

Abstract
Recent studies have highlighted characteristics of T regulatory cells (Tregs) that underlie their suppressive function. However, mechanisms that override their suppressive function in the context of an adaptive immune response are not well understood. In the lungs of mice undergoing allergic inflammation appreciable numbers of Tregs were identified that possessed suppressive function when assayed ex vivo. We investigated whether the Th2-promoting cytokine interleukin-4 (IL-4) played a permissive role that superseded Treg function thereby allowing the development of allergic inflammation. IL-4 signaling via the IL-4Rα-STAT6 axis was required to maintain Foxp3 expression in Tregs and promote their proliferation. However, the results of both in vivo experiments involving adoptive transfer of Tregs into antigen-sensitized versus naïve animals, and in vitro suppression assays performed with or without exogenous IL-4 showed the ability of IL-4 to compromise Treg-mediated suppression. Use of retrovirally expressed constitutively active STAT6 revealed that the underlying mechanism was not IL-4-mediated dysfunction of Tregs but involved resistance of Th cells to Treg-mediated suppression that would permit the development of an adaptive immune response. Our data suggest that infectious tolerance, mediated by membrane-bound TGF-β expressed by Tregs, is compromised by the competing effects of IL4-induced signaling in naïve CD4+ T helper cells.
Keywords: Tregs, T helper cells, IL-4, STAT6, inflammation, lung
Introduction
Asthma is a common disease that affects over 15 million people in the United States and is characterized by mucus hypersecretion, elevated serum IgE, eosinophilic infiltration in the airways, and airway hyperresponsiveness (AHR) (1). The success of immunomodulatory treatments such as steroids and in some instances, antigen-mediated therapy, strongly suggests an immunological contribution to the allergic form of disease (2). The immune responses seen in experimental allergic airway inflammation, as well as in atopic human asthmatics are characterized by a predominance of Th2 cytokines such as IL-4, IL-5, and IL-13. With regard to effects on T cells, IL-4 plays a key role as has been demonstrated in a number of studies. The IL-4R is composed of the IL-4Rα chain and the common gamma chain (γc), which is shared with receptors for IL-2, IL-7, IL-9, IL-15, and IL-21 (3, 4). Signal transducer and activator of transcription 6 (STAT6) mediates many of the functions of IL-4 downstream of the IL-4R. Importantly, mice that lack IL-4Rα or STAT6 are highly resistant to experimentally induced allergic airway inflammation, and show significant defects in Th2 differentiation (5–8). These observations are further supported by the association of altered rates of disease in humans with mutations in their IL-4Rα subunit (9, 10). Specifically, gain of function mutations in IL-4Rα lead to increased signaling which correlates with atopy (10). Conversely, alleles that cause diminished signaling via IL-4Rα lead to reduced serum IgE levels (9). Therefore, the role of IL-4 in Th2 cell differentiation and function, and its importance in airway inflammation are firmly established.
Since allergic asthma in humans essentially represents failure of immune tolerance given that the inciting antigens are ubiquitous, it is also important to consider effects of regulatory T cells (Tregs) on the pathological process. Although a number of variants have been described, one class of Tregs expresses CD4, CD25 (IL-2Rα, part of the high affinity IL-2R), and Foxp3, and is able to inhibit T helper (Th) cell proliferation, which may involve a TGFβ-dependent mechanism (11–13). CD4+CD25+Foxp3+ Tregs found in the peripheral circulation are thymically-derived, are commonly called “naturally occurring” Tregs, and typically require activation by cytokines such as IL-2 in order to show maximal inhibitory function in vitro, a cytokine also shown to promote Treg proliferation and survival (14–19). Activation and/or generation of Tregs in vivo can also occur as our laboratory has demonstrated, by repeated exposure of mice to aerosolized ovalbumin (OVA) in the absence of adjuvant (11). CD25-expressing cells isolated from the spleens of tolerized animals when adoptively transferred can provide protection against experimentally induced airway inflammation in recipient animals (11). However, the mechanisms by which Tregs fail to control asthma remain to be fully elucidated. In the present investigation, we examined the role of IL-4, and demonstrate that although this cytokine has stimulatory effects on Tregs, its effects on Th cells predominate in an IL-4-rich environment such as allergic airway inflammation via a STAT6-mediated pathway.
Materials And Methods
Mice
All mice were obtained from The Jackson Laboratory and were maintained in the Department of Laboratory Resources (DLAR) at the University of Pittsburgh. Male animals were used at 6–8 weeks of age. Maintenance and use of laboratory animals were approved by the Institutional Animal Care and Use Committee (IACUC).
Flow cytometric analysis
Single-cell suspensions obtained from lung tissue were used for flow cytometric analysis. Frequencies of effector T cells (CD4+CD25+Foxp3−) and regulatory T cells (CD4+CD25+Foxp3+) were examined with three-color flow cytometry using αCD4 (Pharmingen), αCD25 (Pharmingen) and αFoxp3 (eBioscience) labeled with peridinin chlorphyll protein (PerCP), phycoerythrin (PE), and fluorescein isothiocyanate (FITC) respectively. Before staining, cells were blocked for 15 min on ice with Fc-block (10% normal mouse serum). Cell populations (2 ×105 events) were assessed using a FACS calibur flow cytometer with CellQuest software (BD PharMingen) and analysis was performed using FlowJo (TreeStar, San Carlos, CA). The frequency of positive cells was calculated by subtracting the value obtained with the respective isotype controls.
Cell isolation and stimulation
Mice were sacrificed and lungs were removed and minced. Minced lungs were incubated in RPMI medium (Gibco) supplemented with 10% fetal bovine serum (Gemini Bioproducts), DNAse I (30 μg/ml of type IV bovine pancreatic DNase I; Roche), and collagenase I (0.7 mg/ml; Sigma) for 45 min at 37°C in a shaking water bath. Single cell suspensions of lungs were obtained by passing the digested lung tissue through a 70 μm nylon strainer (BD Falcon). Mononuclear cells were enriched by performing discontinuous Percoll (Sigma) gradient centrifugation (20 to 55%). Cells collected on the 55% and 45% layers were passed through a 35 μm nylon strainer and counted by trypan blue exclusion. In the case of Treg isolation from Foxp3EGFP knock-in mice, after passing cells through a 70 μm nylon strainer, CD4+ T cells were isolated from the suspension using anti-CD4 microbeads (Miltenyi Biotec). GFP+CD4+ Tregs were isolated from the positively selected CD4+ cell population by cell sorting. In some experiments, splenic CD4+CD25+ Tregs were isolated by enrichment of mononuclear cells through density gradient followed by cell sorting. Tregs were suspended in PBS and adoptively transferred (2×105 cells per recipient) to animals.
