

Abstract
Objective
To investigate the interaction of SAA and SR-BI in remodeling of acute phase HDL (AP HDL).
Methods and Results
We used SAA and SR-BI adenoviral vector expression models to study the interaction between these entities. SR-BI processing of mouse AP HDL generated progressively smaller discreet HDL particles with distinct apolipoprotein compositions. SR-BI actions segregated apolipoproteins with the smallest particles containing only apoA-I. Larger remnants contained apoA-I, apoA-II and SAA. Small apoA-I only particles failed to associate with preformed HDL whereas larger remnants readily did. The presence of SAA on SR-BI processed HDL particles propelled apoA-I to a small lipid-poor form and accelerated apoA-I catabolism.
Conclusions
Data indicate that after core and surface HDL lipid perturbation by SR-BI, SAA propels apoA-I to a small lipid-poor form while accelerating HDL metabolism.
Keywords: SAA, HDL, Inflammation, SR-BI, Metabolism
Intimate associations exist between atherosclerosis and inflammation. The atherosclerotic process itself has features of chronic inflammation (1). Furthermore, the process of atherosclerosis is markedly accelerated by chronic inflammatory disease states such as rheumatoid arthritis (2). Perhaps most notable among the plethora of metabolic changes that affect lipid and lipoproteins during inflammation are the structural and metabolic alterations of HDL (3).
Serum amyloid A protein (SAA) becomes a major apolipoprotein of HDL during the acute phase and can replace apoA-I as the major HDL apolipoprotein (3). Concomitant with SAA induction is a decline in plasma HDL cholesterol and apoA-I levels (3). Notable lipid changes occur and HDL becomes generally enriched in triglycerides (4).
The scavenger receptor class B type I (SR-BI) plays an important role in the metabolism of high density lipoprotein (HDL) (5). It binds HDL with high affinity and by selective lipid uptake mediates the movement of cholesterol ester from the hydrophobic HDL core to the cell (5). SR-BI-mediated selective lipid uptake generates incrementally smaller and denser HDL particles (HDL remnants) (6). With respect to normal human HDL, these remnants are not rapidly cleared from the circulation but rather remodel to form large HDL particles by association with existing lipoprotein particles, preferably HDL (6, 7, 8). A portion of the HDL remnant apolipoproteins is directed toward catabolism (9).
We used two model systems to study the interactions between SR-BI and SAA-containing acute phase HDL (AP HDL). The first is an established model where the metabolic fate of radiolabeled AP HDL in mice over expressing SR-BI is evaluated (6, 7, 8). The second involves monitoring the effect of dual SR-BI and SAA adenoviral expression on endogenous HDL metabolism. The latter approach mimics the acute phase as SAA is predominantly produced in the liver. Data indicate that SR-BI action segregates AP HDL apolipoprotein catabolism. Discrete AP HDL remnant particles with distinct apolipoprotein compositions are generated. The presence of SAA on AP HDL remnants tends to propel apoA-I to small lipid poor forms potentially impacting efflux whilst at the same time accelerating apoA-I catabolism.
Methods
Animals
C57BL/6 and apoA-I-/- (C57BL/6 background) mice were obtained from Jackson Laboratories. An acute phase response was elicited by intra-peritoneal injection of 25 or 100 μg lipopolysaccharide (LPS) (Escherichia coli 0111:B4, Sigma Chemical Company). After 24 hours the mice were humanely killed and AP HDL isolated from plasma by sequential ultracentrifugation. The Veterans Administration Medical Center Institutional Animal Care and Use Committee approved all procedures.
Lipoprotein isolation and radiolabeling
HDL (d = 1.063 to 1.21 g/ml) was isolated from fresh mouse plasma by density gradient ultracentrifugation (3). HDL was dialyzed against 150 mM NaCl, 0.01% EDTA, sterile filtered, and stored under argon at 4° C. Protein concentrations were determined by the method of Lowry et al (10). The apolipoprotein content of HDL fractions was determined by densitometric scanning of Coomassie stained SDS-gels. HDL was radiolabeled by the iodine monochloride method (11).
