Δ strains were grown to mid-log phase in SMD medium and analyzed by immunoblot using antiserum against Pho8. C, GFP-Pho8 reaches the vacuoles of the ccz1Δ and mon1Δ strains. Wild type, ccz1Δ, mon1Δ, and vam3Δ (CWY40) strains were transformed with pGFP-Pho8 (426) and grown in SMD medium to mid-log phase followed by fluorescence microscopy. D, endocytic and MVB vesicles accumulated in the ccz1Δ and mon1Δ cells outside of their vacuoles. The wild type, ccz1Δ, and mon1Δ strains expressing the endocytosis pathway marker Ste3-GFP (316), or the MVB pathway marker Sna3 GFP(416) were grown to mid-log phase followed by fluorescence microscopy. DIC, differential interference contrast.
Next, we examined the delivery of the vacuole integral membrane protein Pho8 through the ALP pathway. Under steady-state conditions, both the mon1Δ and ccz1Δ strains showed an ∼50% block of Pho8 processing, while the wild type strain accumulated mature Pho8 (Fig. 2B and Ref. 11). To further examine the delivery of Pho8 in these two mutant strains, we followed the localization of GFP-Pho8. We used a vam3Δ strain as a control because the v-SNARE Vam3 is required for the ALP pathway. Wild type, ccz1Δ, mon1Δ, and vam3Δ strains expressing GFP-Pho8 were grown to mid-log phase and examined by fluorescent microscopy. Similar to the severe vacuole fragmentation in the vam3Δ strain, both ccz1Δ and mon1Δ also displayed a fragmented vacuole phenotype, although a substantial population of cells exhibited some relatively larger vacuoles (Fig. 2, C and D). In wild type cells, GFP-Pho8 was detected at the vacuole membrane indicating proper delivery of this hydrolase to the vacuole. In contrast to the wild type cells, GFP-Pho8 accumulated in multiple punctate structures in the vam3Δ strain (Fig. 2C). Although vacuoles in the vam3Δ strain are highly fragmented, we were able to conclude that none of these fluorescent dots were inside of the fragmented vacuoles. We found that both the ccz1Δ and mon1Δ strains accumulated GFP-Pho8 on the vacuole membrane but also displayed some punctate GFP-Pho8 dots outside of their fragmented vacuoles, suggesting only a partial block in the delivery of Pho8 (Fig. 2C). Similar results were observed by examining cells expressing Nyv1-GFP, which is also delivered to the vacuole by the ALP pathway (data not shown). These data suggest a partial block in the ALP pathway in the mon1Δ and ccz1Δ strains.
In addition to the Cvt/Apg, CPY, and ALP pathways, proteins destined for the vacuole also transit through the endocytic and MVB pathways. We monitored endocytosis by looking at the localization of Ste3-GFP in the mon1Δ and ccz1Δ strains. Ste3 is the a factor receptor and is down-regulated by both ligand-dependent and ligand-independent modes of endocytosis (35). In this study, we examined the ligand-independent mode. In the wild type strain, Ste3-GFP was diffusely accumulated in the vacuole (Fig. 2D). In contrast, Ste3-GFP was localized to multiple punctate structures outside of the vacuole in the mon1Δ and ccz1Δ strains. These structures may represent endocytic vesicles. These data indicate an endocytic defect in the mon1Δ and ccz1Δ strains. Finally, we examined the localization of Sna3-GFP through the MVB pathway (24). In contrast to the vacuole lumen staining seen in the wild type cells, Sna3-GFP in the ccz1Δ and mon1Δ cells displayed a large population of small punctate structures outside of vacuoles (Fig. 2D), which may represent the late endosome/MVB compartments. Similar results were seen using other MVB pathway marker proteins including Phm5-GFP and GFP-CPS (data not shown).
Ccz1 and Mon1 Are Required for Vesicle Fusion with the Vacuole
The majority of cvt, apg, and aut mutants identified previously were specific to the Cvt and autophagy pathways and did not show defects in other vacuole delivery pathways. These mutants all appear to function at the stage of vesicle induction and/or formation. However, the cvt4 and cvt8 mutants were found to be allelic with VPS39/VAM6 and VPS41/VAM2, respectively (25), indicating a possible overlap with genes whose products play a more general role in vacuole protein localization. Because the mon1Δ and ccz1Δ mutants are defective in multiple vacuole delivery pathways, we propose that Mon1 and Ccz1 have general roles for protein trafficking pathways presumably through their requirements for the vesicle fusion step with the vacuole.
To carefully examine the proposed role of Ccz1 and Mon1 for the fusion of vesicles with the vacuole, we utilized biochemical assays that monitor the block in the transport of prApe1 (17). To determine whether prApe1 was able to bind membrane, we performed a flotation analysis. A total membrane fraction from lysed spheroplasts was subjected to centrifugation through a Ficoll step gradient. In the mon1Δ strain, a portion of prApe1 and the integral ER membrane control protein Dpm1 were pelletable and separated into the float (F) fraction in the absence of detergent (Fig. 3A). In contrast, the cytosolic protein Pgk1 was found exclusively in the supernatant (S) fraction. A similar result was seen with the ccz1Δ strain (data not shown). This result suggests that prApe1 is able to bind to its target membrane.
FIG. 3. Mon1 and Ccz1 are required after completion of Cvt vesicles.