Tregs (CD4+CD25+), Th cells (CD4+CD25−), and antigen-presenting cells (APCs) were isolated from the spleens of 6–8 wk old male Balb/cByJ mice. Spleens were minced, filtered through 70 μM nylon screens, and mononuclear cells were enriched by density gradient centrifugation (LSM, MP Biomedicals). Tregs and Th cells, were isolated using a regulatory T cell magnetic bead isolation kit (Miltenyi Biotec). CD4+ T cells were obtained as a byproduct of the Treg isolation, and CD4−Cd− T cell-depleted were used as APCs. Unless indicated, all cells were cultured in complete medium: Click’s medium supplemented with 10% FBS (Gemini), gentamicin (50 mg/L, Gemini), L-glutamine (2 mM, Gibco), and 2-mercaptoethanol (55 μM, Gibco). T cells were activated with agonistic soluble anti-CD3ε monoclonal antibody (2 μg/ml; BD Biosciences) and gamma irradiated (2000 rad) APCs as a source of costimulation, in most experiments. Some experiments included addition of exogenous IL-2 (50 U/ml) or IL-4 (20 ng/ml) as indicated.
Adoptive transfer of Tregs during experimentally-induced allergic airway inflammation
A model for the inhibition of experimentally-induced airway inflammation was described in detail previously (11). Briefly, mice received i.p. injections of Tregs from OVA tolerized mice followed by an i.p. injection of OVA/alum to prime the animals 15 minutes later (11, 20). After 6 d, animals received a booster i.p. injection of OVA/alum. Mice were rested for 1 wk, then subjected to challenge with 1% aerosolized OVA (20 min/day) for 7 consecutive days and analyzed 20–24 h later. In a variation of this approach, recipient animals received Tregs from tolerized mice after 2 i.p. immunizations with OVA/alum before the first day of the 7 d challenge with 1% aerosolized OVA. Control mice received sham injections of PBS and were subjected to the full inflammation model involving 2 i.p. injections of OVA/alum followed by challenge with aerosolized OVA. A minimum of 4 mice was included in each group in each experiment.
Bronchoalveolar lavage (BAL)
At 20–24 h after the last aerosol challenge in the models described above, BAL was conducted on the animals, and cell differentials were completed, as previously described (11).
[3H]-thymidine incorporation assay
Cells were cultured in round bottom, 96 well plates with soluble anti-CD3ε (2 μg/ml) and 3×104 gamma-irradiated APCs per well (if used in an assay, Th cells were added at 1 Th cell: 1 APC). Freshly isolated Tregs were added at various to Th cells as indicated in the figures with Th cell numbers maintained constant. Cells were cultured for 48 h, and then pulsed with [3H]-thymidine (1 μCi/well, Perkin Elmer) and incubated for an additional 18 h. Incorporation of [3H]-thymidine was measured by harvesting cell debris and DNA onto glass fiber filters, followed by liquid scintillation counting (Wallac). All conditions were performed in triplicate, and data is presented as mean counts per min (cpm) plus or minus the standard deviation (SD).
CFSE dilution assay
Carboxyfluoroscein succinimidyl ester (1 μM, CFSE, Invitrogen) was used as described previously (21). The percentage of divided cells was determined by dividing the number of Th cells undergoing at least one cell division by the total number of live CFSE-labeled cells and multiplying the result by 100.
Western blotting
Nondenaturing cell lysis buffer with 1% Triton (Cell Signaling) was used to make total cell extracts, and Western blotting techniques were used to analyze equal amounts of protein, as described previously (22). Membranes were probed with anti-STAT6 monoclonal antibody (Cell Signaling) at a 1:1000 dilution. Expression of β-actin (Novus) was used to confirm equal protein loading.
Retroviral infection of Tregs and Th cells
A retroviral construct encoding constitutively active STAT6 (caSTAT6) was used (23). The retroviral construct containing caSTAT6 cDNA and the empty vector were purified, and a packaging cell line (Phoenix cells) was infected to generate retrovirus. Infection of Tregs and Th cells was performed as previously described (21).
Statistics
Results shown are mean values ± S.D. Wilcoxon matched pairs test, Mann Whitney test or student’s t test were used to assess statistical significance and differences were considered significant where p≤0.05.
Results
Similar numbers of Tregs in the lungs of tolerized mice and those with airway inflammation
Although T regulatory cells (Tregs) can act as potent inhibitors of the immune system, the failure to maintain immunological tolerance to essentially ubiquitous antigens in patients with autoimmune and allergic diseases suggests that Tregs may be ineffective in these situations. We used a murine model of allergic airway inflammation utilizing antigen sensitization followed by challenge. The model of tolerance involves repeated exposure to aerosolized OVA followed by testing mice for tolerance by subjecting the mice to the inflammation-inducing regimen as previously described by us (11). In each case, we examined the relative numbers of CD4+CD25+Foxp3− (the majority of these cells most likely being T helper (Th) cells) versus CD4+CD25+Foxp3+ (the majority of these cells most likely being Tregs) in the lung. We have noted, however, that Foxp3 expression does not always endow suppressive functions (24) just as Foxp3− cells may include other types of Tregs such as Th3 and Tr1 cells (25, 26). As expected, in naïve as well as in tolerized animals, the number of Foxp3− cells was lower than that of Foxp3+ cells (Fig. 1). In mice immunized for inflammation, the relative number of Foxp3− (hereafter called Th cells) cells was higher than that in naïve or tolerized mice but, surprisingly, the number of Foxp3+ CD4+ T cells (termed Tregs hereafter) was similar to that observed in tolerized mice (Fig. 1). It should be noted that in each case, a Wilcoxon matched pair test was conducted to assess statistical significance between Tregs and Th cells in the same mice, which is why the difference is statistically significant even in inflammation. However, the difference was not statistically significant between Treg numbers under conditions of tolerance and inflammation (Mann and Whitney U test, p>0.38). Similar parallel expansion of Th cells and Tregs in areas of inflammation has been described in multiple studies (27–33). The increase in Th cells in inflammation despite seemingly similar numbers of Tregs prompted us to address first the functional competence of Tregs when isolated from the lungs of mice with allergic airway inflammation.
