


Abstract
AT-rich minisatellites (AT islands) are sites of genomic instability in cancer cells and targets for extremely lethal AT-specific drugs, such as bizelesin. Here we investigated the AT islands in the FRA16B fragile site region for their possible roles in the organization of DNA on the nuclear matrix. The FRA16B AT island nominally spans ∼3 kb of mostly >90% A/T DNA. In silico analysis indicates that this domain exhibits characteristics of nuclear matrix attachment regions (MARs): an exceptionally intense computed ‘MAR potential’ and profound duplex destabilization and flexibility. FRA16B repeats specifically bind to isolated nuclear matrices, which indicates their in vitro MAR function. This binding is several-fold greater than that of a known MAR in the c-myc gene. AT islands in fragile sites FRA16B and FRA16D are significantly more abundant in CEM cells that are hypersensitive to bizelesin compared to normal WI-38 cells. FRA16B overabundance in CEM is due to an ∼10-fold expansion of FRA16B repeats. The expanded FRA16B minisatellites in CEM cells preferentially localize to the nuclear matrix-associated DNA indicating their in vivo MAR function. The unexpanded repeats in WI-38 cells localize to the loop DNA. The c-myc MAR is also matrix-associated in CEM cells while localizing to loop DNA in WI-38 cells. These results are the first to demonstrate that AT islands in fragile sites can function as MARs both in vitro and in vivo. The ability of FRA16B-mediated MAR sites to rearrange depending on the repeat expansion status could be relevant to both genomic instability of cancer cells and their sensitivity to AT-island targeting drugs.
INTRODUCTION
The human genome contains a unique class of minisatellites, referred to as AT islands, which consist of 200–1000+ bp of repetitive sequences (1,2). Long AT islands are extremely A/T-rich (up to 100%) and relatively infrequent (∼3000 in the human genome), which distinguishes them from various shorter and/or less AT-enriched tracts that occur in millions of sites. Damage to AT islands by certain AT-specific anticancer drugs (bizelesin and U78799) is lethal to the cell, which suggests that these domains can be critically important (1,3–6).
Like minisatellites, in general AT islands are hypervariable elements due to polymerase slippage during replication of the repeats and/or unequal recombination events (7,8). The hypervariability of AT islands, in particular those in AT-rich fragile sites, leads to rearrangements, expansion and amplification (2,9–12). Importantly, various AT-rich sites of genomic instability, including fragile sites, have been implicated in the formation of certain tumors, particularly leukemias and lymphomas (9,10,12–18).
AT island sequences are uniquely flexible, thermodynamically destabilized, and exceptionally prone to superhelical stress-induced duplex destabilization (1,19). These attributes are consistent with the idea that AT islands may serve as anchorage sites for DNA on the nuclear matrix (matrix attachment regions, MARs) (1). MARs are vital elements of nuclear organization that are critical for correct replication, cell type-specific transcription, mitosis and other nuclear events (19–22). For some AT islands, experimental evidence confirms their ability to interact with isolated nuclear matrices (1,2). The in silico sequence attributes suggested that AT islands in cancer-relevant AT-rich fragile sites, such as FRA16B, could also function as MARs (1,2,5,9). However, no published study has ever confirmed the ability of AT islands in fragile sites to associate either with isolated nuclear matrices or the nuclear matrix in intact cells.
This investigation explored the MAR properties of FRA16B repeats in silico, in vitro and in vivo, including the comparison of tumor and normal cells. The results show that FRA16B repeats have potent MAR attributes and can specifically interact with isolated nuclear matrices. Abnormally expanded FRA16B repeats were identified in human leukemic CEM cells and compared to unexpanded repeats in normal WI-38 cells. The expanded FRA16B repeats are preferentially associated with the nuclear matrix, which indicates their in vivo MAR function. In contrast the unexpanded (normal) FRA16B repeats localize mainly in the loop DNA.
MATERIALS AND METHODS
In silico sequence analyses
DNA sequences were analyzed using the human genome data at the UCSC Genome Bioinformatics Site (http://www.genome.ucsc.edu/, freeze date of June 2002) (23) using the Blat algorithm (24) at the same site for sequence alignments. The segment of FRA16B locus containing the AT island is covered by DDBJ/EMBL/GenBank AC009055 and overlapping DDBJ/EMBL/GenBank AC123909.
The percentage of A/T bases and duplex stability were calculated for 250 and 25 bp sliding windows, respectively, and DNA flexibility was calculated for a 250 bp stepping window (25 bp step) as described previously (1). MAR potential was assessed using the MAR Finder sequence analysis tool (25) as described elsewhere (1), using default settings. The hypothetical sequences of expanded FRA16B contained multiple copies, arranged in tandem, of the 1200 bp telomeric section of the FRA16B AT island (positions 168 354–169 553 in DDBJ/EMBL/GenBank AC009055) within a segment spanning ∼80 kb of the FRA16B locus (unexpanded size). The results are plotted as MAR potential normalized to the highest peak (Fig. 1D) and as non-normalized integrated MAR potential (Fig. 1E) (1).
Figure 1.



The organization and in silico MAR attributes of the AT island in human chr16q22.1 in the fragile site FRA16B region. (A) Percentage of A/T bases. The broken line corresponds to average content of A/T in the genome (60%). Position 1 corresponds to position 158 261 in DDBJ/EMBL/GenBank AC009055. (B) Potential binding sites for the AT-island-targeting drug bizelesin T(A/T)4A and restriction sites of SalI, BamHI, PvuII and EcoRI used in some experiments. (C) The map of sub-domain 2. Horizontal bars indicate the alignment of the previously reported FRA16B single allele sequence [DDBJ/EMBL/GenBank U85253 (29)] and the 33 bp consensus repeats ATATATTATATATTATATCTAATAATATATA/CTA derived from that sequence and some other repeats identified. All features are plotted to scale. (D) Duplex flexibility (the possible variations in propeller twist angle) and stability (the sequence-dependent free energy of the helix-to-coil transition) calculated for 250 bp moving windows. Horizontal broken lines indicate average flexibility and duplex stability of ∼180 kb surrounding the FRA16B AT island. Sequence positions are numbered as in (A). (E) Top. Calculated MAR potential (normalized to the highest value) of unexpanded (1×) FRA16B AT island and a hypothetical sequence with expanded FRA16B AT island (6×) in a stretch of ∼80 kb sequence of the FRA16B locus. Positions of sub-domains 1 (166 961) and 2 (168 911) of the FRA16B AT island in DDBJ/EMBL/GenBank AC009055 are indicated. Bottom. Integrated MAR potential of unexpanded sub-domain 2 of FRA16B AT island (arrow) and its hypothetical expanded homologs.
Genomic DNA preparations
Leukemic CEM cells and WI-38 normal lung fibroblast cells, cultured as described previously (26), were labeled overnight with 0.1 µCi/ml [14C]thymidine followed by DNA purification using standard procedures. 14C-labeled DNA from normal colon mucosa NCM460 cells was provided by Dr B. Woynarowska. The amounts of DNA from these cell lines were expressed as cell equivalents based on 14C radioactivity of DNA preparations (3,27). Unlabeled total genomic DNA from the following tumor and normal cell lines was purchased from ATCC (Manassas, VA): HL-60 and K562 leukemic cells, H2126 and H1395 lung carcinomas and BL2126 and BL1395 lymphoblast cells.