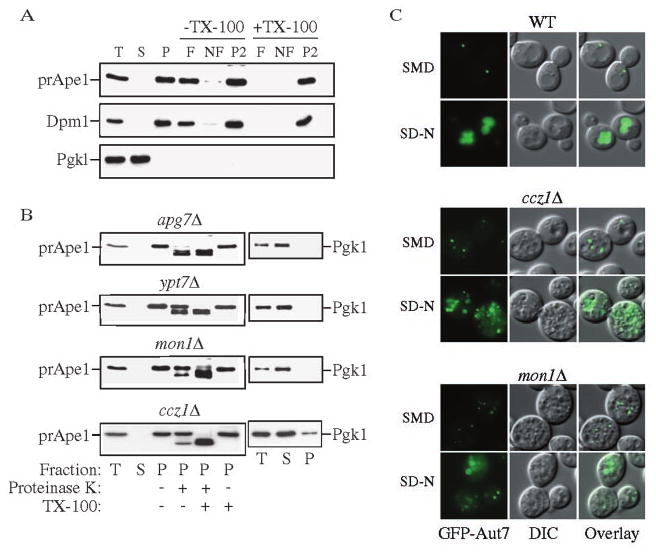
A, precursor Ape1 is membrane associated in the mon1Δ strain. The mon1Δ (JSY1) strain was grown to mid-log phase and converted into spheroplasts. The spheroplasts were lysed osmotically and centrifuged through a Ficoll step gradient with or without Triton X-100 as described under “Experimental Procedures.” Membrane-containing float (F), nonfloat (NF), and pellet (P2) fractions were collected and subjected to immunoblot using antisera or antibodies to Ape1, Dpm1, and Pgk1. B, precursor Ape1 is protease-protected in the mon1Δ and ccz1Δ strains. The apg7Δ (VDY101), ypt7Δ (WSY99), mon1Δ, and ccz1Δ (CWY3) strains were grown to mid-log phase and converted into spheroplasts followed by osmotic lysis. The total lysate (T) was resolved into supernatant (S) and pellet (P) fractions by a 13,000 × g centrifugation, and a portion analyzed by immunoblot using antiserum to Ape1 and Pgk1. The remaining pellet fractions were subjected to protease treatment in the absence or presence of Triton X-100 and subjected to immunoblot using antiserum to Ape1. C, Cvt pathway marker GFP-Aut7 accumulated outside of the vacuole in the mon1Δ and ccz1Δ strains. The wild type, ccz1Δ, and mon1Δ strains were transformed with pCuGFPAut7 (22). The strains were grown to mid-log phase, and images were taken with a fluorescent microscope.
To determine if prApe1 is sequestered within completed Cvt vesicles, we next carried out a protease-sensitivity analysis. Spheroplasts were osmotically lysed as described under “Experimental Procedures,” and the low speed pellet fractions were subjected to exogenous proteinase K treatment in the absence or presence of detergent. The apg7Δ strain is defective in the conjugation of Apg12 to Apg5 and is unable to form completed Cvt vesicles/autophagosomes (36, 37). This strain accumulates prApe1 in a protease-sensitive state in the absence of detergent (Fig. 3B). Ypt7 is a Rab protein that is required for the fusion of Cvt vesicles/autophagosomes with the vacuole (37), and ypt7Δ cells accumulate protease-protected prApe1. Precursor Ape1 in the mon1Δ and ccz1Δ strains was also protease-protected in the absence of detergent (Fig. 3B), suggesting that it accumulated within completed vesicles. The separation of Pgk1 into the supernatant fraction verifies that accumulation of protease-protected prApe1 was not due to inefficient spheroplast lysis.
To determine whether prApe1 was present within cytosolic or subvacuolar vesicles, we extended our analysis of the Cvt pathway by looking at GFP-Aut7 in vivo. Aut7 is required for Cvt vesicle and autophagosome formation and remains associated with these vesicles following completion (22, 38). Thus, it serves as a vesicle marker. Consistent with previously published data, GFP-Aut7 was seen as a single punctate structure accumulating outside the vacuole in the wild type cells grown in rich medium (Fig. 3C, SMD). Under starvation conditions, Aut7 is induced, and we observed a bright vacuole lumen staining of GFP-Aut7 in the wild type strain (Fig. 3C, SD-N). In contrast, GFP-Aut7 in the mon1Δ and ccz1Δ strains displayed multiple punctate dots similar to that seen in ypt7Δ cells (Fig. 3C and Ref. 39). By overlaying the fluorescent and DIC images, we could determine that the multiple punctate structures in these two strains were located outside of the fragmented vacuoles. Under starvation conditions, we detected a stronger GFP-Aut7 signal in the two mutant strains suggesting that they are not defective in Aut7 induction. Some larger double membrane structures that might represent autophagosomes were detected outside of vacuoles in the two mutant strains but none of the GFP-Aut7 appeared to be coincident with the vacuole. Overall, these data suggest that prApe1 is accumulated within completed cytosolic vesicles in both the mon1Δ and ccz1Δ strains. Thus, we conclude that Ccz1 and Mon1 are required for the fusion step of these vesicles with the vacuole.
Ccz1 and Mon1 Are Membrane-associated Proteins
In order to study the localization of Ccz1 and Mon1, we tagged both proteins with the HA epitope. The COOH-terminal HA tagging did not cause dysfunction of Ccz1 or Mon1, because the respective constructs on plasmids complemented the prApe1-sorting defect of null cells and rescued the fragmented vacuole phenotypes (data not shown). It has been shown that Ccz1 is enriched in the P13 and P100 fractions (11). We decided to check Mon1-HA localization together with Ccz1-HA. A strain with Mon1-HA tagged at the chromosomal locus was transformed with pCCZ1-HA(416), grown to mid-log phase, and converted to spheroplasts followed by osmotic lysis. The lysed spheroplasts were subjected to velocity sedimentation as described under “Experimental Procedures.” The cytosolic protein Pgk1 was recovered primarily from the S100 fraction, while the vacuole membrane protein Pho8 was located exclusively in the P13 fraction indicating efficient separation (Fig. 4A). We also examined the localization of Ypt7 and found it was mostly in the P13 fraction. Mon1-HA was recovered in the P13 and P100 fractions; however, we found that Ccz1-HA was not only detected in the P13 and P100 fractions but that a substantial amount also appeared in the S100 fraction indicating a cytosolic population of this protein (Fig. 4A). Next we extended our analyses for these two proteins by examining the stability of their membrane binding. We found that both Ccz1-HA and Mon1-HA were largely stripped from the membrane by treatment with 0.1 m Na2CO3 (pH 10.5) and 3 m urea, while approximately half of each protein remained membrane bound in the presence of 1 m KCl and 1% Triton X-100 (Fig. 4B). Taken together, these data suggest that Ccz1-HA and Mon1-HA are peripherally attached to a membrane compartment(s) that are relatively detergent insoluble. The lack of solubility in the presence of detergent may indicate that both proteins associate with a large protein complex.