Generation of HDL remnants in apoA-I-/- mice
The production of SR-BI-generated HDL remnants in apoA-I-/- mice was described (6, 7, 8). ApoA-I-/- mice weighing at least 25 g were tail vein injected with 1.5 × 1011 particles AdSR-BI, a replication-defective adenoviral vector expressing mouse SR-BI, or Adnull, a control vector containing no transgene. Three days after adenovirus infusion, mice were injected via the jugular vein with a bolus of 700 μg AP HDL traced with 50 μg 125I-AP HDL in 100 μl saline (specific activity of HDL mix 20-43 cpm/ng). Plasma aliquots (∼20 μl) were obtained for analysis at selected times up to 5 hours after bolus administration. By analyzing aliquots from each of the time-points by native gel electrophoresis, we assessed the kinetics of remnant generation in two separate experiments. In this model apoA-I-/- mice contain virtually no endogenous lipoproteins (6). The concentration of HDL remnants in plasma was determined from the counts recovered in the mouse plasma and the known specific activity of the injected 125I-HDL.
In vitro remodeling reactions
Plasma containing HDL remnants, collected from mice at selected times after bolus injection, was mixed 1:10 (v:v) with mouse HDL (1.7mg/ml). After incubations at 37° C, the remodeling reactions were transferred to ice, mixed with an equal volume of saturated sucrose, and immediately applied to a non-denaturing gradient gel and visualized by autoradiography (7, 8).
The apolipoprotein composition of remnant particles or remodeled particles was determined by excising bands from non-denaturing gels, and analyzing by SDS-PAGE and autoradiography (7, 8).
SR-BI and SAA dual adenoviral vector model
Replication-defective adenoviral vectors expressing SR-BI or SAA and a control vector containing no transgene (Adnull) were utilized. AdSR-BI expressed mouse SR-BI and was used at 0.5 × 1011 particles, titred to increase hepatic SR-BI expression approximately 3 fold. This resulted in a 50% decline in HDL levels. The SAA adenoviral vector (AdSAA) expressed the CEJ isotype of mouse SAA and was used at 1 × 1011 particles (12). This increased SAA levels approximately 8 fold without impacting HDL levels. Four groups of C57BL/6 mice (n=4) were injected with identical total amounts of adenoviral vector particles (1.5 × 1011) in one of four combinations, consisting of AdSR-BI plus Adnull, AdSAA plus Adnull, AdSR-BI plus AdSAA, or Adnull only. Blood was collected at 24 and 48 hours by retro-orbital bleeding and the experiment terminated at 72 hours. Western blot analyses were used to verify consistent SR-BI and SAA expression at 72 hours. Plasma HDL levels were measured (Wako Diagnostics, Richmond, VA). The apoA-I and SAA distribution with respect to HDL was analyzed at 72 hours by non-denaturing gradient gel electrophoresis followed by immunoblot analysis for either mouse apoA-I (Biodesign, Saco, ME) or mouse SAA (Rabbit anti-mouse SAA, F.C.D. lab).
Statistical Analysis
Data are presented as mean ± SEM. Statistical analyses to compare differences between plasma HDL levels were carried out using one-way ANOVA with the Holm-Sidak multiple comparisons test (SigmaStat 3.5,GmbH, Germany). Significance was set at p < 0.05.
Results
SR-BI and AP HDL apolipoprotein clearance
To study AP HDL apolipoprotein metabolism after processing by SR-BI, we initially used an established model in which SR-BI was over expressed in apoA-I-/- mice by means of an adenoviral vector (6). In our model a bolus of mouse AP HDL traced with 125I was injected into mice 3 days after viral vector and plasma samples were collected at selected intervals. ApoA-II is drastically reduced when SAA is a major apolipoprotein of HDL. In order compare the clearance of SAA and apoA-II during SR-BI processing, we chose to use AP HDL from mice injected with 25 μg LPS that contained moderate amounts of SAA and apoA-II (SAA∼ 33%; apoA-II ∼ 14% of total protein respectively) (Fig. 1A). AP HDL from mice injected with 100 μg LPS in which SAA constitutes ∼43% of total protein and apoA-II only ∼6% of total protein was employed for subsequent experiments to focus on SAA (Fig. 1A). The apolipoprotein composition of HDL with moderate SAA content (25 μg injected LPS), when processed by SR-BI, was determined by direct SDS PAGE and autoradiography of plasma samples collected at the indicated times after bolus AP HDL injection. This analysis showed a very rapid loss from the circulation of SAA and apoA-II compared to apoA-I when SR-BI was over expressed (Fig. 1B and 1C).