Preparation of nuclear matrices
Nuclear matrices were prepared using the high-salt procedure (28) with slight modifications. Briefly, cells were washed in PBS and then resuspended at 2 × 107 cells/ml in cold 50 mM Tris–HCl pH 7.8, 5 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, 0.2% Triton X-100. All solutions were supplemented with 0.1 mM PMSF and 1/100 dilution of Protease Inhibitor Cocktail for Mammalian Tissues (Sigma-Aldrich). The cell suspension was homogenized in a Dounce homogenizer, nuclei pelleted by centrifugation and resuspended in 10 mM NaCl, 10 mM Tris pH 7.0, 2 mM MgCl2. The suspension was gradually brought up to 1.6 M NaCl, followed by digestion with DNase I, washing by centrifugation, and resuspension of the matrices in the Binding Buffer (50 mM NaCl, 10 mM Tris–HCl pH 7.4, 1 mM MgCl2, 0.25 M sucrose, 0.25 mg/ml BSA) (28). Aliquots of matrix suspension were stored at –20°C prior to the binding assay.
Radiolabeled probes for in vitro MAR assay
FRA16B probes for nuclear matrix binding assay were generated by PCR using cloned FRA16B repeats as template. To clone FRA16B repeats, sequences of sub-domain 2 (Fig. 1) were first amplified using primers 29 and 38 described by Yu et al. (29) and total DNA from CEM cells. The PCR products were cloned into pGEM-T Easy vector system (Promega, Madison, WI) using standard procedures. A clone designated 1B1.33, which contains a 1076 bp insert of FRA16B repeats, was used as template in radiolabeled PCR reaction with d(GGGAATTCGATTGTACTATA) and d(CTCAAGCTATGCATCCA) as the forward and reverse primers, respectively, to give a product of 1164 bp. PCR reactions comprised 20 pg template 1B1.33 clone plasmid DNA, 10 mM Tris–HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.125 µM each primer, 0.16 mM [α32P]dATP (0.016 µCi/µl) and 0.2 mM of dTTP, dGTP and dCTP, in addition to buffer and Taq polymerase (Epicentre, Madison, WI). Thermocycling in a Perkin Elmer model 9600 machine consisted of: initial denaturation for 60 s at 95°C followed by 28 cycles of 15 s at 94°C, 30 s at 45°C and 45 s at 60°C, and a final extension for 5 min at 60°C. To generate the 947 bp c-myc probe, PCR reactions were carried out under the same conditions using the previously described primers (27) and total DNA from CEM cells as template. Both probes were purified on G50 spin columns.
Binding of radiolabeled probes to isolated nuclear matrices (in vitro MAR assay)
Nuclear matrices were washed three times in Binding Buffer. Binding reactions typically consisted of 0.25 × 106 nuclear matrices preincubated for 10 min with excess of unlabeled competitor DNA (typically 10 µg), and ∼1.5 × 104 c.p.m. of radiolabeled probe, all in 50 µl of Binding Buffer. After a 4 h incubation at 37°C, matrices were washed twice by centrifugation at 10 000 g at 4°C with the Binding Buffer and matrix pellets were solubilized overnight at 37°C in 0.5% SDS with proteinase K (0.4 mg/ml). Matrix-associated DNA probes were visualized by electrophoresis in 3% agarose and autoradiography, and quantitated using densitometry and IQ software (Molecular Dynamics). Signals were normalized to the signals of input probes that were co-run on the same gels.
Hybridization probes
The FRA16B probe corresponded to the 33 bp repeat unit in the FRA16B AT island region, d(ATATATTATATATTATATCTAATAATATATA/CTA) (29). The FRA16D probe, d(GTTAAGACTATCTAAGATTCAGATCTCCCT), corresponded to positions 147 547–147 576 in DDBJ/EMBL/ GenBank AF217491S3. The previously established repeat d(ATATATATTTATATATATATTTATATTT) was used as a probe for an AT island in DDBJ/EMBL/GenBank Z79699 (1). A segment of the β-globin gene, d(AGGTTGGTATCAAGGTTA), served as a probe for the non-AT island domain. The specific probes and a degenerated probe d(A/T)60 were obtained as 5′-digoxygenin (DIG)-labeled oligonucleotides from MWG Biotech (High Point, NC). The c-myc MAR was probed using the same primer system resulting in a 947 bp product (1) as described for the nuclear matrix binding assay, except that the PCR DIG labeling kit (Roche, Indianapolis, IN) was used. All the probes produced specific signals for the intended target sequences and negligible cross-hybridization to non-targets.
Dot blot hybridizations
Dot blot analysis was performed in triplicate using three DNA amounts (cell equivalents). Briefly, DNA samples denatured at 95°C for 10 min and rapidly quenched on ice were spotted onto positively charged nylon membranes (Roche) using S&S Minifold I dot blot apparatus (Schleicher & Schuell, Keene, NH) as recommended by the manufacturer. DNA was fixed to membranes by UV crosslinking. Membranes were hybridized overnight with 10 pmol/ml DIG-labeled probe in the hybridization solution [5× SSC (saline sodium citrate buffer: 20× corresponding to 3 M NaCl, 0.3 M sodium citrate, pH 7.0), 0.1% N-lauroylsarcosine, 0.02% SDS, 1% blocking reagent, Roche]. The following hybridization temperatures were used: 30°C for FRA16B and Z79699, 42°C for FRA16D and AT60, 35°C for β-globin and 69°C for c-myc MAR. Following washes at room temperature for oligonucleotide probes and at 65°C for the longer c-myc MAR probe, bound DIG-labeled probes were detected using DIG luminescent detection kit (Roche) according to the manufacturer’s instructions.
Dot signal intensities were quantitated by densitometry. Response (signal versus cell equivalents) was calculated by averaging the data for individual cell equivalent levels. For each probe these signals were averaged and normalized to the respective signals for WI-38 DNA from the same membrane. Normalized data from separate dot blot experiments were again averaged to yield the final results shown in Figure 1B.
Restriction analysis and field inversion gel electrophoresis (FIGE)
DNA from CEM, WI-38 and NCM460 cells was digested overnight at 37°C with SalI (20 U/106 cell equivalents, Invitrogen, Carlsbad, CA) or a combination of BamHI/EcoRI (Promega, Madison, WI) each at ∼30 U/106 cell equivalents DNA. After a phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation, digested DNA samples were separated by FIGE on a 1% agarose gel in TBE buffer in a FIGE Mapper (Bio-Rad Laboratories, Hercules, CA, program numbers 4 and 2 for SalI and BamHI/EcoRI, respectively). Gels were stained with Sybr green I for total DNA, followed by capillary transfer of DNA onto nitrocellulose membranes and hybridization to the FRA16B probe as described for the dot blot hybridization assay.
Preparation of matrix-associated and loop DNA
Matrix-associated and loop DNA fractions were prepared using a modification of the high-salt procedure (28). Harvested [14C]thymidine-labeled cells were resuspended at 2 × 107 cells/ml for 15 min in cold 10 mM Tris–HCl pH 7.0, 2 mM MgCl2, 1/100 dilution of Protease Inhibitor Cocktail for Mammalian Tissues (Sigma-Aldrich) and next homogenized in a Dounce homogenizer. Following centrifugation (1900 g for 30 min) over 45% sucrose in homogenization buffer, nuclear pellets were resuspended at 2 × 107 nuclei/ml in enzyme buffer (6 mM Tris–HCl pH 7.5, 6 mM MgCl2, 100 mM NaCl and 1 mM DTT). Typically, 1 × 107 nuclei were digested with a combination of restriction nucleases that do not cleave AT islands (BamHI, PvuII, EcoRI) at 4 U/µg DNA for 12–16 h overnight at 37°C. The digests were placed on ice and 3 M NaCl (in 10 mM Tris–HCl pH 7.0 and 0.2 mM MgCl2) was slowly added to a final concentration of 1.5 M NaCl. Centrifugation at 1500 g for 15 min separated nuclear matrices in the pellet from digested (loop) DNA in the supernatant. Nuclear matrices were washed once with 1.5 M NaCl in 10 mM Tris–HCl pH 7.0, 1 mM MgCl2, followed by two washes in 10 mM Tris–HCl pH 7.0, 1 mM MgCl2, 10 mM NaCl. The pooled supernatant fractions and the pelleted nuclear matrices were subjected to standard DNA purification protocols to yield loop and matrix-associated DNA fractions, respectively.