FIG. 4. Ccz1 and Mon1 are peripheral membrane proteins.
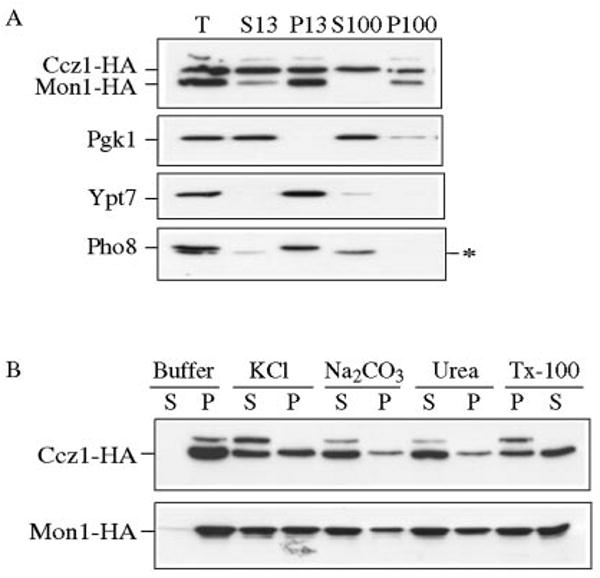
A, Ccz1-HA and Mon1-HA are pelletable. A strain with an HA tag at the Mon1 locus (PSY35) transformed with pCCZ1-HA(416) was grown to mid-log phase and converted into spheroplasts, followed by osmotic lysis in PS200 buffer containing 5 mm MgCl2. The total (T) fraction was separated into low speed supernatant (S13) and pellet (P13) fractions by a 13,000 × g centrifugation step. The S13 fraction was further separated into high-speed supernatant (S100) and pellet (P100) fractions by centrifugation at 100,000 × g. The collected fractions were subjected to immunoblot using antisera to HA, Pgk1, Ypt7, and Pho8. The asterisk marks a cross-reacting band that migrates below Pho8. B, biochemical characterization of pelletable Ccz1-HA and Mon1-HA. Spheroplasts from the Mon1-HA and Ccz1-HA (PSY36) strains were osmotically lysed and spun as described under “Experimental Procedures.” The pellet fractions were resuspended in buffer alone or buffer containing 1 m KCL, 0.1 m Na2CO3, pH 10.5, 3 m urea, or 1% Triton X-100 and separated into supernatant (S) and pellet (P) fractions. Samples were resolved by immunoblot with anti-HA antiserum.
In Vivo Localization of Ccz1-GFP and GFP-Mon1
To investigate the site of action of Ccz1 and Mon1 in vivo, we constructed strains where GFP was fused to the COOH terminus of the MON1 and CCZ1 ORFs at the chromosomal loci. These strains displayed a normal vacuolar phenotype indicating that the expressed fusion proteins are functional (Fig. 5). When cells grown in YPD to mid-log phase (and washed in minimal medium) were examined, Ccz1-GFP was detected in 2–5 perivacuolar dots per cell and also displayed a faint vacuole membrane staining (Fig. 5A). These GFP-staining dot structures were very mobile and could be seen to move around the vacuoles (data not shown). Mon1-GFP had a similar staining pattern to Ccz1-GFP, although the fluorescent signal was weaker.
FIG. 5. In vivo localization of Ccz1 and Mon1.
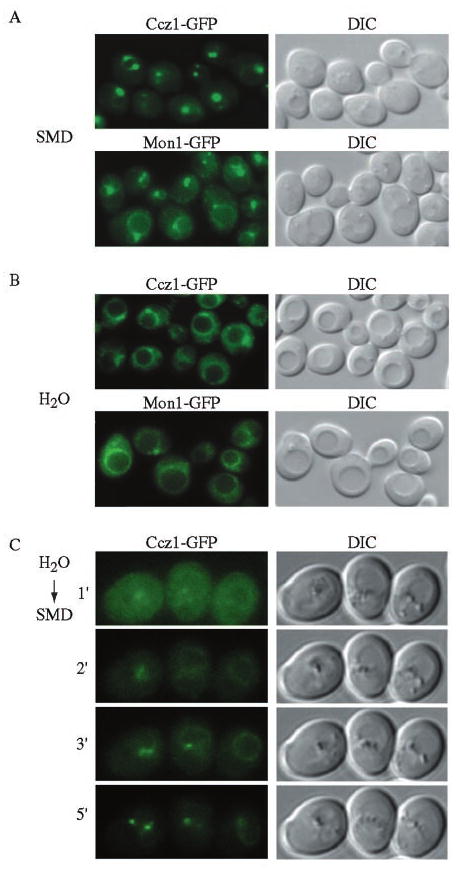
Yeast strains with Ccz1-GFP (PSY46) and Mon1-GFP (PSY47) integrated at the chromosomal loci were grown to mid-log phase in YPD, then washed and resuspended in SMD medium (A) or H2O (B) before being examined by fluorescence microscopy. Ccz1 and Mon1 localize to punctate perivacuolar structures and osmotic shock results in a redistribution to the vacuolar rim. DIC, differential interference contrast. C, a yeast strain with chromosomal Ccz1-GFP (PSY46) was grown in YPD to mid-log phase, washed, and resuspended in water for 5 min, followed by a shift to SMD conditions prior to fluorescence microscopy. Images were taken at minute intervals after the SMD treatment as indicated. Ccz1-GFP gradually redistributed to the punctate structures within 5 min based on time-lapse microscopy. The vacuolar rim staining is difficult to detect due to photobleaching resulting from the time-lapse exposures. Essentially identical results were obtained for Mon1-GFP.
