

The sequencing of the human genome and genomes of several model organisms has revolutionized the ability of researchers and clinicians to study the genetic basis of disease, relationships between genes and phenotypes, and the molecular basis of normal development and physiology. This information has led to better diagnosis of traits and diseases, improved treatment for patients, an ability to conduct genetic testing for many inherited diseases, and novel drug development. The benefits of the genomics revolution have been extended to companion animals. Most unique is the enormous variation among breeds, which is typified in dogs as the result of human-driven selective breeding. In addition to the selected positive traits, breeds often are susceptible to certain diseases, in part as a result of inbreeding. Considerable progress has been made in identifying mutations that cause simple Mendelian traits and diseases in domestic animals, but dissecting the molecular basis of complex or polygenic traits and diseases is a challenge to those involved in genetic mapping of animals.
A draft 7.6X sequence of the genome of Boxer dogs was developed through funding provided by the National Institute of Health; this project was completed in 2005 at the Broad Institute of the Massachusetts Institute of Technology and Harvard University. A map of 500,000 SNPs was developed as part of the National Institute of Health–funded project by comparing sequences between the 1.5X sequence for a single Poodle that had been placed in the public domain and 100,000 random sequences of 12 other dog breeds. Because the dog genome is approximately 2.4 gigabases, this yielded a SNP every 10 kb across the genome for LD–based mapping. A SNP is a variation in a single nucleotide sequence. At a single nucleotide locus, a dog could have 1 of 3 genotypes (homozygous for either alternate nucleotide or heterozygous). Sometimes a mutation results in a polymorphism that affects the encoding of an amino acid, which results in a protein abnormality that affects function of the protein or gene expression when the polymorphism is in a regulatory region.
The breed structure of dogs places them in the unique position of serving as a bridging organism between the phenotypic complexity in humans and ease of genetic manipulation in laboratory mice. Furthermore, there is high interest in canine health. Most modern dog breeds were developed within the past 300 years. Modern purebred dogs represent a limited genetic pool, with disease predispositions derived from 1 (or a small number) of recent mutations. Modern dog breeds offer all the advantages of geographically isolated human populations but with a higher degree of isolation and narrower bottlenecks that make them amenable to LD or association mapping.1 Sequencing the dog genome and surveying SNPs within and among breeds has revealed that their haplotype (the joint genotype of groups of physically linked genetic loci) structure makes them ideal for mapping complex or polygenic traits at both a coarse (within a breed) and fine (among breeds) level.1 This strategy enables identification of the smallest haplotype associated with a trait among 2 or more breeds, thus narrowing the search interval for candidate genes. This approach is called LD mapping. The objective of the information reported here was to encourage clinicians and geneticists to make optimum use of animals admitted to teaching hospitals for association studies of simple and complex traits.
Genetic Tools
The tools for genome-wide screening are currently available to enable investigators to pursue the identification of genes that underlie Mendelian or complex traits and diseases in dogs and, to a lesser extent, other companion animals (Figure 1). Once the most likely mode of inheritance is identified, a power analysis can be used to indicate whether a sufficient number of dogs are available or can be acquired to proceed with a mapping experiment. Currently, genome-wide microsatellite screening sets (minimal screening sets) are the cheapest method to screen the entire dog genome for loci underlying these diseases, but such analysis will not enable sufficiently fine localization to identify suitable candidate genes.2 Fine mapping to narrow or refine these loci based on SNP genotyping and focused on particular chromosomesa or locus-by-locusb methods can then be peformed.3 The canine SNP array we currently use in our DNA bank at Cornell c is a 5-μm, 100-format, perfect-match only probe designed with 20 probes/SNP that can detect a total of 127,132 SNPs. The array also contains 26,625 SNPs that have been defined as the gold-standard set comprising the loci on the first version of the SNP chip.d Other methods to obtain SNP genotypes are available. In addition to the use of genomic DNA sequence variation associated with a phenotype to discover a contributing mutation, investigators can also examine the transcribed gene profile in the tissue of interest. The second-generation canine genome microarraye used at our facility for gene expression profiling (ie, messenger RNA profiling) in dogs incorporates oligonucleotides and expressed sequence tags to comprehensively represent the dog transcriptome. The array contains > 42,800 Canis familiaris probe sets to monitor gene expression for > 18,000 Canis familiaris mRNA- or expressed sequence tag–based transcripts and > 20,000 nonredundant predicted genes. For all gene discovery methods, adequate and appropriate control materials are essential for the experimental study design.