Dot blot, FIGE and PCR analysis of matrix-associated and loop DNA
Matrix-associated, loop and total DNA were analyzed by dot blotting and hybridization to specific probes as described for total DNA. The amounts of DNA fractions loaded were equalized as ‘cell equivalents’ based on 14C radioactivity. The results were expressed as the average ratio of signal intensities of matrix DNA to loop DNA.
PCR analysis to detect FRA16B sequences in matrix-associated and loop DNA fractions was performed using the same FRA16B PCR system (primers 29 and 38) described in the previous section. The reactions were subjected to an initial denaturation at 95°C for 2.5 min, followed by one cycle of 30 s at 95°C, 1.5 min at 45°C, 6 min at 58°C, followed by 32 cycles of (1 min at 88°C, 30 s at 45°C, 4 min at 58°C) and a final 4 min extension at 58°C. The reactions were analyzed by FIGE, followed by Sybr green I staining and Southern transfer and hybridization to the FRA16B probe as described in the previous sections.
Quantitative PCR for c-myc MAR/β-globin
Primers for the MAR region of the c-myc gene and a non-AT rich region in the β-globin gene were described previously (27). Quantitative PCR reactions for c-myc MAR/β-globin, carried out in a multiplex format, comprised (in 20 µl) template DNA, 10 mM Tris–HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.4 µM each c-myc MAR primer, 0.15 µM each β-globin primer, 0.4 mM of dATP and dTTP, 0.2 mM of dGTP and dCTP, 1.2 µCi/reaction [α-32P]dATP (NEN) and 0.8 U MasterAmp™ Taq polymerase (Epicentre, Madison, WI). Total, matrix and loop DNA were used as templates, each in duplicate reactions. The reactions were subjected to initial denaturation for 30 s at 95°C followed by 26 cycles of 20 s at 94°C, 30 s at 55°C and 45 s at 68°C, and a final extension for 3 min at 68°C. PCR products were analyzed by agarose electrophoresis, followed by autoradiography (3,27). The c-myc MAR signals in each reaction were normalized to the signal of β-globin in the same reaction and the resulting ratios averaged for all replicates and template amounts in a given experiment. The reported results are average values of individual means (±SE) from two independent experiments.
RESULTS
The first part of this study addresses the in silico attributes of the FRA16B locus that are relevant to MAR function and verifies the MAR properties of FRA16B sequences in vitro using isolated nuclear matrices. The second part evaluates the abundance and expansion status of FRA16B AT islands in cancer and normal cells and determines the association of these domains with the nuclear matrix in vivo.
AT island minisatellite in the FRA16B locus
As per the recently completed ‘final’ status of chr16q22.1 DNA sequence, the FRA16B AT island spans >3 kb and comprises two adjacent areas of extreme AT-richness (Fig. 1). The A/T content of both sub-domains exceeds 90%, and the linker between them is only slightly less A/T-rich. With this size and AT richness, FRA16B AT island is one of the most expansive human AT islands identified thus far (1 and data not shown).
Both sub-domains harbor profound clusters of T(A/T)4A motifs (Fig. 1B), which are preferred binding sites for the AT-island targeting drugs, such as bizelesin (1,3,4). Sub-domain 1 of the FRA16B AT island consists of various types of simple A/T repeats, some of which contain an occasional G/C pair (Fig. 1C). The telomeric sub-domain 2 is composed of similar repeats but its sections conform to the 33-bp repeat identified previously by Yu et al. as the element responsible for the expansion of FRA16B sites (29).
In silico MAR potential of the FRA16B AT island
Whereas MAR domains do not have a single consensus sequence, the ‘MAR potential’ of a given region can be assessed based on the presence of various features that are known to be associated with functional MARs. Duplex destabilization and increased duplex flexibility are characteristic markers of prominent MARs (1,19). FRA16B AT island shows dramatically elevated flexibility that co-maps to a profound depression in local duplex stability (Fig. 1D). In the magnitude of these effects and the length of the affected sequences, the FRA16B AT island remains unmatched by any other section of the chr16q22.1 locus (Fig. 2A and data not shown).
Figure 2.

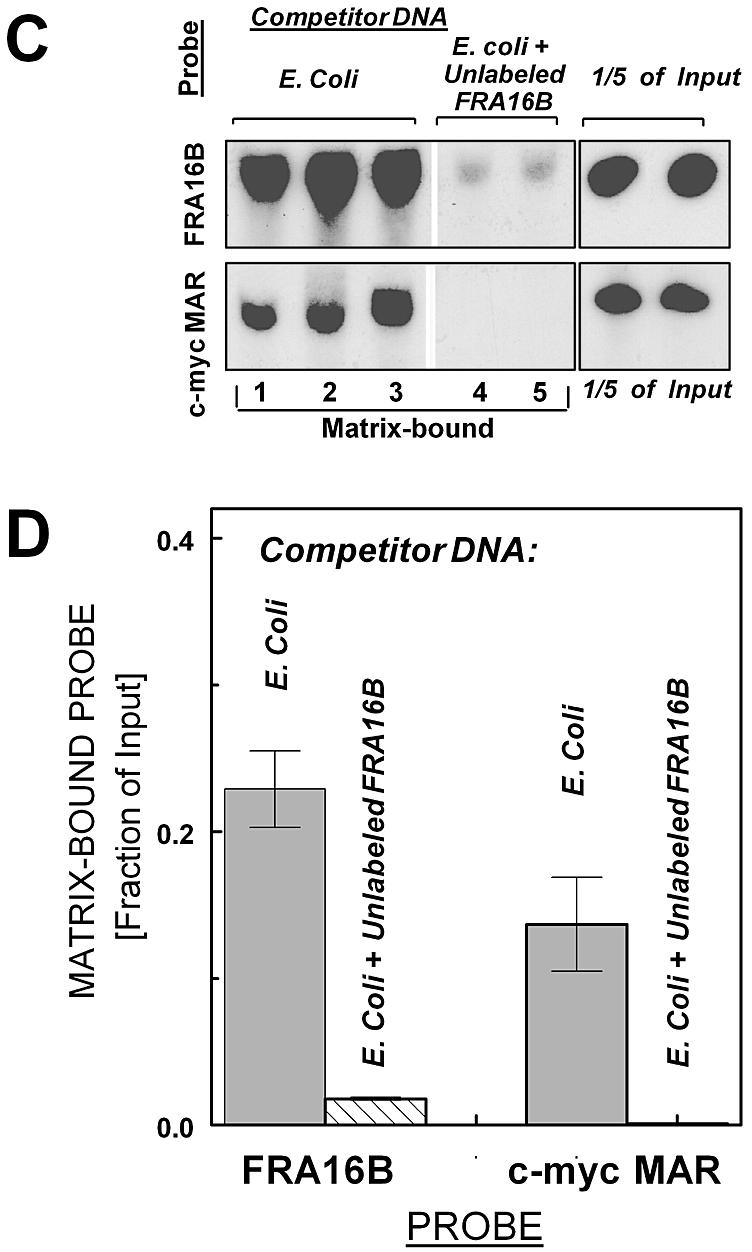
Specific binding of FRA16B and c-myc MAR AT islands to nuclear matrices isolated from leukemic CEM cells (A, B) and normal WI-38 cells (C, D). (A) Binding of radiolabeled probes for FRA16B and c-myc MAR to isolated nuclear matrices from CEM cells in the presence of an ∼2000-fold excess of the indicated unlabeled competitor DNA: E.coli (lanes 1–3), poly(dG.dC) (lanes 4–6) or salmon sperm DNA (lanes 7 and 8). Lanes 9 and 10 for the FRA16B probe contain ∼8-fold excess of unlabeled FRA16B probe, in addition to 2000-fold excess of E.coli DNA. Additional lanes 11 and 12 contain one-fifth of the input amount for each probe. N.D., not determined. (B) Nuclear matrix-bound fraction of input probe for assay with matrices from CEM cells (average from 2–6 reactions ± SE from two independent experiments). (C) Binding of radiolabeled FRA16B and c-myc probes to isolated nuclear matrices from WI-38 cells in the presence of unlabeled E.coli DNA (lanes 1–3) or E.coli DNA + ∼8-fold excess of unlabeled FRA16B probe (lanes 4 and 5, upper panel) or E.coli DNA + unlabeled c-myc probe (lanes 4 and 5, lower panel). (D) Nuclear matrix-bound fraction of input probe for assay with matrices from WI-38 cells (average from 2–4 reactions ± SE from 1–2 independent experiments).