FIGURE 1.
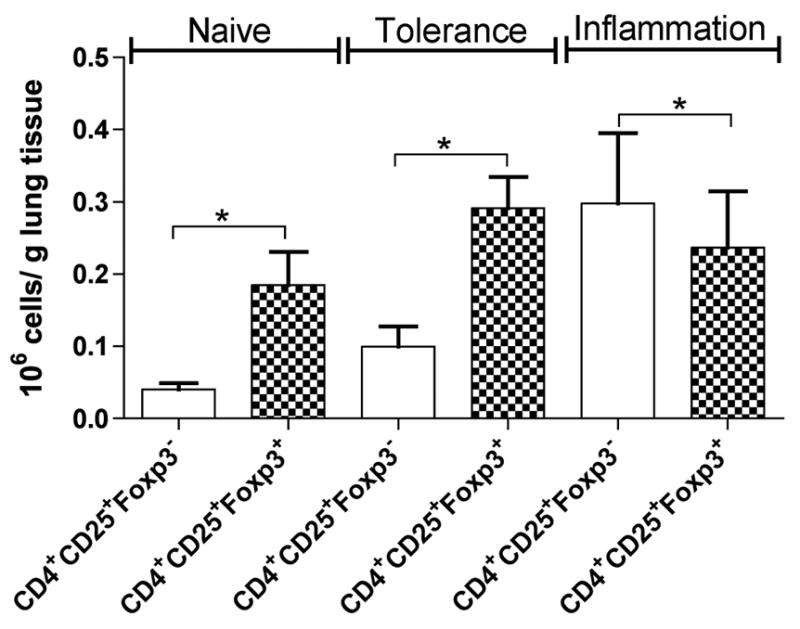
Treg numbers are similar under conditions of tolerance and inflammation. Mice were tolerized by exposure to aerosolized OVA on 10 consecutive days and were then subjected to the inflammation model to test for tolerance by 2 intraperitoneal (i.p.) administrations of OVA/alum followed by OVA aerosol challenge for 7 d. Lung T cells were isolated, stained for CD4, CD25, and Foxp3, and analyzed by flow cytometry. Numbers of CD4+CD25+Foxp3− and Tregs (CD4+CD25+Foxp3+) in lungs of naive mice, mice tolerized to OVA, and mice with OVA-induced allergic airway inflammation were determined as shown (n=6 mice per group). *: p<0.016, 0.016 and 0.031 in naïve, tolerized animals and mice with inflammation as tested with a Wilcoxon matched pairs test. Treg (CD4+CD25+Foxp3+) cell numbers in tolerance versus inflammation revealed a p value of < 0.38 as determined by Mann Whitney test. Results shown are representative of 2 independent experiments.
Tregs isolated from the lungs of mice with allergic airway inflammation retain their immunosuppressive functions
Given that inflammation was induced in mouse lungs despite presence of adequate numbers of Tregs, we wondered whether these Tregs possessed suppressive function. Towards this end, we used Foxp3EGFP-knockin mice to be able to identify Foxp3+ CD4+ T cells in the lungs under conditions of inflammation. EGFP+Foxp3+ Tregs were isolated by cell sorting from the lungs of mice with airway inflammation and splenic CD4+ Th cells from naïve mice were used as target Th cells to assess the suppressive function of the Tregs. When assayed in the absence of any exogenous IL-4, the EGFP+ T cells isolated from the lungs were suppressive in a dose-dependent fashion (Fig. 2). The suppressive function was, however, compromised when IL-4 was added to the coculture. The result of this experiment showed the lung-derived Tregs were not dysfunctional per se since they were able to exercise suppressive function in the absence of IL-4. However, when present in an IL-4-rich environment, as would happen under conditions of allergic inflammation, their suppressive function could not be realized. This phenomenon of coexistence of large numbers of Tregs with Th cells in areas of inflammation has been noted in other studies as well (27–33). Thus the logical next question was why Tregs were rendered ineffective under conditions of inflammation.
FIGURE 2.
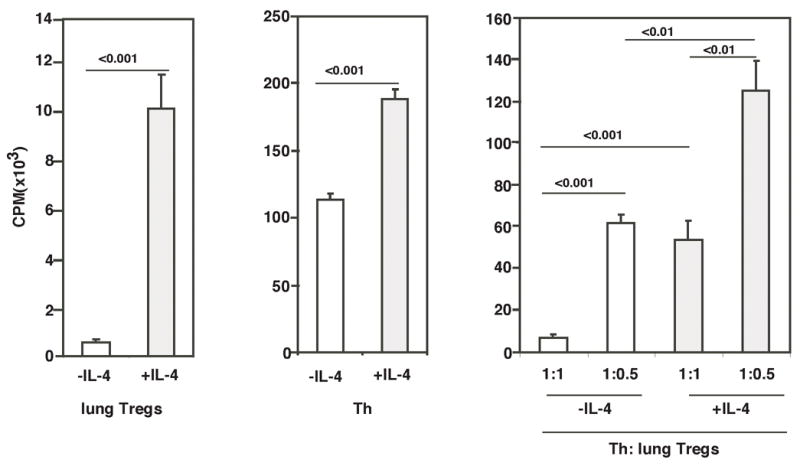
Tregs isolated from lungs of mice undergoing allergic airway inflammation possess suppressive function in the absence of IL-4. Tregs isolated from lung tissue of Foxp3EGFP knock-in mice that were subjected to the inflammation model were cultured with or without IL-4 to assess how Tregs isolated from inflamed tissue responded to IL-4 ex vivo. In suppression assays using these cells, 50×103 or 25×103 Tregs with 50×103 T helper cells (1:1 or 1:0.5 Th/Treg ratio respectively) were stimulated with APC (50×103) and soluble αCD3e (2μg/ml) for 96 h in a 96-well flat-bottom plate with or without IL-4 (20ng/ml). 1 μCi of [3H]-thymidine was added to each well for the final 18 h of incubation. Data shown are means of triplicates ± SD and are representative of 2 independent experiments.