Figure 1A.
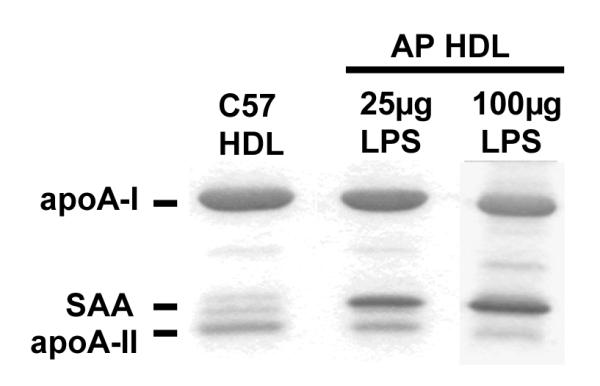
Apolipoprotein composition of mouse HDL.
HDL from mice injected with 25 or 100 μg LPS was subjected to SDS-PAGE analysis on a 5-20% acrylamide gradient gel and stained with Coomassie blue. HDL loading — 5 μg/lane. Quantitation was by densitometric scanning.
Figure 1B.

Preferential loss of apoA-II and SAA after SR-BI processing.
ApoA-I-/- mice were injected with 1.5×1011 particles of adenoviral vector AdSR-BI. After three days when SR-BI expression was maximal, the mice were injected with a bolus of 750 μg 125I-AP HDL (25μg LPS). Plasma samples containing remnants were collected from individual mice at the indicated times after bolus injection and 5 μl aliquots were analyzed by SDS-PAGE (5-20% acrylamide gradient). An aliquot of the injected 125I-AP HDL is shown for comparison. Samples were visualized by autoradiography.
Figure 1C.

No preferential loss of apoA-II and SAA without SR-BI processing. ApoA-I-/- mice were injected with a bolus of 750 μg 125I-AP HDL and plasma samples collected and analyzed as in 1B.
AP HDL remodeling in vivo
The size of mouse AP HDL particles after SR-BI processing was determined by direct analysis of plasma samples by means of non-denaturing gel electrophoresis and autoradiography (Fig. 2A). In this model system, exogenously administered mouse AP HDL (from mice injected with 100 μg LPS) became progressively smaller with the formation of HDL remnants occurring in incremental steps (Fig. 2A). No lipid free apolipoproteins were detected. Short term SR-BI processing (∼1 hour) reduced the size of exogenous AP HDL from 10.2 nm to a remnant particle (R1) with a diameter of approximately 9.8 nm (Fig. 2A). Intermediate processing (3 hours) resulted in the formation of two distinct remnant particles R2 and R3 with diameters of approximately 9.4 and 7.6 nm, respectively (Fig. 2A). Prolonged processing (5 hours) led to the formation of a predominant population of R3 particles (Fig. 2A). These dense, small R3 particles could not be re-floated by density gradient centrifugation, but were larger than “free” apoA-I (6). They appear to be the final stage of SR-BI processing, but can be detected early in this process (0.5 hours). The apolipoprotein composition of the AP HDL remnants was assessed by excising corresponding bands from the gel depicted in Figure 2A and analyzing them by SDS PAGE (Fig. 2B). The injected 125I AP HDL contained predominantly apoA-I and SAA with apoA-II constituting less than 10% of the apolipoproteins (Figs. 1A and 2B). One hour of SR-BI processing generated a R1 remnant that was similar in apolipoprotein content to the starting HDL and a R3 remnant that contained predominantly apoA-I (Fig. 2B). The R2 remnants at 3 and 5 hours contained mainly apoA-I and SAA (Fig. 2B). R3 remnants at these times were enriched for apoA-I with only small amounts of SAA detected. ApoA-II was not detected at these later time points.