Computations using the algorithm of Kramer et al. (25) further underscore the potent MAR attributes of FRA16B AT island, which comprises the only prominent peaks of MAR potential in the entire analyzed stretch of ∼80 kb (Fig. 1E). Moreover, the MAR potential of normal length FRA16B may markedly increase as a result of repeat expansion, as suggested by the analysis of hypothetical sequences with expanded FRA16B sub-domain 2 (Fig. 1E, lower panel). Expanded FRA16B sites are also characterized by more extensive duplex destabilization than the initial length sites (data not shown). These analyses strongly suggest that FRA16B AT islands are likely candidates for strong interaction with the nuclear matrices, in particular if they become expanded.
FRA16B AT island binds specifically to isolated nuclear matrices (in vitro MAR function)
To verify the MAR properties of FRA16B, we utilized a standard in vitro MAR binding assay using isolated nuclear matrices. The assay utilized a 1076 bp probe derived from FRA16B sub-domain 2. Another AT island of known MAR properties (in the 3′ region of the c-myc gene) was used for comparison. The results demonstrate that the FRA16B probe binds specifically to nuclear matrices from leukemic CEM cells (Fig. 2A and B). In the presence of ∼2000-fold excess of unlabeled Escherichia coli DNA or poly(dG/dC) as non-specific competitors, ∼37 and 28%, respectively, of the input labeled FRA16B probe was matrix-associated [Fig. 2A (top lanes 1–3 and 4–6) and B]. Unlike E.coli DNA or poly(dG/dC), salmon sperm DNA contains multiple MAR domains typical of higher eukaryotic organisms. Accordingly, FRA16B binding to the nuclear matrices was significantly reduced (to ∼5.5% input) in the presence of ∼2000-fold excess of salmon sperm DNA. Furthermore, the preincubation of the matrices with ∼8-fold excess of unlabeled FRA16B probe (in addition to E.coli DNA) nearly eliminated the signal of radiolabeled matrix-bound probe.
The binding of the c-myc MAR was also clearly detectable in the presence of unlabeled E.coli DNA or poly(dG/dC) [<5 and 7% of input probe, respectively; Fig. 2A (bottom lanes 1–3 and 4–6) and B], but its magnitude was several-fold lower, compared to the binding of FRA16B probe. Moreover, the c-myc MAR signal was essentially eliminated by salmon sperm DNA, whereas the FRA16B signal was reduced but remained significant under the same conditions. Both probes also interacted specifically with matrices obtained from normal WI-38 cells (Fig. 2C and D). Compared with the matrices from CEM cells, the magnitude of FRA16B binding was somewhat less profound and that of the c-myc probe more profound. Still, the fraction of matrix-bound FRA16B probe exceeded that of the c-myc probe. These results demonstrate that the FRA16B AT island is a potent MAR in vitro, which is capable of more efficient matrix binding than the known MAR in the c-myc gene.
Abnormal abundance of AT islands in FRA16B and FRA16D in leukemic CEM cells
The cellular organization of AT-rich fragile sites was addressed in acute lymphoblastic leukemia CEM and WI-38 normal fibroblast cells. These cell lines are, respectively, hypersensitive and non-hypersensitive to AT island-targeting drugs (GI50 for bizelesin of 2 and 80 pM, respectively) (5,30). Given the inherent instability of bizelesin-targeted AT islands in fragile sites, it was possible that the organization of these domains might be altered in cancer cells. The abundance of specific AT islands was determined in CEM and WI-38 cells using dot blot hybridization of total cellular DNA and probes for FRA16B and other specific AT islands: FRA16D, Z79699 and c-myc MAR.
The quantitation of raw dot blot hybridization data (Fig. 3A) as a function of input DNA (expressed as cell equivalents) indicates minimal differences between DNA from CEM and WI-38 in their overall AT-richness [monitored using (A/T)60 probe] and in the amounts of the non-AT island sequence derived from the β-globin gene. The CEM/WI-38 abundance ratio for (A/T)60 and the β-globin segment amounted to 0.9 ± 0.1 and 1.6 ± 0.2, respectively (Fig. 3B). In contrast, the signal for AT islands in FRA16B and FRA16D was clearly stronger for CEM than for WI-38 DNA. The increase in FRA16D was smaller but also significant (2.3–0.2-fold, P < 0.001). The differences between CEM and WI-38 cells do not extend to all AT islands, as signals for the AT island in Z79699 were only marginally increased in CEM DNA (1.6–0.3-fold, P > 0.05).
Figure 3.


The abundance of AT islands and non-AT islands in total DNA from cancer CEM (filled circles) and normal WI-38 (open squares) cells by dot blot hybridization. (A) The intensity of dot blot hybridization signals for the indicated probes versus input DNA (expressed as cell equivalents). Points are mean ± SE values from a single representative dot blot experiment carried out in duplicate. Error values were smaller than the size of symbols. Inserts show actual dot blots for these data. (B) Averaged dot blot hybridization signals normalized to respective signals for WI-38 cells (mean values ± SE from 5–10 independent experiments).
Several other human cancer and normal cell lines were screened for abnormal FRA16B and FRA16D signals. A significant 4-fold increase (P < 0.001) in the normalized signal of FRA16B was observed in colon carcinoma Colo320DM. No significant differences in FRA16B and FRA16D abundance over WI-38 cells were found for HCT116 colorectal carcinoma, leukemic Jurkat, HL-60 and K562 cells, MO59J glioblastoma, LNCaP-Pro5 prostate cancer, A2780 ovarian carcinoma cells, NCM460 normal colon mucosa, H1395 and H2126 lung adenocarcinomas and normal cells from the same individuals, BL1395 and BL2126, respectively (data not shown). Thus, the overabundance of AT repeats in fragile sites seems specific for some types of cancer cells.
Differential organization of AT-rich fragile site FRA16B in normal and tumor cells
The observed increased abundance of AT-rich fragile sites in CEM tumor cells could, in theory, originate either from markedly amplified loci of unchanged normal length or from a few loci with a profound repeat expansion. To distinguish between these two possibilities, we analyzed the length distribution of restriction fragments generated by BamHI/EcoRI, whose cleavage sites tightly frame the FRA16B AT island, and by SalI, which has one cleavage site 5′ from the AT island with the next site at the nominal distance of ∼39 kb on the 3′ side (see Fig. 1B for the position of these cleavage sites).
FIGE analysis indicates that total DNA from CEM and WI-38 cells is similarly cleaved by BamHI/EcoRI (Sybr green I staining, Fig. 4A). Consistent with the greater abundance of FRA16B repeats, the signal of hybridized FRA16B probe is much stronger for CEM DNA. Moreover, this signal clearly has high molecular weight components of at least ∼50 kb. These large FRA16B BamHI/EcoRI fragments are absent from the DNA of normal WI-38 cells, whose signal centers at ∼6–10 kb, in agreement with the nominal length of FRA16B BamHI/EcoRI fragments (8.1 kb) based on DDBJ/EMBL/GenBank sequence.
Figure 4.