Tregs are able to inhibit the sensitization phase of airway inflammation regardless of their ability to respond to IL-4, but require IL-4 receptor signaling to suppress responses to antigen challenge
Given that the data shown in Fig. 2 showed that IL-4 prevented Tregs from exerting suppressive effects on Th cells, we asked how IL-4 provided a permissive role during the adaptive immune response to an allergen. Our focus on IL-4 and its influence on Th cells versus Tregs in the context of allergic inflammation was stimulated by the recent finding with IL-2 which showed that the crucial role that IL-2 provides in the peripheral maintenance of Tregs under homeostatic conditions stems from the selective expression of IL-2 in the niche in which Tregs reside (34, 35). Thus, by the same token, if Tregs and inflammation can co-exist, one or more cytokines such as IL-4 that are present under conditions of inflammation may contribute to both induction of inflammation as well as proliferation and maintenance of Tregs in inflammation. To address each of these issues individually, we first investigated the function of Tregs when adoptively transferred into mice before the induction of airway inflammation versus transfer when inflammation was already underway. In these experiments, it was not practical to use Tregs from the EGFP-knockin mice because of the sheer numbers required in adoptive transfer experiments. Our laboratory has previously shown that Th2-mediated airway inflammation can be prevented in a murine model of allergic airways disease by the adoptive transfer of CD25+Foxp3+ T cells isolated from the spleens of tolerized animals when the recipients receive the Tregs prior to the beginning of the inflammation protocol (11). In order to determine whether IL-4 influences the in vivo development and function of Tregs, we tolerized WT and IL-4Rα−/− mice by repeated exposure to aerosolized OVA, isolated CD25+ cells from spleens, and transferred these cells into WT recipients. We have previously shown that CD25+ cells from WT animals isolated and transferred in this fashion strongly inhibit induction of experimental airway inflammation in the recipients even though the population may contain some activated effector Th cells which also express CD25 (as with the EGFP+ cells isolated for the experiment shown in Fig. 2) (11). Immediately following the adoptive transfer of CD25+ cells, the recipient animals were subjected to the model of allergic inflammation and were analyzed for inflammatory cell recruitment in the airways of the mice. As shown in Fig. 3A, Tregs from both WT and IL-4Rα−/− mice significantly inhibited airway inflammation. Consequently, IL-4 signaling did not appear to be necessary in order for Tregs to be able to block sensitization to OVA.
FIGURE 3.
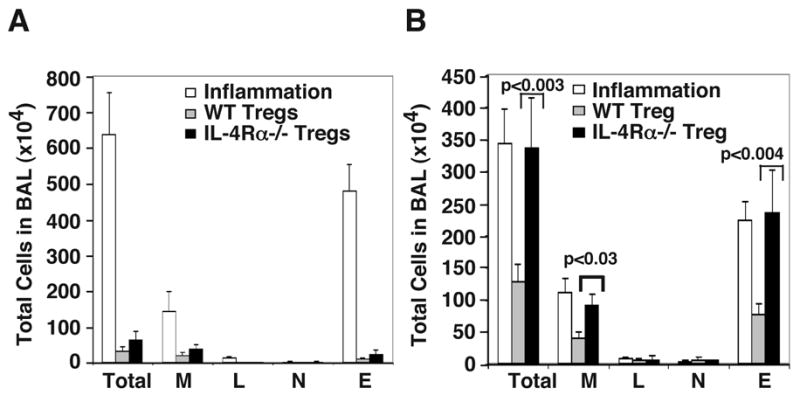
Differential effects of Tregs from WT and IL-4Rα−/− mice on allergic airway inflammation when adoptively transferred before or after antigen-sensitization. WT or IL-4Rα−/− mice were tolerized, and 2×105 Tregs isolated from these mice were transferred i.p. to each WT recipient animal either A, before antigen sensitization or B, after 2 i.p. immunizations with OVA/alum and before antigen challenge. Control mice (shown as inflammation-unfilled bars) received sham injections of PBS and were immunized with OVA/alum. Following 7 d of OVA aerosol challenge of all animals, animals were subjected to BAL, and cell differentials were enumerated. Inflammation: sham injected WT recipients; WT Tregs: WT recipients of WT Tregs; IL-4Rα−/− Tregs: WT recipients of IL-4Rα−/− Tregs. Abbreviations: Total- total leukocyte number; M- macrophages; L- lymphocytes; N- neutrophils; E- eosinophils. Results shown are mean ± SD (n=4 per group) and are representative of 2 independent experiments.
We next investigated whether the same cells could inhibit a recall response to antigen in previously sensitized animals. Recipient animals intraperitoneally received priming and boosting injections of OVA/alum, and then prior to the first OVA aerosol challenge, CD25+ Tregs from tolerized animals were adoptively transferred. Unlike the situation where Tregs drastically reduced inflammation when transferred before sensitization (Fig. 3A), transfer of WT CD25+ cells post-sensitization and prior to antigen challenge reduced the number of eosinophils by about 50%, whereas Tregs isolated from tolerized IL-4Rα−/− mice had no discernible inhibitory effect on eosinophil numbers as assessed in the BAL fluid (Fig. 3B). Therefore, the ability of Tregs to respond to IL-4 appears to be important for inhibition of responses to antigen challenge in previously sensitized animals (Fig. 3B) although it is irrelevant for their ability to block antigen sensitization (Fig. 3A). It is important to note that in previously sensitized animals, a robust Th2 response including the production of IL-4 would have been already underway, which was not the case prior to antigen sensitization. These results led us to investigate whether IL-4 inhibited Treg function directly or made Th cells more resistant to the inhibitory effects of Tregs.