Figure 1.
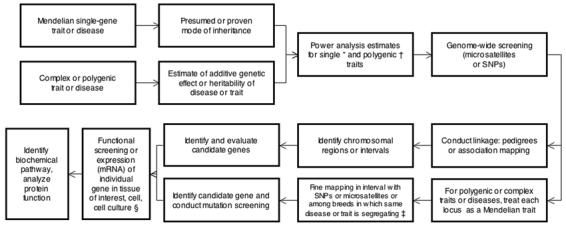
Flow diagram illustrating possible paths from a phenotype for an inherited trait or disease to discovery of a gene or genes underlying the condition and the main features of the process. *Mendelian traits - for diseases attributable to an autosomal recessive trait, 20 to 30 affected animals and 20 to 30 unaffected animals are needed. For diseases attributable to a dominant trait, 30 to 40 affected animals and 30 to 40 unaffected animals are needed. †Complex traits - at extreme distributions for unrelated animals, 100 affected animals and 100 unaffected animals are needed. For pedigrees, 200 to 400 (or more) animals are empirically needed. ‡For this step, 20 to 30 affected animals and 20 to 30 unaffected animals for each breed are needed. §An alternative is to analyze genome-wide expression (by use of microarrays) and consider each expression level as a QTL.
The aforementioned genomic tools can be used to investigate the molecular genetic basis of disease resistance and susceptibility in animals without pedigrees. Linkage mapping is based on the assumption that segregation of alleles for disease genes is random and independent (or in equilibrium), and any recombinations can be detected in known pedigrees that usually span at least 3 generations. However, it is not always possible to acquire complete phenotype information with accompanying DNA on full 3-generation pedigrees that are ideal for linkage mapping. The closer 2 genes or markers are to each another, the less likely that there will be recombination between them. Association or LD mapping relies on historical recombinations. Recombinations over many generations break up the region surrounding the causative or contributing mutation; thus, a small group of linked genes and genetic markers segregate together or in a nonrandom (disequilibrium) manner. At this stage, localization of the chromosomal interval may be refined to the point that immediate screening of candidate genes for mutations can be initiated for a Mendelian trait or disease. Exclusion of candidate genes in the associated interval may be pursued. However, after linkage mapping for complex and some Mendelian traits, fine mapping will need to be conducted before candidate genes can be selected (Figure 1). This can be accomplished by associating phenotypes and SNP genotypes of unrelated, affected animals through use of appropriate control animals.
Association mapping can also be conducted on the entire genome without prior linkage mapping. There is increasing evidence that careful selection of animals used in the evaluation can yield fine mapping results within a breed that are comparable to results obtained by use of multiple breeds.4-6 Sophisticated genetic models can also be used to combine information from pedigrees and unrelated animals. After a mutation in a candidate gene is identified, investigators may need a tissue source to measure gene expression with mRNA to confirm that mutations are functional (Figure 1). Finally, a gene likely to contribute to the phenotype, as determined on the basis of comparative mapping in another species or its known biologic aspects e.g an extracellular matrix encoding gene for a musculoskeletal disease, can be screened for mutations.
Ready access and timely acquisition of sufficient numbers of control and affected animals for the traits of interest are important practical barriers that prevent investigators from being able to fully use dogs in genetic studies. Furthermore, accurate phenotypic ascertainment is essential for all genetic mapping studies (ie, a diagnosis must be accurate). This reduces or eliminates heterogeneity when very similar phenotypes are assumed to be indicators of the same disease process or phenocopies that have the same phenotype but representing different underlying loci. As complex traits become better understood, a phenotype initially perceived as representing an entire disease process can be dissected into distinct subtypes in different populations. Ideally, phenotyping animals for a disease should be conducted by veterinarians with specific expertise in medical diagnostic testing. The genetic archive at the Cornell University Hospital for Animals is an example of a DNA repository that could be developed by a veterinary medical facility. Because dogs represent a unique opportunity for evaluating many inherited diseases in humans, and canine genomics resources are similar to those for human genetic research, dogs are the focus of the information reported here. However, the archive at our facility stores DNA and phenotypes on all animal species admitted to the teaching hospital.