Differential organization of FRA16B region in cancer CEM and normal WI-38 cells. (A) Restriction analysis of BamHI/EcoRI fragments using total DNA from CEM (lanes 1) and WI-38 cells (lanes 2). The amounts of DNA loaded were 24 000 and 32 000 cell equivalents, respectively. Sybr green I staining of FIGE agarose gels for total DNA and a Southern blot hybridization to FRA16B oligonucleotide probe are shown. The positions of BamHI/EcoRI cleavage sites in FRA16B locus are shown in Figure 1. (B) FIGE analysis of SalI restricted DNA from CEM (lanes 1 and 2) and WI-38 (lane 4) cells. NCM460 (lane 3) represents an additional type of normal cell. The amounts of DNA loaded for CEM, NCM460 and WI-38 were 7000, 10 000 and 14 000 cell equivalents, respectively. The second CEM lane is a re-digest of the digested sample in lane 1 with an excess of SalI (500 U SalI/106 cell equivalents). The position of the SalI cleavage site in the vicinity of the FRA16B AT island is indicated in Figure 1. Size markers are indicated in lanes M.
Hybridization to FRA16B probe after FIGE analysis of SalI digests (Fig. 4B) corroborates the striking differences between CEM and two normal cell lines, WI-38 and NCM460. The FRA16B signal for SalI fragments from WI-38 and NCM460 normal cells peaks at <20 kb and virtually no signal is detected above ∼25 kb. SalI fragments of that size are also present in DNA from CEM, but the prominent FRA16B signal is observed at much larger fragment sizes between 50 and 100 kb. This pattern was fully reproducible using several independently digested DNA preparations. Also, any possibility that the large-size ‘smear’ of restricted CEM DNA reflected incomplete digestion could be ruled out, since the re-digestion of this material using a large excess of SalI resulted in a virtually identical fragment size distribution (Fig. 4B). The presence of abnormally long SalI fragments, detected using the same FRA16B probe has been previously reported as indicative of expanded FRA16B repeats (29).
It needs to be noted that the FRA16B signals for WI-38 cells are comparable, but not identical, to the nominal sizes of restriction fragments for unexpanded FRA16B (8.1 and 39.2 kb for BamHI/EcoRI and SalI, respectively). These nominal sizes are for single normal alleles, as listed in the current version of the human genome sequence. However, AT islands are known to be polymorphic and are typically present in several distinct sizes. In addition, the quality of the repetitive sequences in databases, specifically regarding the number of repeats, is known to be limited (31). The polymorphism and overlapping hypervariability are probably responsible for the FRA16B signal in the restriction fragments, indicative of a distribution rather than discrete fragments. In particular, the sizes of SalI fragments may be affected by DNA sequences in the vicinity of the FRA16B AT island. The entire FRA16B locus is rich in other repetitive sequences that are also likely to be hypervariable and present in a range of sizes in both WI-38 and CEM cells. Nonetheless, it is clear that FRA16B fragments in CEM cells are markedly longer than those in WI-38 cells. Collectively, the consistent appearance of large molecular weight restriction products indicates that FRA16B repeats in CEM cells are profoundly expanded compared to their counterparts in normal WI-38 or NCM460 cells.
This interpretation is supported by the indications of long PCR products for CEM DNA using a primer system reported previously by Yu et al. (29). Even though this system is known to be inefficient in amplifying highly expanded FRA16B repeats, products of 10–30+ kb were evident when CEM DNA was used as a template (data not shown). These products are markedly larger than the signals of normal length (∼1.2–1.4 kb) generated with WI-38 or NCM460 DNA.
In vivo MAR function of FRA16B repeats in CEM cells, but not in WI-38 cells
To verify in vivo the MAR properties of the FRA16B AT island, we determined the MAR status of these domains in CEM and WI-38 cells. The digestion of nuclear DNA with a mix of restriction enzymes that cleave in the vicinity of, but not within, AT islands (Fig. 1), followed by the standard preparation of nuclear matrices allowed for the separation of the matrix-associated DNA from loop DNA. Under the conditions used, the matrix-associated DNA fraction typically comprised 2–5% of total DNA. The matrix-associated and loop DNA fractions were next analyzed using dot blot hybridizations for the distribution of AT islands and control sequences.
Hybridizations with the FRA16B probe revealed a highly non-uniform loop/matrix distribution. For CEM cells, FRA16B repeats were 7.1 ± 1.3-fold more abundant in matrix-associated DNA than in loop DNA (Fig. 5). In WI-38 DNA fractions, however, the FRA16B signal seemed relatively depleted in the matrix DNA, with matrix/loop ratio of 0.6 ± 0.3 indicating that FRA16B is largely in the loop DNA in that cell line. The c-myc MAR was also preferentially found in the matrix-associated DNA fraction of CEM cells (nearly 6-fold enrichment), consistent with the previously reported analogous findings in leukemic HL-60 cells (32,33). These results strongly suggest that FRA16B repeats have an in vivo MAR function in intact CEM cells but not in WI-38 cells.
Figure 5.

The partitioning of FRA16B repeats and other model sequences between matrix-associated and loop DNA fractions in cancer CEM and normal WI-38 cells. Matrix versus loop nuclear localization of specific DNA sequences in CEM and WI-38 DNA by dot blot hybridization analysis. Matrix and loop DNA fractions were prepared as described in Materials and Methods and analyzed by dot blot hybridization to the FRA16B oligonucleotide probe. The ratios of signal intensities in the matrix and loop DNA fractions shown are average values from 2 to 5 experiments using at least two different matrix/loop DNA preparations for each cell line.
In contrast, the signals for the loop versus matrix fractions vary only slightly when probed for FRA16D, Z79699, AT60 and β-globin (Fig. 5). In CEM DNA, the matrix/loop signal ratios ranged from 1.1 ± 0.03 to 1.8 ± 0.3 for Z79699 and β-globin, respectively. Hence, these sequences are more or less uniformly distributed between the loop and matrix-associated DNA. In addition, no significant differences were seen for these probes between the two cell lines tested.
Association of FRA16B AT island with the nuclear matrix in CEM cells via expanded sub-domain 2
Consistent with dot blot analysis, the FRA16B signal was barely detectable for the loop DNA (Fig. 6A). In contrast, a strong FRA16B signal was observed in matrix-associated DNA. Since the cleavage sites by the restriction enzymes used to prepare matrix-associated DNA narrowly frame the FRA16B AT island (Fig. 1B), these fragments should reflect well the size of FRA16B AT islands that are matrix bound. This FRA16B signal spans a 10–50+ kb range, clearly demonstrating that the expanded repeats are the ones that confer the in vivo MAR function.
Figure 6.

Size analysis and PCR amplification of FRA16B repeats in matrix-associated and loop DNA fractions from CEM cells. Matrix and loop DNA fractions were prepared as described in Materials and Methods. The positions of cleavage sites of BamHI, PvuII and EcoRI used in the preparation of matrix-associated and loop DNA relative to the FRA16B AT island are indicated in Figure 1A. (A) Size distribution of matrix-associated and loop DNA fractions from CEM cells (lanes 2 and 3, respectively, each at 3 × 104 cell equivalents). Total undigested CEM DNA is also shown (lanes 1, 1 × 104 cell equivalents). DNA samples were analyzed by FIGE agarose electrophoresis followed by Sybr green I staining (left) and Southern transfer and hybridization to the FRA16B oligonucleotide probe (right). (B) PCR amplification of matrix and loop fractions of CEM DNA (each at 3 × 103 cell equivalents) using FRA16B primers 29 and 38. The PCR conditions were identical to those denoted in Figure 3. PCRs were analyzed by FIGE agarose electrophoresis followed by Southern transfer and hybridization to the FRA16B oligonucleotide probe. Lanes M1 and M2 show size markers detected by Sybr green I.