Washing yeast cells under low osmotic conditions causes multilobed vacuoles to fuse together and swell. When the Ccz1-GFP and Mon1-GFP yeast were washed with water prior to microscopy, the vacuoles could be seen to enlarge (Fig. 5B). The punctate staining pattern of Ccz1-GFP and Mon1-GFP was largely lost and was replaced by an increased signal on the vacuolar rim. Furthermore, by shifting the hypotonic treatment back to SMD, we were able to recover the punctate-staining pattern along with the fainter vacuole ring localization of Ccz1-GFP and Mon1-GFP (Fig. 5C). The punctate pattern appeared rapidly and became saturated within 5 min of reversing the osmotic conditions. Therefore, we conclude that the majority of Ccz1-Mon1 complexes localize to several membrane structures right next to the vacuole and could possibly attach to the vacuole membrane to achieve their function.
Because both proteins displayed a similar subcellular distribution by fluorescent microscopy, we extended the analysis by examining the co-localization of YFP- and CFP-tagged proteins. Because Mon1 tagged chromosomally at the COOH terminus with CFP or YFP showed a very weak fluorescent signal, we replaced chromosomal MON1 with YFP-MON1 under the control of the CUP1 promoter. The resulting strain showed normal vacuolar morphology (Fig. 6A). Ccz1-CFP and YFP-Mon1 showed multiple punctate dots similar to the pattern seen with the GFP-tagged constructs. Furthermore, the two proteins co-localized (Fig. 6A). The staining pattern seen with GFP-Mon1 and Ccz1-GFP was different from the single punctate structure observed with most Apg/Cvt proteins that localize to the pre-autophagosomal structure (20). To determine whether Mon1 and Ccz1 localized to a distinct compartment, we compared the distribution of Ccz1-YFP to Cvt19-CFP. Cvt19 is a receptor or adaptor for prApe1 (40) and localizes to the pre-autophagosomal structure (20). We found that the punctate dots corresponding to Ccz1-YFP did not co-localize with the pre-autophagosomal structure represented by Cvt19-CFP (Fig. 6B).
FIG. 6. Ccz1 and Mon1 co-localize to a perivacuolar compartment different from the pre-autophagosomal structure.
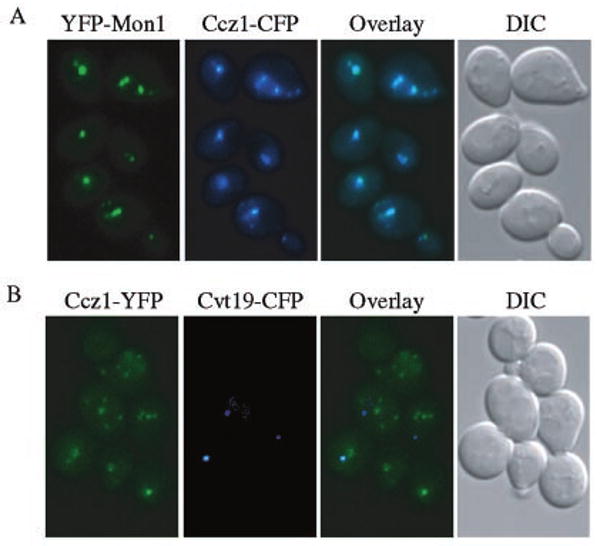
A, strain PSY45 expressing YFP-Mon1 from the CUP1 promoter and Ccz1-CFP was grown to mid-log phase in YPD. YFP-Mon1 expression was induced with 50 μm CuSO4 for 1 h prior to microscopy. B, strain PSY42 expressing Ccz1-YFP from the chromosomal loci and Cvt19-CFP from a plasmid, was grown to mid-log phase in SMD and then for 1 h in YPD. All cells were washed once in SMD before being examined by fluorescence microscopy.
Ccz1 and Mon1 Form a Stable Protein Complex
We have shown that the ccz1Δ and mon1Δ strains have similar vacuole protein transport defects, and that Ccz1 and Mon1 are both pelletable and that their association with membranes has similar biochemical properties. In addition, both proteins co-localized by fluorescent microscopy (Fig. 6A). To further investigate the subcellular localization of Ccz1-HA and Mon1-HA, we resolved the membrane compartments on an OptiPrep density gradient as described under “Experimental Procedures.” After centrifugation, fractions were collected from the top of the gradient and analyzed by immunoblot. Ccz1-HA and Mon1-HA were both detected in fractions 8 through 13 (Fig. 7A). We compared their distribution with endomembrane markers Dpm1 (ER), Anp1 (Golgi), Pep12 (endosome), and Pho8 (vacuole). All these proteins displayed fractionation patterns that were distinct from Ccz1-HA and Mon1-HA. We also checked the localization of Ypt7 that has been suggested to interact with Ccz1, and found that a population of these proteins overlapped in fractions 8 and 9, but that the peaks were distinct (Fig. 7A).
FIG. 7. Ccz1 and Mon1 physically interact.