Canine Patient Base
Only DNA of purebred dogs is collected to reduce and refine the total number of case and control acquisitions, which reduces handling and tracking costs. In addition, purebred dogs are better suited for LD-based mapping because the longer LD attributable to inbreeding minimizes the number of markers needed for initial screening, and a related breed can be used for fine mapping to reduce the candidate genes in the chromosomal interval. Furthermore, genetic heterogeneity should be lower in purebred dogs because most modern purebred dogs were developed from a few base breeds. In addition, > 1 inherited disease or trait will often segregate within any given breed, so dogs affected with a condition in 1 evaluation could be used as control animals in the evaluation of another inherited disease or condition.
Public Relations
It is essential to earn the trust, goodwill, and enthusiasm of dog owners when attempting to obtain blood samples for DNA isolation and information about their dogs. At our facility, we developed an informational website (www.vet.cornell.edu/research/dnabank) to explain the rationale for DNA archiving and provide a list of the diseases for which DNA is stored. We also created a dedicated e-mail address for use in educating owners and to enable us to answer their questions and receive information from them subsequent to their pets' visits to our hospital. Owners of pedigreed dogs are provided a pamphlet that describes the DNA archiving system and are requested permission to obtain a blood sample from their pet.
Phenotyping and Selection of Control Animals
Accurate phenotypes are critical for genetic evaluations and decisions regarding the composition of a DNA bank. During the initial appointment, when diagnostic tests performed indicate a presumptive diagnosis of a disease of interest for the DNA archive, the owner is asked to sign a consent form to include a DNA sample in the archive. Diagnostic test results for that animal for the visit in question are checked by use of an on-line medical record system or hard copy of the medical record. The diagnostic results are confirmed as being consistent with the final diagnosis by interpretation of clinical pathology results, radiology reports, and other test results. An important characteristic of the DNA archive at our facility is that all diagnoses are made by board-certified specialists or residents in training under the supervision of the board-certified specialists; thus, phenotype ascertainment is as accurate as possible. A common vocabulary (clinical and pathologic criteria) used for diagnostic screening of the diseases of interest was developed by each specialty service. All samples submitted from external sources (ie, other veterinary medical teaching hospitals) must fit the defined criteria, and diagnostic tests performed at other venues are reevaluated by the board-certified specialists at our facility before final inclusion in the archive.
Prospectively sampled control dogs are phenotyped by use of diagnostic tests identical to those used for dogs with the specific diseases. A second group of older dogs (ie, > 6 or 7 years old) are used as control animals to study diseases for which they were screened and had proven negative results. The final group of control dogs for diseases with rare alleles is chosen at random from among dogs of the same breed that have been included in the database. In this situation, more control than affected dogs will be genotyped to account for any false negative results in the control group.
Blood Collection, DNA Purification, and Storage of DNA
Blood samples are collected by venipuncture into tubes that contain sodium EDTA. Standard operating procedures are in accordance with guidelines for collecting a blood volume on the basis of the body weight of each animal; therefore, volume of blood samples ranges from 0.2 to 30 mL. The DNA is extracted by use of a DNA purification kit.f The DNA obtained by use of this protocol is not degraded, as determined by evaluation with gel electrophoresis, and can be used successfully in PCR assays and restriction enzyme digestions. It also has been successfully used for microarray analysis. Typically, size of the DNA is 100 kb, with a range of 50 to 200 kb. Before a DNA sample is stored, the concentration is estimated (on the basis of the absorbance at 260 nm on a spectrophotometer). The number of micrograms of DNA in each tube is recorded in the database, which maintains a current total of the number of micrograms available for each animal. Quality of the DNA is assessed by use of a value of ≥ 1.80 for the ratio of the absorbance at 260 nm to the absorbance at 280 nm. To ensure adequate amounts of DNA are available for future analysis, the goal for storage concentration is a minimum of 200 ng/μL. Stocks of DNA are stored at −20° in freezers used only for this purpose; these freezers have an emergency backup power supply.