The presence of long FRA16B repeats in matrix-associated DNA was corroborated by the PCR experiments using FRA16B primers. As mentioned previously, this PCR system is likely to overemphasize unexpanded repeats. Accordingly, both matrix and loop DNA fractions gave rise to short PCR products of ∼1–1.4 kb that appear to reflect the unexpanded FRA16B (Fig. 6B). Still, the very long PCR products ranging from 15 to 30 kb were clearly discernible with CEM matrix DNA. Such products were virtually absent from PCR reactions using CEM loop DNA. Since these PCR primers were designed for sub-domain 2, repeats in this sub-domain appear to be responsible for the expansion of the FRA16B AT island.
c-myc MAR AT island: differential MAR function in CEM but not in WI-38 cells without AT island expansion
The c-myc MAR domain can be either matrix-associated or localized in loop DNA in various cell lines (32–34). Unlike FRA16B, however, c-myc MAR is not known to be subject to size expansion. Thus, for comparison to FRA16B data, we simultaneously determined both the actual in vivo MAR status of the c-myc MAR and its domain size in CEM and WI-38 cells using quantitative PCR. A model non-AT island/non-MAR β-globin sequence was concurrently examined as a negative control.
The identical size of PCR products confirms that the c-myc MAR domain remains unchanged in both CEM and WI-38 cells (Fig. 7). The signals for c-myc MAR, however, were markedly enhanced relative to the β-globin signal, when the matrix CEM DNA was used as template, and was depleted with the loop DNA as template. Thus, quantitative PCR analysis confirms the in vivo MAR status of the c-myc MAR domains in CEM cells. The opposite distribution of c-myc MAR was displayed in normal WI-38 cells strongly suggesting that c-myc MAR is not associated with the nuclear matrix in normal WI-38 cells. The β-globin segment clearly remains in the loop DNA (no MAR function) in both cell lines. Given the identical sizes of the c-myc MAR PCR products, the acquired in vivo MAR function of this domain in CEM cells most probably reflects a mechanism other than repeat expansion.
Figure 7.

Quantitative PCR analysis of the distribution of the c-myc MAR and the β-globin non-AT island region between matrix-associated and loop DNA in CEM and WI-38 cells. Total, matrix and loop DNA fractions, prepared as described in Materials and Methods, were used as templates in duplex PCR reactions that amplified both regions. The PCR conditions were optimized for the linearity of the signal versus template amounts. The top panel shows examples of autoradiograms of agarose electrophoresis of the PCR products at a single level of DNA template. For the quantitation of the products, reactions were performed using three different levels (250, 500 and 1000 cell equivalents) of each template DNA. The bottom panel shows the average ratio of c-myc MAR/β-globin signals (mean ± SE) from two independent experiments.
DISCUSSION
Although long AT islands represent a distinct element of the human genome and a specific target for extremely potent anticancer drugs, their nature and function(s) remain elusive. The underlying organization of AT islands and their impact on cell function are likely to differ between cancer and normal cells, given that these domains include well known elements of genomic instability implicated in various cancers. One of the most expansive AT islands identified is located in the fragile site region FRA16B (Fig. 1). This report uses bioinformatics and biochemical approaches to demonstrate for the first time that FRA16B AT islands: (i) have the in silico attributes of strong MARs, (ii) bind specifically to isolated nuclear matrices (in vitro MAR properties) and (iii) can serve as strong in vivo MARs when present in tumor cells in abnormally expanded forms.
Our previous studies raised the possibility that the critical nature of long AT islands as drug targets might reflect their potential to function as MARs (1,5). Moreover, the diverse sensitivity of various cell lines to AT-island targeting drugs, such as bizelesin (5,30) suggested possible differences in the structure/organization of AT islands. AT islands in fragile sites, including the prominent FRA16B domain, were prime candidates to consider in terms of both MAR function and likely cell type variability.
Although human MARs do not share any specific consensus sequence, they reveal several common characteristics. The in silico analysis shows that the FRA16B AT island exhibits such fundamental MAR attributes as computed MAR potential, profound duplex destabilization and markedly increased duplex flexibility. In the magnitude of these attributes, FRA16B AT islands overwhelm their adjacent sequences (Fig. 1). The high MAR potential of AT islands in general has been proposed to reflect their propensity to form unwound partially base-unpaired structures, especially under conditions of superhelical stress (1,19). These properties depend strongly on the length of the destabilized repeats (1,19). Accordingly, the hypothetical expansion of FRA16B repeats further enhances their computed MAR potential. The experimental determinations corroborate the in silico predictions by demonstrating potent and specific interaction of FRA16B repeats with nuclear matrices isolated from CEM and WI-38 cells (in vitro MAR properties, Fig. 2). Of note is the fact that FRA16B repeats show greater ability to bind the nuclear matrices than a known MAR and a less prominent AT island in the c-myc gene.
The examination of bizelesin-hypersensitive CEM cells by dot blot hybridization indicates that AT islands in two fragile sites, FRA16B and FRA16D, are abnormally abundant when compared to non-hypersensitive cells, such as WI-38 (Fig. 3). The increased abundance is not a common trait of all AT islands. Other AT islands examined (in DDBJ/EMBL/GenBank Z79699 and in the c-myc gene) are present in similar levels in both CEM and WI-38 cells.
The observed overabundance of FRA16B repeats in CEM cells probably reflects a major expansion of this AT island well beyond the normal span of ∼3.5 kb (in a DDBJ/EMBL/GenBank consensus sequence). Various experimental approaches consistently suggest the expansion of FRA16B repeats by one order of magnitude, ranging from ∼5+-fold (restriction analysis, Fig. 4), through ∼7-fold (size distribution of matrix-associated FRA16B DNA, Fig. 6), to 11-fold and 12–20-fold (the abundance ratio and the size of FRA16B PCR products, Figs 3 and 6, respectively). The expansion of the FRA16B AT island seems to originate specifically from the telomeric sub-domain 2. Our results are thus in agreement with those of Yu et al., who have proposed the expansion of these 33 bp repeats in individuals expressing the FRA16B fragile site (29). Whereas CEM cells clearly show abnormally expanded FRA16B repeats, they seem also to retain the normal, unexpanded FRA16B allele, as suggested by the signals corresponding in size to those in the normal cell lines.
Consistent with the in silico predictions, the expansion of FRA16B repeats profoundly affects their ability to serve as MARs in the cellular settings. The expanded repeats (in CEM cells) are found preferentially in the matrix-associated DNA, whereas the unexpanded repeats (in WI-38 cells) localize mainly to the loop fraction of DNA (Figs 5 and 6). The expanded length of FRA16B repeats in CEM cells might possibly enhance their non-specific recovery in the matrix-associated fraction of CEM DNA. However, the employed high-salt method of preparing matrix-associated DNA is known to be stringent in removing non-MAR and weaker MAR sequences and leaving only the most tightly matrix-bound DNA segments (35). Moreover, longer length fragments should be more susceptible to mechanical shearing, which would decrease rather than increase their recovery. Therefore, the enhanced matrix association of expanded FRA16B AT islands most likely reflects the intrinsic matrix association of these sequences in the cell. Collectively, the data are consistent with the idea that the expansion of FRA16B repeats drives the reorganization of matrix attachment sites with the abnormally expanded FRA16B domains becoming strong, permanent in vivo matrix anchorage sites.
The unexpanded FRA16B repeats are still likely to serve as transient MARs. Weaker and/or transient interactions are expected to remain unrevealed by the stringent in vivo assay. The ∼1.1 kb FRA16B probe, which corresponds to the unexpanded repeats, is clearly able to bind to the isolated matrices from normal cells. Hence, the lack of strong association of the unexpanded FRA16B repeats with the nuclear matrix in intact WI-38 cells cannot be attributed to any peculiar properties of their nuclear matrices. These findings underscore the need to exercise caution and use both in vitro and in vivo determinations to define MAR properties of a specific region.