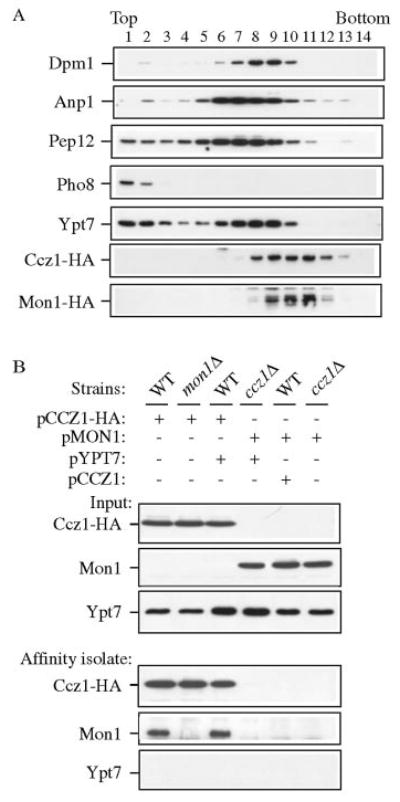
A, Ccz1 and Mon1 co-fractionated but were separated from endomembrane marker proteins by OptiPrep density gradients. The Mon1-HA strain (PSY35) expressing pCCZ1-HA(416) was analyzed by density gradient separation as described under “Experimental Procedures.” Fractions were subjected to immunoblot using antisera or antibodies to Dpm1 (ER), Anp1 (Golgi), Pep12 (endosome), Pho8 (vacuole), Ypt7, and HA. B, Ccz1-HA co-precipitates Mon1 by native immunoprecipitation. Wild type, ccz1Δ (CWY3), and mon1Δ (JSY1) strains were transformed with pCCZ1-HA(426), pMON1(426), and/or pYPT7(424), and were grown to mid-log phase followed by glass bead lysis in HEPES native immunoprecipitation buffer. An aliquot (10 μl) of lysate was used as the loading control. Lysates were incubated with anti-HA antibody and protein A-Sepharose as described under “Experimental Procedures” and subjected to immunoblot against HA, Mon1, and Ypt7.
To extend this analysis we examined whether Ccz1 physically interacts with Mon1, using a co-immunoprecipitation assay. We expressed a combination of pCCZ1-HA(426), pMON1 (426), and pYPT7(424) in several strain backgrounds (Fig. 7B). Overexpression of the respective proteins did not cause any significant effect for the wild type or mutant strains with regard to prApe1 maturation (data not shown). Cells were lysed with glass beads, and the crude cell lysate was subjected to Western blot as the loading control (Fig. 7B, input). We generated polyclonal antiserum against Mon1 as described under “Experimental Procedures.” The antiserum detected a very weak band of ∼70 kDa in the wild type strain and showed a greatly increased level of this band in cells expressing a multicopy MON1 plasmid (Fig. 7B and data not shown). Cells were subjected to a native immunoprecipitation with antiserum against HA as described under “Experimental Procedures.” The precipitated immune complexes (affinity isolate) were then subjected to SDS-PAGE and Western blots using antibodies or antiserum against HA, Mon1, and Ypt7. In the wild type strain containing overexpressed Ccz1-HA, the immune complex pulled down a substantial amount of the chromosomal Mon1 (Fig. 7B). The immunoaffinity signal is specific against Mon1 because no Mon1 signal was detected in the mon1Δ strain under the same immunoprecipitation conditions. When Mon1 was overexpressed in the absence of Ccz1-HA, none of the Mon1 could be detected in the immune complex indicating that the isolation of Mon1 was dependent on Ccz1 (Fig. 7B). The reverse interaction was also observed when we carried out the immunoprecipitation using antiserum against Mon1 (data not shown). It has been published previously that Ccz1-HA co-immunoprecipitates with Ypt7 (12). However, we were unable to detect any Ypt7 signal in this experiment. It is possible that the interaction between Ypt7 and Ccz1 (and maybe also Mon1) is transient, whereas the interaction between Ccz1 and Mon1 is abundant and stable.
Discussion
Ccz1 and Mon1 Are Required in Multiple Pathways to the Yeast Vacuole
The biosynthetic Cvt pathway that delivers the precursor form of the vacuolar hydrolase Ape1 from the cytoplasm to the yeast vacuole is the subject of our investigation. We have identified many cvt mutants and found that most of them exhibited an extensive genetic, biochemical, and morphological overlap with apg and aut mutants that are defective in the degradative autophagy pathway (reviewed in Refs. 5 and 28). To gain a comprehensive understanding of the Cvt pathway, we further identified mutants defective in prApe1 sorting by screening the yeast deletion library. Using this strategy, we have identified two new proteins, Ccz1 and Mon1, required for the Cvt, autophagy and pexophagy pathways (Fig. 1). To further understand the precise role(s) of these two proteins we first performed an extensive analysis to determine whether the ccz1Δ and mon1Δ strains had pleiotropic effects in other vacuole transport pathways. The CPY pathway involves transport through a portion of the secretory pathway, and its itinerary includes the prevacuolar compartment (PVC)/endosome. We found that the CPY pathway cargo protein Prc1 was missorted as the p2 form in the mon1Δ strain. A similar secretion phenotype has been shown previously for the ccz1Δ strain (11).
The ALP pathway diverges from the CPY pathway in the late Golgi; cargo proteins such as Pho8 do not pass through the PVC before reaching the vacuole. We showed a partial defect in Pho8 processing in these two mutant strains (Fig. 2B). When we further studied the localization of GFP-Pho8, we observed an appearance of the chimera on fragmented vacuoles in these two mutants. In contrast, a vam3Δ strain accumulated a large population of small vesicles containing GFP-Pho8 that were not observed on the fragmented vacuoles. Although we do see some GFP-Pho8 on intermediate vesicle structures in the ccz1Δ and mon1Δ strains, the majority of GFP-Pho8 targeted to their fragmented vacuoles (Fig. 2C). Thus, it appears that the ALP pathway that bypasses the endosome is relatively unaffected in the ccz1Δ and mon1Δ strains. The partial Pho8 processing defect might reflect reduced processing capacity of the vacuole resulting from the missorting of Prc1, Pep4, and other hydrolases that utilize the CPY pathway. This possibility is supported by the observation of 50% precursor Pho8 in purified vacuoles from the ccz1Δ and mon1Δ strains (data not shown). Analysis of Ste3-GFP and Sna3-GFP revealed blocks in the endocytosis and MVB pathways (Fig. 2D). These data indicate that mon1Δ and ccz1Δ have pleiotropic defects in multiple vacuole-sorting pathways.