Figure 2A.

Size analysis 125I-AP HDL (100 μg injected LPS) after SR-BI processing. Plasma samples (7 μl) containing HDL remnants, collected from a SR-BI-overexpressing mouse at the indicated times after injection of a bolus of 125I-AP HDL, were separated on a 4-18% acrylamide non-denaturing gel. 125I-AP HDL injected into the mouse is shown for comparison. Samples were visualized by autoradiography. The mobility of standards of known diameter is indicated. R1, R2, R3: Remnant HDL particles of varying sizes.
Figure 2B.

Apolipoprotein composition of HDL remnant particles.
HDL remnant particles R1, R2, and R3, generated by 1, 3 and 5 hr of SR-BI processing of injected 125I-AP HDL, were excised from the non-denaturing gel depicted in Fig. 2A and subjected to SDS-PAGE (5-20% acrylamide) and autoradiography to determine their apolipoprotein composition. 125I-AP HDL excised from the same non denaturing gel is shown for comparison.
AP HDL remnant association with HDL
We have reported that similarly produced remnants from normal human HDL associate with preformed HDL (7). We determined whether the AP HDL remnants varied in their ability to associate with HDL. For these studies, plasma samples collected at selected intervals after bolus injection of 125I AP HDL were incubated with unlabeled C57BL/6 mouse HDL, analyzed by non-denaturing gradient gel electrophoresis and autoradiography to visualize apolipoprotein originally present in SR-BI-processed remnants (Fig. 3A). AP HDL remnants obtained after 1 hour of SR-BI processing readily associated with C57BL/6 mouse HDL (band T) (Fig. 3A). By 3 hours, SR-BI processing generated a subpopulation of AP HDL remnants that appeared to be resistant to HDL association, (band B). Longer SR-BI processing (5 hours) resulted in the generation of increasing amounts of the resistant remnant (band B) (Fig. 3A). Bands corresponding to T and B were excised from the non-denaturing gel (Fig. 3A) and subjected to SDS PAGE (Fig. 3B). The 125I-AP HDL remnant that was resistant to HDL association (band B) contained only apoA-I. The larger remodeled remnant (T) contained apoA-I, SAA and apoA-II. As AP HDL is processed by SR-BI, progressively more remnant particles are generated that are resistant to associating with HDL. These remodeling-resistant remnants contain mainly apoA-I, whereas remnants capable of remodeling by association of HDL contain apoA-I, SAA and apoA-II.
Figure 3A.

Size change of mouse AP HDL remnants that remodel by associating with mouse HDL.
Plasma samples containing HDL remnants, collected from a SR-BI-over expressing mouse 1, 3 and 5 hr after bolus 125I-AP HDL injection were mixed 1:10 (v/v) with C57BL/6 mouse HDL (1.7mg /ml) for 2 hr at 37° C to allow for remnant association with HDL. The resultant products were subjected to non-denaturing gel electrophoresis using a 4-18% acrylamide gradient and visualized by autoradiography. Mouse AP HDL is shown for comparison. T: remodeled remnant particles and B: remodeling-resistant particles.
Figure 3B.

Apolipoprotein composition of remodeled HDL remnant particles.
125I-AP HDL remnant particles (1, 3, and 5 hr) that remodeled by association with C57BL/6 HDL (T) as well as remodeling-resistant remnant particles (B) were excised from the non-denaturing gel depicted in Fig. 3A and subjected to SDS-PAGE (5-20% acrylamide gradient) followed by autoradiography. AP HDL excised from the same non-denaturing gel is shown for comparison.