This notion is further substantiated by the results with the c-myc MAR region, which provides a useful reference for the differential organization of the FRA16B AT island. Even though the c-myc region is a weaker MAR in vitro than FRA16B repeats (Fig. 2) and is not a subject of expansion, the c-myc domain also becomes matrix associated in CEM cells, while localizing to loop DNA in WI-38 cells (Figs 6 and 7). Importantly, the reorganization on the nuclear matrix can reprogram the c-myc expression. The overexpression of c-myc is implicated in the proliferative capacity of CEM cells, in contrast to minimal c-myc expression in normal cells (36,37). In Namalva cells with two c-myc alleles, only the allele associated with the nuclear matrix was actively transcribed, while the other allele, located in the chromatin loops, was not (34). Given the vital roles of MARs, it is tempting to speculate that aberrant rearrangements of other MAR-capable AT islands, exemplified by the acquired in vivo MAR function demonstrated for the FRA16B locus, may also have a profound impact on cancer cell functions.
Changes in discrete sites of genomic instability, including AT-rich fragile sites, have been postulated to contribute to the tumorigenic phenotype rather than being merely accompanying events, although further efforts are needed to definitively substantiate this idea (9,11,38). Repeat expansion has been previously linked to the fragilities of the FRA16B locus and another AT-rich fragile site region, FRA10B (9,29,39). Our data enhance the previous findings by demonstrating the rearrangements of FRA16B fragile sites in cancer CEM cells. Moreover, the FRA16D fragile site locus, which possibly is also abnormal in CEM cells, is well implicated as a localized site of DNA instability in cancer cells (12,16–18). It is also worth noting that various genetic studies pinpointed enhanced DNA helix flexibility and nadirs of duplex stability as empirical markers for mammalian sites of genomic instability (16,40,41). Exactly the same attributes characterize prominent MARs and AT islands. The possible overlap between the sites of genomic instability and MARs has been suggested (1,9), but, to the best of our knowledge, such an overlap has never been previously documented. The evidence that abnormal FRA16B fragile sites can be strong MARs in CEM cells warrants further study in that direction.
Finally, the differences in organization of AT islands, such as those in FRA16B, may affect cell responses to AT-island-targeting drugs. Cytotoxicity of these drugs differs profoundly among various cell lines (30 and Woynarowski,J.M., Trevino,A.V. and Herzig,M.C., unpublished data). Several cancer cell lines are clearly hypersensitive with GI50 values in the low pM range (1.8–30 pM). Two normal and several other cancer cell lines were 1–2 orders of magnitude less sensitive. Simple explanations that such diverse cytotoxicities might arise from different formation or repair of drug adducts have been ruled out. While the nature of the hypersensitivity to AT-island-reactive drugs remains to be determined, our collective findings are consistent with a critical role of genome organization. This possibility is directly implied by a 15-fold hypersensitivity to bizelesin of Colo320HSR compared to its isogenic variant Colo320DM, despite a similar level of drug adducts. These two lines differ only in the organization of their massively amplified sequences (integrated into the genome and present in double minute chromosomes, respectively).
Abnormal organization of bizelesin targets—AT islands, as exemplified by the expanded FRA16B locus in CEM cells, could sensitize cells to this drug. An expanded AT island offers proportionately more potential drug binding sites than its unexpanded variant. Less than 10 drug adducts are sufficient for killing a CEM cell compared to ∼200 adducts needed for an equitoxic effect in WI-38 (30). Moreover, drug adducts could be more lethal if located in a region playing a more crucial role for the cell when associated with the matrix, compared to the same repeats in a non-MAR location. Consistent with the MAR roles in replication, bizelesin is a potent inhibitor of DNA synthesis causing a rapid onset of the potent S-phase block in CEM cells, while only a mild S-phase accumulation is observed at much longer times in WI-38 cells (30). Further studies are underway to investigate the possible connection between the differential organization of AT islands and the observed large differences in sensitivity to AT-island-targeting drugs.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by a Pilot Project grant from Children’s Cancer and Research Institute at the UTHSCSA.
REFERENCES
- 1.Woynarowski J.M., Trevino,A.V., Rodriguez,K.A., Hardies,S.C. and Benham,C.J. (2001) AT-rich islands in genomic DNA as a novel target for AT-specific DNA-reactive antitumor drugs. J. Biol. Chem., 276, 40555–40566. [DOI] [PubMed] [Google Scholar]
- 2.Woynarowski J.M. (2003) AT islands—their nature and potential for anticancer strategies. Curr. Cancer Drug Targets, in press. [DOI] [PubMed] [Google Scholar]
- 3.Woynarowski J.M., Napier,C., Trevino,A.V. and Arnett,B. (2000) Region-specific DNA damage by AT-specific DNA-reactive drugs is predicted by drug binding specificity. Biochemistry, 39, 9917–9927. [DOI] [PubMed] [Google Scholar]
- 4.Herzig M.C.S., Rodriguez,K.A., Trevino,A.V., Dziegielewski,J., Arnett,B., Hurley,L. and Woynarowski,J.M. (2002) The genome factor in region-specific DNA damage: The DNA-reactive drug U-78779 prefers mixed A/T-G/C sequences at the nucleotide level but is region-specific for long pure AT islands at the genomic level. Biochemistry, 41, 1545–1555. [DOI] [PubMed] [Google Scholar]
- 5.Woynarowski J.M. (2002) Targeting critical regions in genomic DNA with AT-specific anticancer drugs. Biochim. Biophys. Acta, 1587, 300–308. [DOI] [PubMed] [Google Scholar]
- 6.Woynarowski J.M. (2002) Preferential damage to defined regions of genomic DNA by AT-specific anticancer drugs. In Jones,G.B. and Chapman,E.J. (eds), Advances in DNA Sequence Specific Agents, Elsevier Press Inc., Vol. 4, pp. 1–27. [Google Scholar]
- 7.Debrauwere H., Gendrel,C.G., Lechat,S. and Dutreix,M. (1997) Differences and similarities between various tandem repeat sequences: Minisatellites and microsatellites. Biochimie, 79, 577–586. [DOI] [PubMed] [Google Scholar]
- 8.Bois P. and Jeffreys,A.J. (1999) Minisatellite instability and germline mutation. Cell. Mol. Life Sci., 55, 1636–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards R.I. (2001) Fragile and unstable chromosomes in cancer: causes and consequences. Trends Genet., 17, 339–345. [DOI] [PubMed] [Google Scholar]
- 10.Smith D.I., Huang,H. and Wang,L. (1998) Common fragile sites and cancer (Review). Int. J. Oncol., 12, 187–196. [PubMed] [Google Scholar]
- 11.Rockwell S., Yuan,J., Peretz,S. and Glazer,P.M. (2001) Genomic instability in cancer. Novartis Found. Symp., 240, 133–142; also 142–151. [DOI] [PubMed] [Google Scholar]