Data Management
At our facility, signalment, DNA information, and disease characterization for each animal resides in a server database designed and developed by a programmer-analyst in the information technology group at our veterinary medical college who worked closely with administrators of the DNA bank. It is updated and searched through a custom Web application by use of active server pages. Users can access the program through a Web browser. Administrators of the DNA bank log patients and samples into the database and record demographic information (which may be automatically downloaded from our veterinary medical teaching hospital database) and sample information (such as date of collection, source, and diagnoses). The DNA technician records additional information relating to DNA collection, including sample amount and storage location in the freezers. A search function enables researchers to find samples by diagnosis, breed, sex, and age. Currently, the search function is restricted to internal users (ie, those at our university) and their collaborators. An interface to the database through an access data project also enables administrators of the DNA bank to submit queries and to download data from any of the data fields.
Status of Mapping of Inherited Traits in Dogs
It is currently estimated that there are 400 to 500 discrete genetic traits (most of which correspond to disease phenotypes) recognized in dogs. In a report in 2004,7 334 genetically transmitted diseases in purebred dogs were listed, and diseases in each of 148 purebred dogs were identified. In 2007,8 an on-line database of inherited disorders and other single-locus traits in farm and companion animals (ie, Online Mendelian Inheritance in Animals) listed 461 genetic traits in dogs. The responsible mutations for 58 mostly single-locus diseases have been identified in dogs by the use of candidate genes and classical mapping strategies. The high relevance of diseases in dogs to diseases in humans, coupled with the intrinsic importance of dogs to humans as companions with a shared environment, makes a compelling argument for improving resources that facilitate research into the genetic basis of diseases, physiology, and anatomy of dogs. However, dogs are also highly suitable for use in the analysis of complex disease traits.
Several investigators recently reported9-19 successful mapping or identification of a causative gene for genetic traits in dogs that ranged from eye diseases to coat color. Other reports20-37 indicate at least 18 genetic diseases in dogs have been used in the investigation of gene therapy. Investigators have successfully mapped the chromosomal regions (ie, QTLs) that contain genes that contribute to complex traits.38-44 The challenge is to uncover genes or quantitative trait nucleotides within each QTL that influence the trait or disease process. It has been reported45 that polymorphisms in the insulin-like growth factor-1 gene are responsible for a major proportion of the variation in size of Portuguese Water Dogs and size differences among breeds. Investigators in that study used the principal of mapping the locus of a complex trait in a single breed and refining the locus region by mapping among breeds.
Power Analysis
Power to map disease alleles by association will primarily be a function of mode of disease transmission, number of case and control animals, allele frequency, relative risk conferred by the allele, allelic interaction, and LD between the disease and marker allele. We have simulated Mendelian diseases with varying degrees of penetrance and multigenic inheritance in which a locus accounts for a 2- to 5-fold increase in the odds of developing a disease and in which we also varied the amount of total genetic variation in the trait. We identified 2 typical results from those simulations (Figure 2). For our simulations, power was plotted on the y-axis as a function of the number of case animals (and an equal number of control animals), assuming a Mendelian recessive disease with 80% penetrance and a critical threshold of P < 0.0001. With an LD between the disease and marker allele of r2 = 0.8 that should be attainable with a dense SNP screen within a breed, the power to map the disease approached 99% for approximately 20 case animals and was approximately 60% when there were 10 case animals. Even in genomic scans in which marker coverage was not as complete or when considering LD mapping among multiple breeds with r2 = 0.5, power to map these diseases will be high. In practice, investigators mapped hyperparathyroidism, an autosomal dominant trait, by the use of 30 affected and 40 unaffected Keeshonds and identified the causal mutation.g
Figure 2.