In vitro, exogenous IL-4 maintains Foxp3 expression and promotes Treg proliferation but inhibits Treg-mediated suppression of Th cells
Since in data shown in Fig. 3, introduction of Tregs to in the lungs after allergen sensitization decreased the ability of the Tregs to suppress inflammation and further lack of IL-4Rα signaling totally compromised the ability of the Tregs to suppress inflammation, we first determined the effect of IL-4 on Foxp3 expression in Tregs which is required for the suppressive function of Foxp3-expressing Tregs. Approximately 90% of freshly isolated spleen CD25+ cells from naïve mice express Foxp3 (data not shown), but after 3 d in culture, the percentage decreases substantially to a frequency of 37% (Fig. 4A), in agreement with results reported previously (36). In the absence of IL-4, the overall viability of cells was 30.5 % after 3d in culture. The Foxp3- cells shown in the illustration were all viable based on their FSC/SSC properties. Propidium iodide staining could not be performed to identify live cells as Foxp3 staining necessitated cell permeabilization. Addition of IL-4 allowed for maintenance of Foxp3 expression and also increased viability of WT cells from 30.5 to 61.2% and of STAT6-deficient Tregs from 36.1 to 49.8% but, as expected, did not improve the viability of IL-4Rα-deficient Tregs (data not shown). IL-4-mediated increase in Foxp3+ cells was not observed with IL-4Rα- or STAT6-deficient cells, showing that STAT6-dependent IL-4 signaling is required in the maintenance of Foxp3 expression in naturally occurring Tregs (Fig. 4A). However, this observation is different from the negative effect of STAT6 on de novo Foxp3 expression in CD4+ T cells (37). Similar to the maintenance of Foxp3 expression in the presence of IL-4, the frequency of CD25hi cells was also higher when IL-4 was included in the culture and was partially maintained in STAT6−/− cells (Fig. 4A). It should be noted that in the presence of IL-4, the frequency of CD25hi population was 86.3% but that of Foxp3+ cells was slightly less (74%), which suggests that the small population of CD25hi cells that was not Foxp3+ represented Th cells that were co-purified with Tregs. The maintenance of Foxp3 expression in Tregs by STAT6 signaling seems to be in contrast to the negative effect of STAT6 on de novo induction of Foxp3 in naïve CD4+ T cells (37).
FIGURE 4.

Exogenous IL-4 maintains Foxp3 expression and enhances Treg proliferation but reduces Treg-mediated suppression. A, Tregs and Th cells were isolated from naïve WT, STAT6−/−, and IL-4Rα−/− mice and used as shown. CD4-depleted WT splenocytes were irradiated (2000 rads), and used as APCs (3×104 per well) in conjunction with soluble anti-CD3 (2 μg/ml) to stimulate (A and B) Tregs alone or (C) Th cell/Treg cells in co-culture. In A, Tregs were cultured for 3 d and Foxp3 or CD25 expression was analyzed by flow cytometry. Without IL-4, the frequency of viable cells was 30.5%, 36.1% and 37.3% of total cells for Tregs isolated from WT, STAT6−/− and IL-4Rα−/− mice respectively after 3 d in culture. In the presence of IL-4, the corresponding live cell frequencies were 61.2%, 49.8% and 38.8%. Gating for live cells was based on forward and side scatter, which in routine assays match data obtained with PI/Annexin staining. B and C, cells were stimulated in the presence or absence of IL-4 (20 U/ml), as indicated. B, Tregs were cultured for 2 d, pulsed with [3H]-thymidine for 18 hr, and analyzed by liquid scintillation counting (Treg:APC was 1:1). Data shown are mean±S.D. C, Th cells were labeled with CFSE and cultured with Tregs in the indicated ratios for 3 d. Cells were stained and analyzed by flow cytometry. The Y-axis shows the percentage of Th cells that divided 1 or more times. Numbers in dot plots are the percentage of cells in the quadrant. D, Th cells and Tregs were isolated from WT, STAT6−/−, or IL-4Rα−/− mice. Th cells labeled with CFSE and were cocultued in all possible combinations as indicated for a CFSE dilution assay. Assays were conducted in the presence or absence of exogenous IL-4 (20 ng/ml). Data shown are mean±S.D. and represent average of data from 2 independent experiments.
We noted a population of low CD4 expressors within the Foxp3− or CD25− population. The lower CD4 expressors were not dying cells based on their scatter properties. Without added cytokine, the frequency of the low CD4 expressors in WT Tregs was 45.5 %. However, it was reduced to 14% in the presence of IL-4 (or IL-2-not shown). It is possible that IL-4 and IL-2 upregulate expression of the coreceptor CD4 on Tregs to promote TCR signaling. To distinguish between anti-apoptotic effects of IL-2 on the cells versus effects on proliferation or other parameters, we investigated the effect of a combination of IL-4 and IL-2, which gave essentially similar results obtained with either cytokine alone (data not shown).
To assess the effect of IL-4 on Treg proliferation, CD25+ cells were isolated from the spleens of naïve mice and were cultured with IL-4, APCs and anti-CD3ε in vitro. IL-4 increased Treg proliferation by approximately 23 fold compared to that of untreated cells, and this effect was absent when cells were either from IL-4Rα−/− or STAT6−/− animals (Fig. 4B). In support of our expectations based on the in vivo data, these results suggested that IL-4 might actually enhance Treg-mediated suppression of airway inflammation since both Treg proliferation and Foxp3 expression are associated with enhancement of suppressive function (21).