SR-BI and SAA co-expression
Data presented in Fig. 4 indicate that expression of Adnull alone, or in combination with AdSAA, has no significant effect on HDL levels in C57BL/6 mice. A moderate level of SR-BI over expression (3-fold over control) by AdSR-BI, decreased HDL levels by approximately 50% to 35 mg/dl at 72 hours. Interestingly, over expression of SAA together with SR-BI significantly further reduced HDL levels to 8 mg/dl. Plasma from the 4 experimental groups at 72 hours was examined by non-denaturing gradient gel electrophoresis and immunoblot analysis for apoA-I or SAA. Most notable is the decreased apoA-I signal with concomitant SR-BI and SAA over expression compared to the respective vectors plus Adnull (Fig. 5A). Further, approximately 30% of the remaining apoA-I is propelled to a smaller (7.7nm) form, which is likely lipid depleted (Fig. 5A). In the AdSR-BI group HDL tends to be somewhat smaller whereas Adnull alone and AdSAA alone have no significant influence on apoA-I distribution. The immunoblot for SAA is positive in the appropriate experimental groups. SAA was only detected associated with normal-sized HDL and no smaller forms were identified even when the blot was over exposed.
Figure 4.

HDL levels of mice after injection of adenoviral vectors expressing SAA, SR-BI or SR-BI plus SAA.
Four groups of mice (n=4) were injected with 1.5 × 1011 particles of adenovirus expressing SR-BI, SAA, or SR-BI plus SAA as set out in Methods. HDL cholesterol was measured at the indicated times after viral injection.
a: 0h vs 48h vs 72h p<0.05
b: 48h vs 72h p<0.05
c: AdSR-BI plus Adnull vs AdSR-BI plus AdSAA 72h p<0.05
Figure 5.

Combined action of SR-BI and SAA generate small HDL particles containing apoA-I but not SAA.
Plasma (3μl) collected from 4 individual mice 72 h after administration of Adnull, AdSAA, AdSRBI or combinations of viruses as set out in Methods were subjected to non-denaturing gel electrophoresis on a 4-20% acrylamide gel, transferred to PVDF and blotted for apoA-I (5A) or SAA (5B).
Discussion
HDL remodeling in plasma plays an important role in regulating HDL levels and metabolism, including the generation of lipid free and lipid poor apolipoproteins that can function as acceptors in cellular cholesterol efflux. Remodeling studies of polydisperse human HDL are difficult. The impact of inflammation on HDL compounds this problem. Mouse HDL is relatively homogeneous and lack particles containing only apoA-I, allowing for simplified interpretation (13). Data presented in this article support the following: (I) SR-BI processing of mouse AP HDL in vivo generated progressively smaller, discreet remnant HDL particles with distinct apolipoprotein compositions. (II) SR-BI processing of mouse AP HDL segregated apolipoproteins in that the smallest particles contained only apoA-I, whereas larger remnant particles contained apoA-I, apoA-II as well as SAA. (III) The small particles that contained only apoA-I failed to associate with mouse HDL, whereas the larger remnants readily associated with pre-formed HDL. (IV) SAA association with SR-BI processed HDL particles moved apoA-I to a small lipid poor form, which holds implications for cholesterol efflux, and also accelerates apoA-1 catabolism.
Our data indicate that SR-BI over expression has a major influence on the rate of apolipoprotein catabolism with the different apolipoproteins of AP HDL clearing at different rates. The implication is that under normal conditions the metabolic segregation of apolipoproteins is mediated by SR-BI, but on a more limited scale. Thus the known difference in clearance rates of HDL apolipoproteins could be the result of SR-BI action. The degree to which inflammation could alter the various structural and functional class B scavenger receptor modalities merits consideration. Studies of the binding kinetics of SR-BI to different ligands, suggests that SR-BI exists in different functional forms with dimerization reported (14, 15). There are further two distinct isoforms (SR-BI and SR-BII) that have different functions and distribution (16). Certain HDL particles could be subjected to more intense SR-BI action, resulting in the apolipoprotein manifestations that we present. Although the experimental conditions are in a non-steady state and detailed kinetic interpretations are not warranted, it is clear that SAA clears more rapidly than apo-AII. Given that SAA was reported to be a ligand for SR-BI and that SAA was internalized by cells in a SR-BI-dependent manner (17), the possibility exists that SAA is directly taken up by hepatocyles from the space of Disse.