Graphs of power analysis as a function of sample size for the detection of disease loci for a Mendelian recessive trait (A) or a multigenic trait (B). The analyses were conducted for r2 values of 0.5 (dotted lines), 0.65 (dashed lines) and 0.8 (dotted and dashed lines); the r2 value is a measure of LD. For each analysis, the number of case and control animals was equal.
Similarly, we performed a simulation with a similar graph for a multigenic trait in which a locus accounts for a 2-fold increase in the odds of developing a disease at a threshold of P < 0.001 (Figure 2). Power to map this locus was high and increased to approximately 99% for a 5-fold increase at P < 0.0001.The number of affected and unaffected animals required to achieve adequate power for Mendelian and complex diseases is still a matter of debate. Use of a simple model of an allele that increases risk of disease by a multiplicative factor of 2 or 5 and genotyping with the same SNP density revealed that the power to detect a locus with a sample size of 100 affected and 100 unaffected dogs was estimated at 50% and 97%, respectively,1 when genotyping at approximately 15,000 evenly spaced SNPs. The frequency of a disease in a breed and its relevance to veterinarians, dog owners, breed clubs, and funding agencies will ultimately determine the diseases or traits that are mapped. In addition, it will be preferable to implement an LD-based fine mapping approach when there are sufficient dogs of other breeds with the same disease.
Statistical Expertise and Genotyping Accuracy
The necessity for collaboration between experimental, genetic, and statistical experts and clinical faculty in designing and analyzing mapping experiments is another important issue to consider. Analysis of whole-genome SNP data requires high quality-control procedures at all stages of data collection. Particularly pressing are issues of DNA quality, handling of missing SNP genotypes, and identification of SNPs with consistent genotyping problems among samples. A mixture of existing and novel methods can be used to address these problems, including comparison of genotypes obtained on different platforms and high thresholds for inclusion of dogs in an evaluation (eg, at least 80% to 90% of genotypes must have been unambiguously determined by at least 2 algorithms). For dogs with extended pedigrees, standard evaluations of Mendelian inheritance should be used to identify potential genotyping errors. Developing algorithms sufficiently robust to evaluate cryptic population structure is an important investment.
Sharing of Resources
Applications for collaborative projects that involve the use of DNA from the archive at our veterinary medical teaching hospital are encouraged under the condition that genotypes (and eventually associated phenotypes) will be made available in a Web-based database and sufficient DNA will be provided to Cornell University for archiving for future use. Requests related to complex disease mapping will be given highest priority. Additionally, genotypes and phenotype information derived from archived DNA samples will be made available to geneticists and statisticians after publication of the data. These genotypes can then be used as control materials for other studies when the animals were screened for diseases of interest. Development of a universal control sample to serve > 1 investigator is a worthy goal. Making the genotypes accumulated on animals in a DNA archive available to all investigators is a critical element in reducing expenses associated with genotyping.
Conclusions
The DNA archives developed at veterinary medical teaching hospitals will be important resources for mapping disease loci and identifying underlying genes. The most important feature of a DNA archive is accurate identification or exclusion of the disease or diseases of each animal. Such archives will be complementary resources to tissue banksh already available to researchers studying the genetic basis of disease in animals and can make a unique contribution to investigators conducting association studies in animals.
Acknowledgments
Supported by NIH grant number R24 GM082910-01A1; by the Baker Institute for Animal Health, Center for Vertebrate Genomics, and the Department of Clinical Sciences, College of Veterinary Medicine, Cornell University; and by Pfizer Incorporated.
The authors thank Drs Kerstin Lindblad-Toh, Philip Reilly, and Jeffrey Murray for technical assistance in establishing the DNA archive at Cornell University.
Abbreviations
- kb
Kilobases
- LD
Linkage disequilibrium
- QTL
Quantitative trait locus
- SNP
Single nucleotide polymorphism
Footnotes
Applied Biosystems Inc. SNPLex or molecular inversion probe technology. Available at: products.appliedbiosystems.com/ab/en/US/adirect/ab?cmd=catNavigate2&catID=600763. Accessed October 1st, 2008.