To address the impact of IL-4 on Treg function, we labeled Th cells with CFSE, which allowed us to exclusively follow Th proliferation in the Treg/Th mix. Th cells were labeled with CFSE and all possible combinations of Tregs and Th cells from WT, STAT6−/− and IL-4Rα−/− mice were included to directly assess the influence of IL-4-signaling in Tregs versus Th cells in Treg-mediated suppression. In data presented in Fig. 4C, each set of three bars represents the suppressive function of all three types of Tregs on a single type of Th cell, which is indicated on the X-axis. Similar heights of bars within each triplicate indicated that the type of Treg had little influence on suppression irrespective of the presence or absence of IL-4. On the other hand, the height of the first triplicate set was greater than the other two, and was increased by the presence of IL-4, demonstrating that IL-4 decreased suppression by Tregs (38, 39) which was dependent on functional IL-4Rα and STAT6 in Th cells. It was interesting to note that in the presence of IL-4, IL-4Rα-deficient Tregs were less efficient than STAT6-deficient Tregs in suppressing Th cell proliferation (Fig. 4D). This was probably because of the lack of the second pathway, IRS-2/Akt, downstream of IL-4Rα in IL-4Rα-deficient Tregs that has been associated with T cell proliferation and survival (40–42). Tregs unable to trigger this pathway in response to IL-4 would have both survival and proliferation compromised in addition to Foxp3 expression (Fig. 4A) and can be expected to be less efficient than STAT6-deficient Tregs with this pathway intact in exercising suppressive effects as we observed. Collectively, our data indicated that while IL-4-induced STAT6 activation promotes proliferation of both Tregs and Th cells, and increases Th cell resistance to suppression, it did not appreciably influence the suppressive function of Tregs when cells were able to respond to IL-4. Rendering Tregs completely unresponsive to IL-4 made Th cells less sensitive to Treg-mediated suppression probably due to the presence of fewer Tregs in culture. Thus, the previous in vitro data of Pace et al on the ability of IL-4 to reduce Treg suppressive function was also evident in these experiments. However, we performed additional experiments to determine whether this impairment of suppressive function of Tregs in the presence of IL-4 was due to effects on both Tregs and on Th cells. This question was particularly relevant since the in vitro data presented in Fig. 4C showed that IL-4 signaling in Tregs was not what all that mattered in the ability of IL-4 to dampen Treg function.
Constitutive STAT6 signaling in Tregs Does Not Impair Treg Function
While STAT6 in Th cells appeared to be necessary for IL-4-mediated resistance to Tregs, it was unclear whether STAT6 activation alone in Th cells would promote their resistance to Tregs. To address this question, cells were infected with a retrovirus encoding a constitutively active form of STAT6 (STAT6VT referred to here as caSTAT6) (43). The bicistronic retroviral construct also contained the gene for GFP, which allowed both monitoring of infection efficiency by flow cytometry as well as isolation of caSTAT6-expressing cells by cell sorting. The functionality of the caSTAT6 protein was evaluated by infection of Tregs from STAT6−/− mice and observation of proliferation. Cells expressing caSTAT6 proliferated significantly better than cells infected with the same retrovirus lacking the caSTAT6 gene (Fig. 5A). This was in agreement with data presented in Fig. 4B showing proliferation-inducing effects of IL-4 on Tregs and additionally demonstrating that activated STAT6 downstream of IL-4-signaling is an inducer of CD4+ T cell proliferation. Since retroviral infection involves prestimulation of cells with IL-2, which also promotes Treg proliferation, as expected, the proliferation-promoting effect of caSTAT6 was less pronounced compared to that of IL-4 added to freshly isolated Tregs from naïve mice (Fig. 4B).
FIGURE 5.
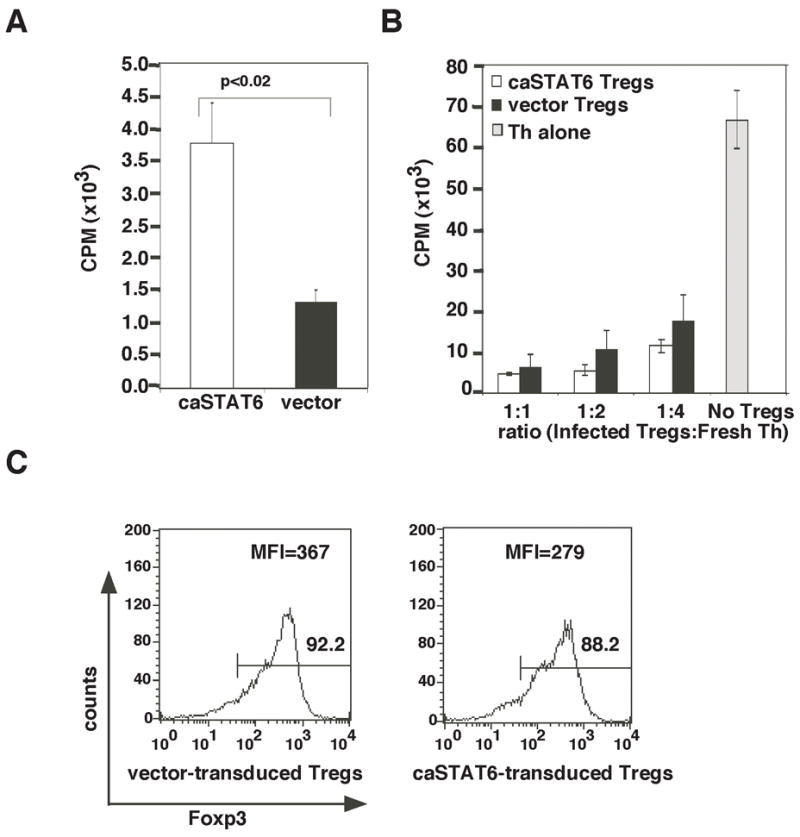
STAT6 signaling promotes Treg proliferation but does not impair Treg suppressive function. (A) Tregs were isolated from STAT6−/− mice, infected with retrovirus expressing caSTAT6 or control retrovirus and examined for proliferation by [3H]-thymidine incorporation. (B) GFP+ Tregs were sorted, rested for 2 d, and stimulated with APCs and anti-CD3ε alone or co-cultured in varying ratios with freshly isolated WT Th cells in a [3H]-thymidine incorporation assay. (C) Tregs isolated from STAT6-deficient mice were infected with control or caSTAT6-expressing retrovirus, sorted based on GFP expression and Foxp3 expression was analyzed by flow cytometry.