SR-BI processing results in progressively smaller AP HDL with 3 discrete small remnants emerging (R1, R2 and R3) that are generated in incremental steps. As SR-BI associates preferentially with large HDL particles, one potential reason for the accumulation of smaller particles of discrete size is the possibility that as HDL become smaller, they lose their ability to interact with SR-BI (18). Accumulated remnants after AP HDL injection may represent subpopulations of HDL that differ markedly in their affinity for SR-BI. An alternative explanation for the incremental changes in AP HDL particle size may be physical constraints in HDL particle structure allowing only for discreet entities to stably exist.
There is a remarkable segregation of apolipoproteins with respect to the remnants formed. It is notable that the small predominantly apoA-I remnant (R3) is rapidly generated, albeit only in small amounts. Central to apoA-I dissociation from HDL during remodeling is a reduction in core lipids, as for instance mediated by cholesteryl ester transfer protein (CETP) (19, 20). Hydrolysis of HDL surface phospholipids by acute phase phospholipases, notably Group IIA secretory phospholipase A2 (IIA sPLA2), reduce HDL size and alter apoA-I epitopal expression but does not per se result in apoA-I liberation from the particle (20). However, concomitant HDL core reduction by CETP and surface hydrolysis by IIA sPLA2 markedly amplified the movement of apoA-I from HDL to lipid poor forms (20). SR-BI selective lipid uptake is similar to the latter in that transfer of cholesterol ester from the core of the HDL particle is associated with movement of surface phospholipids to the cell. The shedding of a predominantly apoA-I remnant is thus not surprising as apoA-I is the least lipophilic of the HDL apolipoproteins with the highly lipophilic SAA promoting this process.
We have previously reported that normal human HDL remnants, when mixed with mouse plasma either in vivo or in vitro, increase in size to HDL-like particles (6, 7). It was of interest to determine whether the AP HDL remnants containing different amounts of apoA-I, SAA and apoA-II vary in their ability to associate with HDL. Data indicate that as for normal human HDL, the apoA-I-only remnant (R3) is resistant to HDL association (8). SAA and apoA-II containing remnants sustain a structural organization that allows re-association with existing HDL. It is remarkable that the small apoA-I only particle (R3) does not re-associate with normal HDL. This clearly distinguishes it from pre-β HDL or free apoA-I (21). It suggests a rather unique conformation of the apoA-I on this remnant, possibly similar to the small apoA-I only HDL particles isolated by anti-human apoA-I immunoaffinity chromatography (21).
The second model utilizing dual adenoviral vector expression of SR-BI and SAA expands our understanding of acute phase HDL remodeling. The SAA expression effected is physiologically relevant and equates to a ”moderate” inflammatory response. SAA is mostly produced in the liver where it could impact the formation of HDL. Data indicate that in the absence of enhanced selective lipid uptake by SR-BI, SAA does not influence apoA-I distribution or HDL levels. However, when selective lipid uptake is increased it has a potent affect to propel apoA-I to a smaller form and decrease HDL levels. Apart from putative alterations in SR-BI function, other factors have been shown to enhance HDL selective lipid uptake during inflammation. Most notable is the acute phase secreted phospholipases such as group IIA sPLA2 and endothelial lipase (EL) (22,23). Modification of HDL by these enzymes result in markedly enhanced selective uptake with subsequent increase in HDL catabolism and decreased plasma HDL levels. When HDL core and surface lipid content is thus perturbed, SAA clearly holds the potential to dramatically alter the equilibrium between HDL bound and lipid poor apoA-I. This holds implications for interpreting the data presented utilizing our first model. The likelihood is that the continual presence of SAA on remnants serves to propel more apoA-I to lipid poor forms. This suggest that SAA could promote cholesterol efflux by not only serving as an extracellular cholesterol acceptor, but also by liberating apoA-I (24).
Cholesterol acceptors need to be available particularly in extracellular spaces where efflux occurs (19). Proteins secreted by cholesterol-loaded macrophages, including acute phase secreted phospholipases, phospholipid transfer protein, cholesterol ester transfer protein, hepatic lipase and lipoprotein lipase, could play a key role in HDL remodeling within the vascular subendothelium, including the space of Disse, that leads in part to the generation of lipid poor apoA-I (19, 25). Numerous cells in these spaces produce SAA under inflammatory conditions where it could act to further propel apoA-I to lipid poor forms from remodeled remnants.