Applied Biosystems Inc. ABI TaqMan. Available at: products.appliedbiosystems.com/ab/en/US/adirect/ab?cmd=catNavigate2&catID=601279. Accessed October 1st, 2008.
Version 2 canine SNP array. Affymetrix. Available at: www.affymetrix.com. Accessed October 1st, 2008.
Broad Institue. Canine array FAQ. Available at: www.broad.mit.edu/mammals/dog/caninearrayfaq.html. Accessed October 1st, 2008.
Canine Genome 2.0 Microarray. Affymetrix GeneChip. Available at: www.affymetrix.com/products/arrays/specific/canine_2.affx. Accessed October 1st, 2008.
Gentra Puregene blood kit, by Qiagen. Available at: www.qiagen.com/Products/GenomicDnaStabilizationPurification/GentraPuregeneBloodKit.aspx. Accessed October 1st, 2008.
Goldstein R. Department of Clinical Sciences, Cornell University, Ithaca, NY: Personal communication, 2007
Modiano Laboratory, University of Minnesota. Available at: www.modianolab.org. Accessed October 1st, 2008.
References
- 1.Lindblad-Toh K, Wade CM, Mikkelsen TS, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 2.Clark LA, Tsai KL, Steiner JM, et al. Chromosome-specific microsatellite multiplex sets for linkage studies in the domestic dog. Genomics. 2004;84:550–554. doi: 10.1016/j.ygeno.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Applied Biosystems SNP genotyping Website. Available at: marketing.appliedbiosystems.com/mk/get/SNP_LANDING. Accessed Aug 18, 2007.
- 4.Bjornerfeldt S, Hailer F, Nord M, et al. Assortative mating and fragmentation within dog breeds. BMC Evol Biol. 2008;8:28. doi: 10.1186/1471-2148-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstein O, Zangerl B, Pearce-Kelling S, et al. Linkage disequilibrium mapping in domestic dog breeds narrows the progressive rod-cone degeneration interval and identifies ancestral disease-transmitting chromosome. Genomics. 2006;88:541–550. doi: 10.1016/j.ygeno.2006.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quignon P, Herbin L, Cadieu E, et al. Canine population structure: assessment and impact of intra-breed stratification on snp-based association studies. PLoS ONE. 2007;2(12):e1324. doi: 10.1371/journal.pone.0001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guide to hereditary and congenital diseases in dogs Website. Available at: www.siriusdog.com/articles/hereditary-congenital-diseases-dog.htm. Accessed Mar 29, 2007.
- 8.Online Mendelian inheritance in Animals (OMIA) Website. Available at www.angis.org.au/Databases/BIRX/omia. Accessed Mar 29, 2007.
- 9.Awano T, Katz ML, O'Brien DP, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006;89:254–260. doi: 10.1016/j.ymgme.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Awano T, Katz ML, O'Brien DP, et al. A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006;87:341–348. doi: 10.1016/j.ymgme.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Beltran WA, Hammond P, Acland GM, et al. A frameshift mutation in RPGR exon ORF15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:1669–1681. doi: 10.1167/iovs.05-0845. [DOI] [PubMed] [Google Scholar]
- 12.Callan MB, Aljamali MN, Margaritis P, et al. A novel missense mutation responsible for factor VII deficiency in research Beagle colonies. J Thromb Haemost. 2006;4:2616–2622. doi: 10.1111/j.1538-7836.2006.02203.x. [DOI] [PubMed] [Google Scholar]