- 12.Driouch K., Prydz,H., Monese,R., Johansen,H., Lidereau,R. and Frengen,E. (2002) Alternative transcripts of the candidate tumor suppressor gene, WWOX, are expressed at high levels in human breast tumors. Oncogene, 21, 1832–1840. [DOI] [PubMed] [Google Scholar]
- 13.Horwitz M., Benson,K.F., Li,F.Q., Wolff,J., Leppert,M.F., Hobson,L., Mangelsdorf,M., Yu,S., Hewett,D., Richards,R.I. et al. (1997) Genetic heterogeneity in familial acute myelogenous leukemia: evidence for a second locus at chromosome 16q21–23.2. Am. J. Hum. Genet., 61, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Popescu N.C. (1994) Chromosome fragility and instability in human cancer. Crit. Rev. Oncog., 5, 121–140. [DOI] [PubMed] [Google Scholar]
- 15.Huebner K., Garrison,P.N., Barnes,L.D. and Croce,C.M. (1998) The role of the FHIT/FRA3B locus in cancer. Annu. Rev. Genet., 32, 7–31. [DOI] [PubMed] [Google Scholar]
- 16.Mangelsdorf M., Ried,K., Woollatt,E., Dayan,S., Eyre,H., Finnis,M., Hobson,L., Nancarrow,J., Venter,D., Baker,E. and Richards,R.I. (2000) Chromosomal fragile site FRA16D and DNA instability in cancer. Cancer Res., 60, 1683–1689. [PubMed] [Google Scholar]
- 17.Ried K., Finnis,M., Hobson,L., Mangelsdorf,M., Dayan,S., Nancarrow,J.K., Woollatt,E., Kremmidiotis,G., Gardner,A., Venter,D., Baker,E. and Richards,R.I. (2000) Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet., 9, 1651–1663. [DOI] [PubMed] [Google Scholar]
- 18.Paige A.J., Taylor,K.J., Stewart,A., Sgouros,J.G., Gabra,H., Sellar,G.C., Smyth,J.F., Porteous,D.J. and Watson,J.E. (2000) A 700-kb physical map of a region of 16q23.2 homozygously deleted in multiple cancers and spanning the common fragile site FRA16D. Cancer Res., 60, 1690–1697. [PubMed] [Google Scholar]
- 19.Benham C., Kohwi-Shigematsu,T. and Bode,J. (1997) Stress-induced duplex DNA destabilization in scaffold/matrix attachment regions. J. Mol. Biol., 274, 181–196. [DOI] [PubMed] [Google Scholar]
- 20.Bode J., Benham,C., Ernst,E., Knopp,A., Marschalek,R., Strick,R. and Strissel,P. (2000) Fatal connections: when DNA ends meet on the nuclear matrix. J. Cell. Biochem., 35 (Suppl), 3–22. [DOI] [PubMed] [Google Scholar]
- 21.Berezney R., Mortillaro,M.J., Ma,H., Wei,X. and Samarabandu,J. (1995) The nuclear matrix: a structural milieu for genomic function. Int. Rev. Cytol., 162A, 1–65. [DOI] [PubMed] [Google Scholar]
- 22.Chernov I.P., Akopov,S.B., Nikolaev,L.G. and Sverdlov,E.D. (2002) Identification and mapping of nuclear matrix-attachment regions in a one megabase locus of human chromosome 19q13.12: long-range correlation of S/MARs and gene positions. J. Cell. Biochem., 84, 590–600. [PubMed] [Google Scholar]
- 23.Kent W.J., Sugnet,C.W., Furey,T.S., Roskin,K.M., Pringle,T.H., Zahler,A.M. and Haussler,A.D. (2002) The Human Genome Browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kent W.J. (2002) BLAT—the BLAST-like alignment tool. Genome Res., 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kramer J.A., Singh,G.B. and Krawetz,S.A. (1996) Computer-assisted search for sites of nuclear matrix attachment. Genomics, 33, 305–308. [DOI] [PubMed] [Google Scholar]
- 26.Woynarowska B.A., Woynarowski,J.M., Herzig,M.C.S., Roberts,K., Higdon,A.L. and MacDonald,J.R. (2000) Differential cytotoxicity and induction of apoptosis in tumor and normal cells by hydroxymethylacylfulvene (HMAF). Biochem. Pharmacol., 59, 1217–1226. [DOI] [PubMed] [Google Scholar]
- 27.Herzig M.C., Trevino,A.V., Arnett,B. and Woynarowski,J.M. (1999) Tallimustine lesions in cellular DNA are AT sequence-specific but not region-specific. Biochemistry, 38, 14045–14055. [DOI] [PubMed] [Google Scholar]
- 28.Fernandes D.J., Smith-Nanni,C., Paff,M.T. and Neff,T.A. (1988) Effects of antileukemia agents on nuclear matrix-bound DNA replication in CCRF-CEM leukemia cells. Cancer Res., 48, 1850–1855. [PubMed] [Google Scholar]
- 29.Yu S., Mangelsdorf,M., Hewett,D., Hobson,L., Baker,E., Eyre,H.J., Lapsys,N., Le Paslier,D., Doggett,N.A., Sutherland,G.R. Richards,R.I. (1997) Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell, 88, 367–374. [DOI] [PubMed] [Google Scholar]
- 30.Jackson J.A., Trevino,A.V., Herzig,M.C. and Woynarowski,J.M. (2002) Differential cytotoxicity of bizelesin in model tumor and normal cells parallels differences in abundance and nuclear organization of AT-rich fragile sites. Proc. Am. Assoc. Cancer Res., 43, 1092. [Google Scholar]
- 31.Park J., Betel,D., Gryfe,R., Michalickova,K., Di Nicola,N., Gallinger,S., Hogue,C.W. and Redston,M. (2002) Mutation profiling of mismatch repair-deficient colorectal cncers using an in silico genome scan to identify coding microsatellites. Cancer Res., 62, 1284–1288. [PubMed] [Google Scholar]
- 32.Chou R.H., Churchill,J.R., Flubacher,M.M., Mapstone,D.E. and Jones,J. (1990) Identification of a nuclear matrix-associated region of the c-myc protooncogene and its recognition by a nuclear protein in the human leukemia HL-60 cell line. Cancer Res., 50, 3199–3206. [PubMed] [Google Scholar]
- 33.Chou R.H., Churchill,J.R., Mapstone,D.E. and Flubacher,M.M. (1991) Sequence-specific binding of a c-myc nuclear-matrix-associated region shows increased nuclear matrix retention after leukemic cell (HL-60) differentiation. Am. J. Anat., 191, 312–320. [DOI] [PubMed] [Google Scholar]
- 34.Djondjurov L.P., Andreeva,M.M., Markova,D.Z. and Donev,R.M. (1994) Spatial and structural segregation of the transcribed and nontranscribed alleles of c-myc in Namalva-S cells. Oncol. Res., 6, 347–356. [PubMed] [Google Scholar]
- 35.Donev R.M. (2000) The type of DNA attachment sites recovered from nuclear matrix depends on isolation procedure used. Mol. Cell. Biochem., 214, 103–110. [DOI] [PubMed] [Google Scholar]
- 36.Goldman J. and McGuire,W.A. (1992) H-ras and c-myc RNA expression in human T-cell ALL and in normal human lymphocytes. Pediatr. Hematol. Oncol., 9, 309–316. [DOI] [PubMed] [Google Scholar]
- 37.McGuffie E.M., Pacheco,D., Carbone,G.M. and Catapano,C.V. (2000) Antigene and antiproliferative effects of a c-myc-targeting phosphorothioate triple helix-forming oligonucleotide in human leukemia cells. Cancer Res., 60, 3790–3799. [PubMed] [Google Scholar]
- 38.Handt O., Sutherland,G.R. and Richards,R.I. (2000) Fragile sites and minisatellite repeat instability. Mol. Genet. Metab., 70, 99–105. [DOI] [PubMed] [Google Scholar]
- 39.Hewett D.R., Handt,O., Hobson,L., Mangelsdorf,M., Eyre,H.J., Baker,E., Sutherland,G.R., Schuffenhauer,S., Mao,J.I. and Richards,R.I. (1998) FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol. Cell, 1, 773–781. [DOI] [PubMed] [Google Scholar]
- 40.Toledo F., Coquelle,A., Svetlova,E. and Debatisse,M. (2000) Enhanced flexibility and aphidicolin-induced DNA breaks near mammalian replication origins: implications for replicon mapping and chromosome fragility. Nucleic Acids Res., 28, 4805–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svetlova E.Y., Razin,S.V. and Debatisse,M. (2001) Mammalian recombination hot spot in a DNA loop anchorage region: a model for the study of common fragile sites. J. Cell. Biochem., 36 (Suppl), 170–178. [DOI] [PubMed] [Google Scholar]