Ccz1 and Mon1 Function at the Vesicle Fusion Step
The defect in multiple vacuole delivery pathways and the observation of vesicle-like transport intermediates accumulated in the two mutants suggested that Mon1 and Ccz1 might act at the stage of fusion with the vacuole. Taking advantage of the established model for the Cvt/Apg pathway (28), we could assess the role of Mon1 and Ccz1 through biochemical analyses that examined the state of prApe1 (17). In the past few years, we have dissected the Cvt/Apg pathway into several discrete steps. These include vesicle nucleation and cargo sequestration, vesicle formation/completion, docking/fusion, and subvacuolar vesicle lysis followed by maturation of prApe1 (Fig. 8). Accordingly, we have developed biochemical tools to assess the stage at which the cargo protein prApe1 accumulates during transport in mutant strains. The membrane flotation analysis indicated that prApe1 in the mon1Δ strain was membrane-associated (Fig. 3A). Furthermore, we found that prApe1 in the mon1Δ and ccz1Δ strains was in a protease-protected form, suggesting that the sequestration step for the Cvt complex was completed. Thus, mon1Δ and ccz1Δ are the first two mutants that have been isolated in several screens for apg, aut, and cvt mutants that act after completion of the sequestering vesicles.
FIG. 8. Working model for the Cvt and autophagy pathways.
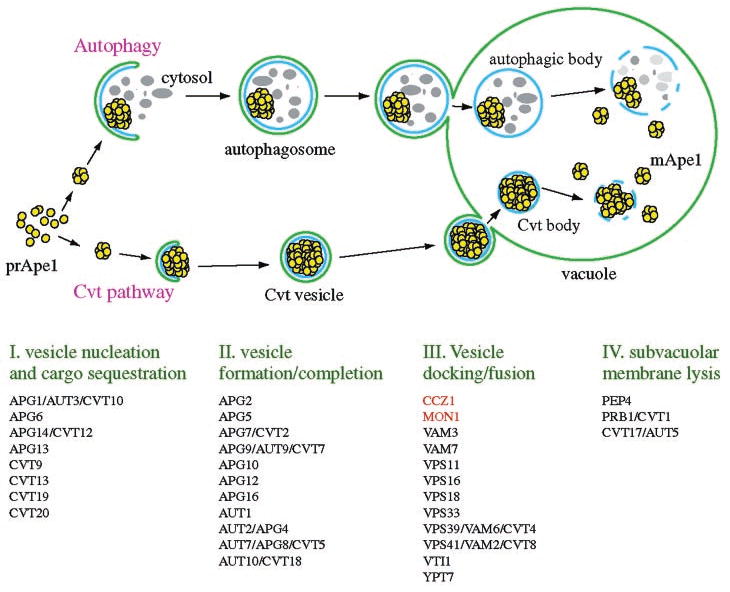
The type of vesicles that are produced depends on the nutrient conditions. Autophagosomes form during autophagy under conditions of nutrient deprivation. Cvt vesicles are generated through the Cvt pathway under nutrient rich conditions. Four general steps of both pathways are indicated below the illustration. Components that are required for the Cvt and Apg pathways are indicated based on their putative roles.
Protease-protected prApe1 could accumulate in subvacuolar vesicles within the vacuole lumen in strains defective in the vesicle lysis step. To verify that the ccz1Δ and mon1Δ strains are defective in delivery to the vacuole, we investigated the distribution of GFP-Aut7 in the two mutant strains. Aut7 is a component that is required for Cvt vesicle formation and is itself localized to Cvt vesicles (41). Accordingly, Aut7 serves as a useful vesicle marker. In contrast to the single perivacuolar (SMD) or luminal (SD-N) dot observed in the wild type strain, GFP-Aut7 is localized outside of the vacuole in multiple punctate structures in the ccz1Δ and mon1Δ strains (Fig. 3C). Furthermore, the localization of GFP-Aut7 in the two mutants was very similar to that seen in the ypt7Δ strain. Ypt7 is a Rab GTPase that is required for the fusion of multiple vesicle types, including Cvt vesicles and autophagosomes, with the vacuole (37). Taken together, our data indicate that Ccz1 and Mon1 function at the stage of fusion of autophagosomes/Cvt vesicles with the vacuole.
Molecular Function of Ccz1 and Mon1 in Fusion
Ccz1 and Mon1 are both peripheral membrane proteins (Fig. 4). There is an additional cytosolic pool of Ccz1 (Fig. 4A). The two proteins co-localized to a relatively higher density compartment that did not peak at the same position as most endomembrane markers (Fig. 7A). Both proteins appeared to function as a stable protein complex (Fig. 7B) termed the Ccz1-Mon1 complex in this study. Similarly, in vivo examination of Ccz1 and Mon1 tagged with fluorescent markers suggested that the Ccz1-Mon1 complex localizes to perivacuolar dot structures that overlap (Fig. 6). The previously published immunofluorescent data suggest that Ccz1 co-localizes with the endosomal marker Nhx1 (11), suggesting that these dots might represent some kind of endosome/PVC structure. Neither protein localized to the pre-autophagosomal structure that is thought to be the site of Cvt vesicle/autophagosome synthesis or the site of the donor membrane (Fig. 6). We have also observed faint vacuole membrane staining for both Mon1 and Ccz1, indicating these proteins might target to the vacuole membrane to achieve their function in fusion. Although we did not observe a vacuole peak of Ccz1 and/or Mon1 in our OptiPrep density gradient, we could not rule out the possibility that their association with the vacuole is relatively weak and is lost during the biochemical procedures. A similar phenotype is seen with Cvt18, which is lost from the vacuole following spheroplast lysis (42).