- 13.Clark LA, Wahl JM, Rees CA, et al. Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc Natl Acad Sci U S A. 2006;103:1376–1381. doi: 10.1073/pnas.0506940103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerns JA, Cargill EJ, Clark LA, et al. Linkage and segregation analysis of black and brindle coat color in domestic dogs. Genetics. 2007;176:1–11. doi: 10.1534/genetics.107.074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGraw RA, Carmichael KP. Molecular basis of globoid cell leukodystrophy in Irish Setters. Vet J. 2006;171:370–372. doi: 10.1016/j.tvjl.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Mellersh CS, Boursnell ME, Pettitt L, et al. Canine RPGRIP1 mutation establishes cone-rod dystrophy in miniature longhaired dachshunds as a homologue of human Leber congenital amaurosis. Genomics. 2006;88:293–301. doi: 10.1016/j.ygeno.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Mellersh CS, Pettitt L, Forman OP, et al. Identification of mutations in HSF4 in dogs of three different breeds with hereditary cataracts. Vet Ophthalmol. 2006;9:369–378. doi: 10.1111/j.1463-5224.2006.00496.x. [DOI] [PubMed] [Google Scholar]
- 18.Pettigrew R, Fyfe JC, Gregory BL, et al. CNS hypomyelination in rat terrier dogs with congenital goiter and a mutation in the thyroid peroxidase gene. Vet Pathol. 2007;44:50–56. doi: 10.1354/vp.44-1-50. [DOI] [PubMed] [Google Scholar]
- 19.Zangerl B, Goldstein O, Philp AR, et al. Identical mutation in a novel retinal gene causes progressive rod-cone degeneration in dogs and retinitis pigmentosa in humans. Genomics. 2006;88:551–563. doi: 10.1016/j.ygeno.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Acland GM, Aguirre GD, Bennett J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12:1072–1082. doi: 10.1016/j.ymthe.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer TR, Jr, Gu YC, Creevy KE, et al. Leukocyte adhesion deficiency in children and Irish Setter dogs. Pediatr Res. 2004;55:363–367. doi: 10.1203/01.PDR.0000111287.74989.1B. [DOI] [PubMed] [Google Scholar]
- 22.Bauer TR, Jr, Hai M, Tuschong LM, et al. Correction of the disease phenotype in canine leukocyte adhesion deficiency using ex vivo hematopoietic stem cell gene therapy. Blood. 2006;108:3313–3320. doi: 10.1182/blood-2006-03-006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casal M, Haskins M. Large animal models and gene therapy. Eur J Hum Genet. 2006;14:266–272. doi: 10.1038/sj.ejhg.5201535. [DOI] [PubMed] [Google Scholar]
- 24.Ciron C, Desmaris N, Colle MA, et al. Gene therapy of the brain in the dog model of Hurler's syndrome. Ann Neurol. 2006;60:204–213. doi: 10.1002/ana.20870. [DOI] [PubMed] [Google Scholar]
- 25.Collins CA, Morgan JE. Duchenne's muscular dystrophy: animal models used to investigate pathogenesis and develop therapeutic strategies. Int J Exp Pathol. 2003;84:165–172. doi: 10.1046/j.1365-2613.2003.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Meyer SF, Vanhoorelbeke K, Chuah MK, et al. Phenotypic correction of von Willebrand disease type 3 blood-derived endothelial cells with lentiviral vectors expressing von Willebrand factor. Blood. 2006;107:4728–4736. doi: 10.1182/blood-2005-09-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellinwood NM, Vite CH, Haskins ME. Gene therapy for lysosomal storage diseases: the lessons and promise of animal models. J Gene Med. 2004;6:481–506. doi: 10.1002/jgm.581. [DOI] [PubMed] [Google Scholar]
- 28.Felsburg PJ, Hartnett BJ, Gouthro TA, et al. Thymopoiesis and T cell development in common gamma chain-deficient dogs. Immunol Res. 2003;27:235–246. doi: 10.1385/IR:27:2-3:235. [DOI] [PubMed] [Google Scholar]
- 29.Fujiki N, Yoshida Y, Ripley B, et al. Effects of IV and ICV hypocretin-1 (orexin A) in hypocretin receptor-2 gene mutated narcoleptic dogs and IV hypocretin-1 replacement therapy in a hypocretin-ligand-deficient narcoleptic dog. Sleep. 2003;26:953–959. doi: 10.1093/sleep/26.8.953. [DOI] [PubMed] [Google Scholar]
- 30.Jacobson SG, Acland GM, Aguirre GD, et al. Safety of recombinant adeno-associated virus type 2-RPE65 vector delivered by ocular subretinal injection. Mol Ther. 2006;13:1074–1084. doi: 10.1016/j.ymthe.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Jiang H, Lillicrap D, Patarroyo-White S, et al. Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood. 2006;108:107–115. doi: 10.1182/blood-2005-12-5115. [DOI] [PubMed] [Google Scholar]