The generation of increased lipid poor apoA-I by SAA during HDL remodeling clearly comes at the potential price of enhanced catabolism and decreased HDL levels. It is likely too limited to view SAA as either pro-or anti-atherogenic. It is the context in which SAA is expressed, particularly with respect to amount, duration, and presence of HDL modifying factors that could have varying effects. Teleologically, the dramatic changes in HDL composition and metabolism during inflammation, likely serves a shorter term purpose to allow the organism to survive a noxious assault. Longer term inflammation could be deleterious in that HDL levels may become so reduced that there is a lack of “substrate” to derive lipid acceptors from.
Acknowledgments
Sources of Funding This work was supported by NIH award PO1 HL086670 and VA Merit Review Funds (FCD).
Footnotes
Disclosures None.
References
- 1.Lee RT, Libby P. The unstable atheroma. Arterioscler. Thromb. Vasc. Biol. 1997;17:1859–1867. doi: 10.1161/01.atv.17.10.1859. [DOI] [PubMed] [Google Scholar]
- 2.Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE. Cardiovascular death in rheumatoid arthritis. Arthritis & Rheumatism. 2005;52(3):722–732. doi: 10.1002/art.20878. [DOI] [PubMed] [Google Scholar]
- 3.Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J. Biol. Chem. 1986;261:9644–9651. [PubMed] [Google Scholar]
- 4.Cabana VG, Lukens JR, Rice KS, Hawkins TJ, Getz GS. HDL content and composition in acute phase response in three species: triglyceride enrichment of HDL a factor in its decrease. J. Lipid Res. 1996;37:2662–2674. [PubMed] [Google Scholar]
- 5.Acton SL, Rigott A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-B1 as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 6.Webb NR, Cai L, Ziemba KS, Yu J, Kindy MS, van der Westhuyzen DR, de Beer FC. The fate of HDL particles in vivo after SR-BI-mediated selective lipid uptake. J. Lipid Res. 2002;43:1890–1898. doi: 10.1194/jlr.m200173-jlr200. [DOI] [PubMed] [Google Scholar]
- 7.Webb NR, De Beer MC, Asztalos BF, Whitaker N, van der Westhuyzen DR, de Beer FC. Remodeling of HDL remnants generated by scavenger receptor class B type I. J. Lipid Res. 2004;45:1666–1673. doi: 10.1194/jlr.M400026-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.de Beer MC, van der Westhuyzen DR, Whitaker NL, Webb NR, de Beer FC. SR-BI-mediated selective lipid uptake segregates apoA-I and apoA-II catabolism. J. Lipid Res. 2005;46:2143–2150. doi: 10.1194/jlr.M500068-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Kozarsky KF, Donahee MH, Rigotti A, Iqbal SN, Edelman ER, Krieger M. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol. Nature. 1997;387:414–417. doi: 10.1038/387414a0. [DOI] [PubMed] [Google Scholar]
- 10.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 11.Bilheimer DW, Eisenberg S, Levy RI. The metabolism of very low density lipoproteins. I. Preliminary in vitro and in vivo observations. Biochim. Biophys. Acta. 1972;260:212–221. doi: 10.1016/0005-2760(72)90034-3. [DOI] [PubMed] [Google Scholar]
- 12.Webb NR, de Beer MC, van der Westhuyzen DR, Kindy MS, Banka CL, Tsukamoto K, Rader DL, de Beer FC. Adenoviral vector-mediated overexpression of serum amyloid A in apoA-I-deficient mice. J. Lipid Res. 1997;38:1583–1590. [PubMed] [Google Scholar]