A summary of the published data of the components involved in the Cvt/Apg pathways is shown in Fig. 8. At the fusion step, SNARE proteins including Vam3 (43), Vti1 (44), and Vam7 (45) are required for both Cvt vesicle and autophagosome fusion with the vacuole. Ypt7, the Rab GTPase (37), and its proposed effector complex, termed the class C Vps/HOPS (homotypic fusion and vacuole protein sorting) complex that comprises Vps11, Vps16, Vps18, Vps33, Vps39, and Vps41 (25, 46), is also essential machinery at this step. Among these identified components required for the Cvt vesicle/autophagosome fusion step, none retain a normal vacuole phenotype when they are deleted from the genome. On the other hand, most but not all mutants that exhibit a vacuole fragmentation phenotype are defective for the Cvt/autophagy pathway. For example, although the kcs1Δ strain showed a fragmented vacuole phenotype, it accumulated the mature form of Ape1 (Ref. 47).2 Similarly, some vps mutants such as vps5 have fragmented vacuoles but are essentially normal for import of prApe1 (15).
In this study we introduce the novel Ccz1-Mon1 complex as acting at the fusion step in the Apg/Cvt pathways, as well as in most other pathways that involve vesicle fusion with the vacuole. Because of its apparently general role in vacuole biogenesis and function, it is important to examine whether the Ccz1-Mon1 complex is part of the basic vacuole fusion mechanism. For example, what is the specific molecular role of the Ccz1-Mon1 complex? Ccz1 has been reported to interact with Ypt7 (12). Is the Ccz1-Mon1 complex also part of the Ypt7 effector complex? Although our co-immunoprecipitation data did not reproduce the published result of Ccz1-HA and Ypt7 interaction, we suggest that this interaction is transient whereas the Ccz1-Mon1 complex is very abundant and stable. We are currently trying to determine the specific role of the Ccz1-Mon1 complex in the Cvt and Apg pathways. An in vitro analysis will provide additional insight into their function in the mechanism of vesicle fusion.
Acknowledgments
We thank Drs. Scott Emr, Mark Longtine, Sean Munro, Jeremy Thorner, and William Wickner and the Yeast Resource Center for supplying antiserum and plasmids. We thank members of the Klionsky laboratory, especially Drs. Fulvio Reggiori and John Kim, for helpful discussions and providing plasmids.
Footnotes
This work was supported by National Institutes of Health Public Health Service Grant GM53396 (to D. J. K.), the Lewis E. and Elaine Prince Wehmeyer Trust (to C.-W. W.), and a research fellowship from the Science and Technology Agency of Japan (to J. S.).
The abbreviations used are: CPY, carboxypeptidase Y; ALP, alkaline phosphatase; Ape1, aminopeptidase I; CFP, cyan fluorescent protein; Cvt, cytoplasm to vacuole targeting; GFP, green fluorescent protein; prApe1, precursor aminopeptidase I; PVC, pre-vacuolar compartment; SMD, synthetic minimal medium with dextrose; SD/-N, synthetic minimal medium with dextrose but lacking nitrogen; YFP, yellow fluorescent protein; ORF, open reading frame; MES, 4-morpho-lineethanesulfonic acid; PIPES, 1,4-piperazinediethanesulfonic acid.
P. E. Stromhaug and D. J. Klionsky, unpublished data.
References
- 1.Klionsky DJ, Herman PK, Emr SD. Microbiol Rev. 1990;54:266–292. doi: 10.1128/mr.54.3.266-292.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim J, Scott SV, Klionsky DJ. Int Rev Cytol. 2000;198:153–201. doi: 10.1016/s0074-7696(00)98005-7. [DOI] [PubMed] [Google Scholar]
- 3.Lemmon SK, Traub LM. Curr Opin Cell Biol. 2000;12:457–466. doi: 10.1016/s0955-0674(00)00117-4. [DOI] [PubMed] [Google Scholar]
- 4.Weisman LS, Wickner W. Science. 1988;241:589–591. doi: 10.1126/science.3041591. [DOI] [PubMed] [Google Scholar]
- 5.Klionsky DJ, Ohsumi Y. Annu Rev Cell Dev Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Klionsky DJ. J Biol Chem. 1998;273:10807–10810. doi: 10.1074/jbc.273.18.10807. [DOI] [PubMed] [Google Scholar]
- 7.Baba M, Osumi M, Scott SV, Klionsky DJ, Ohsumi Y. J Cell Biol. 1997;139:1687–1695. doi: 10.1083/jcb.139.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott SV, Baba M, Ohsumi Y, Klionsky DJ. J Cell Biol. 1997;138:37–44. doi: 10.1083/jcb.138.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klionsky DJ, Emr SD. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harding TM, Hefner-Gravink A, Thumm M, Klionsky DJ. J Biol Chem. 1996;271:17621–17624. doi: 10.1074/jbc.271.30.17621. [DOI] [PubMed] [Google Scholar]
- 11.Kucharczyk R, Dupre S, Avaro S, Haguenauer-Tsapis R, Slonimski PP, Rytka J. J Cell Sci. 2000;113:4301–4311. doi: 10.1242/jcs.113.23.4301. [DOI] [PubMed] [Google Scholar]
- 12.Kucharczyk R, Kierzek AM, Slonimski PP, Rytka J. J Cell Sci. 2001;114:3137–3145. doi: 10.1242/jcs.114.17.3137. [DOI] [PubMed] [Google Scholar]