- 32.McCormack WM, Jr, Seiler MP, Bertin TK, et al. Helper-dependent adenoviral gene therapy mediates long-term correction of the clotting defect in the canine hemophilia A model. J Thromb Haemost. 2006;4:1218–1225. doi: 10.1111/j.1538-7836.2006.01901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarkar R, Mucci M, Addya S, et al. Long-term efficacy of adeno-associated virus serotypes 8 and 9 in hemophilia A dogs and mice. Hum Gene Ther. 2006;17:427–439. doi: 10.1089/hum.2006.17.427. [DOI] [PubMed] [Google Scholar]
- 34.Ting-De Ravin SS, Kennedy DR, Naumann N, et al. Correction of canine X-linked severe combined immunodeficiency by in vivo retroviral gene therapy. Blood. 2006;107:3091–3097. doi: 10.1182/blood-2005-10-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trobridge G, Beard BC, Kiem HP. Hematopoietic stem cell transduction and amplification in large animal models. Hum Gene Ther. 2005;16:1355–1366. doi: 10.1089/hum.2005.16.1355. [DOI] [PubMed] [Google Scholar]
- 36.Wang B, O'Malley TM, Xu L, et al. Expression in blood cells may contribute to biochemical and pathological improvements after neonatal intravenous gene therapy for mucopolysaccharidosis VII in dogs. Mol Genet Metab. 2006;87:8–21. doi: 10.1016/j.ymgme.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Yanay O, Brzezinski M, Christensen J, et al. An adult dog with cyclic neutropenia treated by lentivirus-mediated delivery of granulocyte colony-stimulating factor. Hum Gene Ther. 2006;17:464–469. doi: 10.1089/hum.2006.17.464. [DOI] [PubMed] [Google Scholar]
- 38.Carrier DR, Chase K, Lark KG. Genetics of canid skeletal variation: size and shape of the pelvis. Genome Res. 2005;15:1825–1830. doi: 10.1101/gr.3800005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chase K, Sargan D, Miller K, et al. Understanding the genetics of autoimmune disease: two loci that regulate late onset Addison's disease in Portuguese Water Dogs. Int J Immunogenet. 2006;33:179–184. doi: 10.1111/j.1744-313X.2006.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lark KG, Chase K, Sutter NB. Genetic architecture of the dog: sexual size dimorphism and functional morphology. Trends Genet. 2006;22:537–544. doi: 10.1016/j.tig.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chase K, Lawler DF, Adler FR, et al. Bilaterally asymmetric effects of quantitative trait loci (QTLs): QTLs that affect laxity in the right versus left coxofemoral (hip) joints of the dog (Canis familiaris) Am J Med Genet. 2006;124:239–247. doi: 10.1002/ajmg.a.20363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chase K, Lawler DF, Carrier DR, et al. Genetic regulation of osteoarthritis: a QTL regulating cranial and caudal acetabular osteophyte formation in the hip joint of the dog (Canis familiaris) Am J Med Genet A. 2005;135:334–335. doi: 10.1002/ajmg.a.30719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Todhunter RJ, Mateescu R, Lust G, et al. Quantitative trait loci for hip dysplasia in a cross-breed canine pedigree. Mamm Genome. 2005;16:720–730. doi: 10.1007/s00335-005-0004-4. [DOI] [PubMed] [Google Scholar]
- 44.Zhu L, Zhang Z, Schweitzer PA, et al. Single nucleotide polymorphisms refine quantitative trait loci intervals for hip joint laxity in dogs. Anim Genet2. 2008;39:141–146. doi: 10.1111/j.1365-2052.2007.01691.x. [DOI] [PubMed] [Google Scholar]
- 45.Sutter NB, Bustamante CD, Chase K, et al. A single IGF1 allele is a major determinant of small size in dogs. Science. 2007;316:112–115. doi: 10.1126/science.1137045. [DOI] [PMC free article] [PubMed] [Google Scholar]