- 13.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim Biophys Acta. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 14.de Beer MC, Durbin DM, Cai L, Mirocha N, Jonas A, Webb NR, de Beer FC, van der Westhuyzen DR. Apolipoprotein A-II modulates the binding and selective lipid uptake of reconstituted HDL by scavenger receptor BI. J. Biol. Chem. 2001;276:15832–15839. doi: 10.1074/jbc.M100228200. [DOI] [PubMed] [Google Scholar]
- 15.Williams DL, de La Llera-Moya M, Thauhnai ST, Lund-Katz S, Connelly MA, Azhar S, Anantharamaiah GM, Phillips MC. Binding and cross-linking studies show that scavenger receptor BI interacts with multiple sites in apolipoprotein A-I and identify the class A amphipathic alpha-helix as a recognition motif. J. Biol. Chem. 2000;275:18897–18904. doi: 10.1074/jbc.M002411200. [DOI] [PubMed] [Google Scholar]
- 16.Webb NR, Connell PM, Graf GA, Smart EJ, de Villiers WJS, de Beer FC, van der Westhuyzen DR. SR-B2, an isoform of the scavenger receptor BI containing an alternate cytoplasmic tail, mediates lipid transfer between high density lipoprotein and cells. J. Biol. Chem. 1998;273:15241–15248. doi: 10.1074/jbc.273.24.15241. [DOI] [PubMed] [Google Scholar]
- 17.Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J. Biol. Chem. 2005;280:2954–2961. doi: 10.1074/jbc.M411555200. [DOI] [PubMed] [Google Scholar]
- 18.de Beer MC, Durbin DM, Cai L, Jonas A, de Beer FC, van der Westhuyzen DR. Apolipoprotein A-I conformation markedly influences HDL interaction with scavenger receptor BI. J. Lipid Res. 2001;42:309–315. [PubMed] [Google Scholar]
- 19.Curtiss LK, Valenta DT, Hime NJ, Rye KA. What is so special about apolipoprotein AI in reserve cholesterol transport? Arterioscler Thromb Vasc Biol. 2006;26:12–19. doi: 10.1161/01.ATV.0000194291.94269.5a. [DOI] [PubMed] [Google Scholar]
- 20.Jahangiri A, de Beer MC, Noffsinger V, Tannock LR, Ramaiah C, Webb NR, van der Westhuyzen DR, de Beer FC. HDL Remodeling During the Acute Phase Response. Arterioscler. Thromb. Vasc. Biol. 2009;29:261–267. doi: 10.1161/ATVBAHA.108.178681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JY, Lanningham-Foster L, Boudyguina EY, Smith TL, Young ER, Colvin PL, Thomas MJ, Parks JS. Pre-beta high density lipoprotein has two metabolic fates in human apolipoprotein A-I transgenic mice. J. Lipid Res. 2004;45:716–728. doi: 10.1194/jlr.M300422-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.de Beer FC, Connell PM, Yu J, de Beer MC, Webb NR, van der Westhuyzen DR. HDL modifications by secretory phospholipase A2 promotes scavenger receptor class B type 1 interaction and accelerates HDL catabolism. J. Lipid Res. 2000;41(11):1849–1857. [PubMed] [Google Scholar]
- 23.Nijstad N, Wiersma H, Gautier T, van der Giet M, Maugeais C, Tietge UJF. Scavenger receptor Bi (SR-BI)-mediated selective uptake is required for the remodeling of HDL by endothelial lipase. J. Biol. Chem. 2009 doi: 10.1074/jbc.M807683200. In press. [DOI] [PubMed] [Google Scholar]
- 24.van der Westhuyzen DR, Cai L, de Beer MC, de Beer FC. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J. Biol. Chem. 2005;280(43):35890–35895. doi: 10.1074/jbc.M505685200. [DOI] [PubMed] [Google Scholar]
- 25.Leitinger N, Watson AD, Hama SY, Ivandic B, Qiao JH, Huber J, Faull KF, Grass DS, Navab M, Fogelman AM, de Beer FC, Lusis A, Berliner JA. Role of group II secretory phospholipase A2 in atherosclerosis. 2. Potential involvement of biologically active oxidized phospholipids. Arterioscler. Thromb. Vasc. Biol. 1999;19:1291–1298. doi: 10.1161/01.atv.19.5.1291. [DOI] [PubMed] [Google Scholar]