Despite promotion of Treg proliferation by IL-4, caSTAT6 caused only a minimal change in suppressive function, showing that IL-4 signaling via STAT6 does not directly alter Treg function on a per cell basis (Fig. 5B). The similar suppressive function of vector- and caSTAT6-transduced Tregs despite increased proliferation of the latter suggested more than a numbers game as far as Tregs were concerned and prompted us to focus on Th cells to study the effect of IL-4 signaling. The percentage of Tregs expressing Foxp3 transduced with caSTAT6 was similar to that of vector-transduced Tregs at the end of the culture period. The mean florescence intensity of Foxp3 in caSTAT6-transduced Tregs was slightly lower than that in the control Tregs (Fig. 5C). Thus, in agreement with data shown in Fig. 4, STAT6 expression in naturally occurring Tregs did not compromise Foxp3 expression.
STAT6 signaling is sufficient for enhancement of Th cell resistance to Treg-mediated suppression
We first ensured expression of caSTAT6 in Th cells by infecting splenic STAT6−/− CD4+ Th cells with the retrovirus and analyzing for STAT6 protein expression by western blotting techniques (Fig. 6A). To characterize the Th cells infected with caSTAT6, cells were sorted according to GFP expression and stimulated with anti-CD3 and APC. After 3 days of stimulation, transduced cells were analyzed for GATA-3 and T-bet expression by Western blotting techniques and analyzed cytokine levels in culture supernatant. Both vector- and caSTAT6-transduced Th cells expressed T-bet, although caSTAT6 transduced cells expressed less T-bet. However, GATA-3 was not detected in vector or caSTAT6 transduced cells (Fig. 6B). In retroviral transductions, cells are stimulated to vigorously proliferate for 40 h in the presence of IL-2 before retroviral infection. Since caSTAT6 was introduced only 40 h after cell activation, it was not surprising that T-bet but not GATA-3 was expressed. Interestingly, IL-13 was detected in the culture supernatant the level of which was a little higher from cells expressing caSTAT6. IFN-γ levels were slightly lower when caSTAT6 was expressed (Fig. 6B). It was not surprising that both Th1 and Th2 cytokines were detected in the cultures stimulated under neutral conditions. However, as published previously showing the ability of caSTAT6 to promote IL-4 expression via chromatin remodeling in the 3′ region of the IL-4/IL-13/RAD50 locus independent of GATA-3 (44), in our experiments, caSTAT6 promoted IL-13 expression in the absence of detectable GATA-3 expression.
FIGURE 6.

STAT6 signaling promotes Th cell resistance to suppression. Th cell from STAT6−/− mice were infected with retrovirus expressing caSTAT6 or control retrovirus. (A) Cells were cultured and total cell extract was made and analyzed by immunoblotting techniques for STAT6 protein expression. The blot was reprobed for β-actin expression to determine protein loading. (B) Th cells were sorted based on the expression of GFP and restimulated with anti-CD3 and irradiated APC for 3 days. Nuclear extracts were made from the cells and analyzed for the expression of T-bet, GATA-3 and CREB, the latter serving as a control for protein loading. Also, nuclear extract from polarized Th2 cells was used as a positive control for GATA-3 expression. The culture supernatants were analyzed by ELISA for IFNγ and IL-13 production. (C) Infected Th cells were sorted and combined with WT Tregs in the indicated ratios in a thymidine incorporation assay. Before use in the assay, Tregs were isolated and cultured for 1 d in the presence of IL-2 (50 U/ml) and plate-bound anti-CD3 and anti-CD28 to activate and increase their potency. Th cell proliferation was reproducibly lower in this type of experiment compared to that shown in 5B because in these experiments, Th cells were stimulated for several days during the infection procedure, which reduced their proliferative potential compared to that of freshly isolated Th cells used in Fig. 5B. Data shown are mean ± S.D. and are representative of 2 independent experiments.
Th cells expressing caSTAT6 were more resistant to Treg-mediated suppression compared to cells infected with control retrovirus, which was most appreciable at a low Th:Treg ratio (Fig. 6C). Of note, Th cell proliferation was different in the results shown in Figs. 5B and 6C because of the difference in culture conditions in the two types of experiments. In data presented in Fig. 6C, the cells were stimulated for several days during the infection procedure, which decreased their ability to proliferate upon exposure to additional activating stimuli while in Fig. 5B, freshly isolated Th cells with high proliferative potential were used. Additionally, this most likely also accounted for the minimal difference in proliferation between vector versus caSTAT6-transduced Th cells in Fig. 6C in contrast to that in Fig. 5B because of the more prolonged influence of IL-2 as a proliferating signal to both types of cells in the former instance. In fact this similarity in Th proliferation whether vector or caSTAT6 was introduced was of benefit in this experiment since it allowed us to investigate the effect of Tregs on similarly proliferating Th cells. Taken together, in side-by-side cultures with similarly proliferating Th cells, those with persistent STAT6 activation expressing caSTAT6 were rendered less susceptible to suppression by Tregs. Although IL-4 promoted expansion of Tregs, it did not in a cell intrinsic fashion influence their suppressive effect on Th cells. Since some suppression of Th cell proliferation was still evident despite caSTAT6 expression in the Th cells (Fig. 6C), signaling pathways induced by additional pro-inflammatory cytokines secreted during inflammation may additively or synergistically confer with STAT6 the net resistance to suppression during peak effector cell function.
Discussion
The present study shows that constitutive STAT6 signaling in a Th cell impairs the suppressive potential of T regulatory cells. The biological significance of this finding lies in the scenario of allergen-induced increased Th cell numbers accompanied by eosinophilia in the airways even though Tregs also increase in numbers at the same location. Based on similar observations in other disease models, it is clear that while Th cells mediating immune responses to either foreign or self-antigens can be readily identified at sites of inflammation, Tregs also develop and coexist with Th cells during inflammation (27–33). In experimental autoimmune encephalitis (EAE), a model of multiple sclerosis, while Tregs were found to infiltrate the central nervous system (CNS), similar to our findings and that of others in allergic airway inflammation (Fig. 1 and (33), these Tregs were unable to prevent disease (32). Similarly, in sarcoidosis, a granulomatous disease of the lung, Tregs present in abundance around the granulomas were unable to inhibit TNF-α and IFN-γ production by effector T cells (29). So, why is inflammation induced if inflammatory conditions can promote Treg numbers concurrently with Th cell numbers? We focused our attention on IL-4 to explain this phenomenon. This was due to the fact that IL-4 promotes proliferation of not only Th cells but also of Tregs, as shown previously in in vitro studies (38) and supported by our own data (Fig. 4). This effect is similar to that of IL-2, which has been shown to promote Treg proliferation and boost its suppressor function (14–16). IL-2 is also critical for the peripheral maintenance of Tregs (45,46). STAT5b, downstream of IL-2 receptor signaling, has been shown to be important for Treg accumulation and suppressor function in humans (14). Although IL-4 was described to inhibit Treg suppression in vitro (39), the role of STAT6, which is a major mediator of IL-4 effector function, in causing Th cell resistance to Treg function when both are present together has not been investigated prior to this study.