- 13.Muren E, Oyen M, Barmark G, Ronne H. Yeast. 2001;18:163–172. doi: 10.1002/1097-0061(20010130)18:2<163::AID-YEA659>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, III, Steever AB, Wach A, Philippsen P, Pringle JR. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 15.Klionsky DJ, Cueva R, Yaver DS. J Cell Biol. 1992;119:287–299. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klionsky DJ, Banta LM, Emr SD. Mol Cell Biol. 1988;8:2105–2116. doi: 10.1128/mcb.8.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang CW, Kim J, Huang WP, Abeliovich H, Stromhaug PE, Dunn WA, Jr, Klionsky DJ. J Biol Chem. 2001;276:30442–30451. doi: 10.1074/jbc.M102342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y, Klionsky DJ. J Cell Biol. 2000;148:465–480. doi: 10.1083/jcb.148.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rehling P, Darsow T, Katzmann DJ, Emr SD. Nat Cell Biol. 1999;1:346–353. doi: 10.1038/14037. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Huang WP, Stromhaug PE, Klionsky DJ. J Biol Chem. 2002;277:763–773. doi: 10.1074/jbc.M109134200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urbanowski JL, Piper RC. J Biol Chem. 1999;274:38061–38070. doi: 10.1074/jbc.274.53.38061. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Huang WP, Klionsky DJ. J Cell Biol. 2001;152:51–64. doi: 10.1083/jcb.152.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cowles CR, Snyder WB, Burd CG, Emr SD. EMBO J. 1997;16:2769–2782. doi: 10.1093/emboj/16.10.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reggiori F, Pelham HRB. EMBO J. 2001;20:5176–5186. doi: 10.1093/emboj/20.18.5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harding TM, Morano KA, Scott SV, Klionsky DJ. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Kamada Y, Stromhaug PE, Guan J, Hefner-Gravink A, Baba M, Scott SV, Ohsumi Y, Dunn WA, Jr, Klionsky DJ. J Cell Biol. 2001;153:381–396. doi: 10.1083/jcb.153.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vida TA, Emr SD. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Klionsky DJ. Annu Rev Biochem. 2000;69:303–342. doi: 10.1146/annurev.biochem.69.1.303. [DOI] [PubMed] [Google Scholar]
- 29.Kucharczyk R, Gromadka R, Migdalski A, Slonimski PP, Rytka J. Yeast. 1999;15:987–1000. doi: 10.1002/(SICI)1097-0061(199907)15:10B<987::AID-YEA403>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 30.Abeliovich H, Dunn WA, Jr, Kim J, Klionsky DJ. J Cell Biol. 2000;151:1025–1034. doi: 10.1083/jcb.151.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott SV, Nice DC, III, Nau JJ, Weisman LS, Kamada Y, Keizer-Gunnink I, Funakoshi T, Veenhuis M, Ohsumi Y, Klionsky DJ. J Biol Chem. 2000;275:25840–25849. doi: 10.1074/jbc.M002813200. [DOI] [PubMed] [Google Scholar]
- 32.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. J Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hutchins MU, Veenhuis M, Klionsky DJ. J Cell Sci. 1999;112:4079–4087. doi: 10.1242/jcs.112.22.4079. [DOI] [PubMed] [Google Scholar]
- 34.Piper RC, Cooper AA, Yang H, Stevens TH. J Cell Biol. 1995;131:603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L, Davis NG. J Cell Biol. 2000;151:731–738. doi: 10.1083/jcb.151.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y, Kominami E. Mol Biol Cell. 1999;10:1367–1379. doi: 10.1091/mbc.10.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Dalton VM, Eggerton KP, Scott SV, Klionsky DJ. Mol Biol Cell. 1999;10:1337–1351. doi: 10.1091/mbc.10.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. J Cell Biol. 2000;151:263–276. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. EMBO J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott SV, Guan J, Hutchins MU, Kim J, Klionsky DJ. Mol Cell. 2001;7:1131–1141. doi: 10.1016/s1097-2765(01)00263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang WP, Scott SV, Kim J, Klionsky DJ. J Biol Chem. 2000;275:5845–5851. doi: 10.1074/jbc.275.8.5845. [DOI] [PubMed] [Google Scholar]
- 42.Guan J, Stromhaug PE, George MD, Habibzadegah-Tari P, Bevan A, Dunn WA, Jr, Klionsky DJ. Mol Biol Cell. 2001;12:3821–3838. doi: 10.1091/mbc.12.12.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darsow T, Rieder SE, Emr SD. J Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer von Mollard G, Stevens TH. Mol Biol Cell. 1999;10:1719–1732. doi: 10.1091/mbc.10.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato TK, Darsow T, Emr SD. Mol Cell Biol. 1998;18:5308–5319. doi: 10.1128/mcb.18.9.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato TK, Rehling P, Peterson MR, Emr SD. Mol Cell. 2000;6:661–671. doi: 10.1016/s1097-2765(00)00064-2. [DOI] [PubMed] [Google Scholar]
- 47.Seeley ES, Kato M, Margolis N, Wickner W, Eitzen G. Mol Biol Cell. 2002;13:782–794. doi: 10.1091/mbc.01-10-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson JS, Klionsky DJ, Banta LM, Emr SD. Mol Cell Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wurmser AE, Emr SD. EMBO J. 1998;17:4930–4942. doi: 10.1093/emboj/17.17.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerhardt B, Kordas TJ, Thompson CM, Patel P, Vida T. J Biol Chem. 1998;273:15818–15829. doi: 10.1074/jbc.273.25.15818. [DOI] [PubMed] [Google Scholar]