We show that although IL-4 promotes Treg proliferation and maintains Foxp3 expression, Tregs are functionally compromised under conditions of inflammation. This is an excellent example of how the same cytokine maintains the dynamics of inflammation by exercising its function on both Th cells and Tregs. When an adaptive immune response picks up, it induces Th resistance but keeps Tregs functionally competent so that when T effector cells contract later in inflammation, the Tregs are poised to take over and restore homeostasis. In a similar fashion, spontaneous IL-2 secretion by lung effector T cells in sarcoidosis has been implicated in Treg expansion around the granulomas (29). The balance between the competing effects of IL-4 or IL-2 on Treg versus T effector function likely determines the outcome of antigenic stimulation in terms of tolerance induction or the magnitude of inflammation induced.
So how might chronic activation of STAT6 render cells unresponsive to Tregs? GATA-3 is the master regulator of Th2 differentiation as we and others previously established (22, 47, 48). STAT6 is required during early stages of Th2 differentiation to promote GATA-3 expression but subsequently GATA-3 autoregulates its own expression. STAT6 has been also shown to rescue Th cells from an anergy checkpoint after priming to induce T cell proliferation and differentiation (49). STAT6 activation is promoted by multiple signals during Th cell differentiation, which includes those emanating from CD28 and ICOS. Tregs mediate suppression by multiple mechanisms many of which involve TGF-β (26). We have previously shown that soluble TGF-β inhibits Itk activation and Ca2+ flux in T cells thereby inhibiting GATA-3 activation to cause inhibition of Th2 differentiation (12). More recently, we have shown that membrane-bound TGF-β activates Notch1 in target T helper cells and co-operation between these two pathways causes inhibition of Th2 responses in the lung (11, 20). The question then arises as to how TGF-β-induced suppressive mechanisms are overridden by effector cytokines such as IL-4 and IL-13 secreted by Th cells during allergic inflammation.
In a recent report, Tregs expressing membrane-bound TGF-β were found to induce Foxp3 expression in naïve Th cells that imparted suppressive properties to these cells in a process of infectious tolerance (50). Interestingly, recently in other published work, STAT6 was shown to inhibit TGF-β1-induced Foxp3 induction via direct binding to the Foxp3 promoter (37). Similarly, GATA-3 was also shown to bind to the Foxp3 promoter thereby blocking TGF-β1-induced Foxp3 expression (51). Collectively, these observations provide a molecular basis for our finding that caSTAT6 renders Th cells unresponsive to suppression by Tregs. First, the recent reports would explain why Tregs would be more suppressive irrespective of IL-4Rα signaling when transferred to presensitized animals (Fig. 3A). Under these conditions, the Tregs would be able to proliferate in response to both IL-2 and IL-4 since recently activated Th cells would supply both cytokines and Tregs would have the added advantage of being able to induce infectious tolerance by inducing Foxp3 in Th cells rendering them suppressive as well (50). The induction of Foxp3 would be in competition with the opposing effects of STAT6 and GATA-3 induction in the freshly activated Th cells. In contrast, when the Tregs are administered after sensitization (Fig. 3B), the mice have already at their disposal effector cells with high levels of GATA-3 expression, which would block Foxp3 induction in these already differentiated Th2 cells (51). Secondly, availability of less IL-2 would cause the Tregs to depend more on IL-4 for Foxp3 expression and proliferation which is why the Tregs were still functional in inhibiting the effector cells (albeit less compared to transfer into animals before sensitization) when they were derived from WT mice (Fig. 3B and ref. 48) but their suppressive function was severely impaired in the absence of IL-4 signaling (Fig. 3B). In the in vitro studies shown in Figs. 5 and 6, expression of caSTAT6 would prevent Foxp3 induction in the Th cells (43) but would be of less consequence in Tregs, which already express Foxp3. This suggests that that the process of infectious tolerance competes with the opposing process of Th cell differentiation to different lineages during an adaptive immune response. Given that Tregs can be detected in large numbers not only in the context of allergic inflammation but other types of inflammation as well (27–33), there are at least two explanations for why adaptive immune response is launched despite the power of infectious tolerance. First, once TCR stimulation plus adequate costimulation drives Th cell proliferation during an adaptive immune response, factors such as STAT6 and GATA-3 and possibly additional transcription factors as well prevent de novo Foxp3 induction to allow an adaptive immune response to take hold. Second, under conditions of inflammation, Tregs may not express adequate levels of LAP-membrane-bound TGF-β on their cell surface, which would be interesting to study in the future. Reciprocally, protocols that promote tolerance such as persistent inhalation of aerosolized antigen may be particularly effective in promoting expression of membrane-bound TGF-β as we previously reported (11, 20). These TGF-β-expressing Tregs would be effective in inducing infectious tolerance via induction of Foxp3 in naive CD+ T cells.
Our results also have relevance to cytokine-based therapeutic control of asthma. The IL-4 variant, Pitrakinra, which potently inhibits binding of both IL-4 and IL-13 to IL-4Rα, was recently shown to significantly alleviate asthma symptoms (52). These results may be explained by the ability of Pitrakinra to block STAT6 signaling downstream of IL-4Rα in effector T cells thereby relieving resistance to Treg suppression. Since other cytokines such as IL-6, also secreted by Th2 cells, may supplant IL-4 in maintaining Treg proliferation and Foxp3 expression as we and others have shown (21, 53), the suppressive effect of Tregs concurrent with blockade of effects of IL-4/IL-13 on Th cells by agents such as Pitrakinra may significantly disable the effector function of the latter. Thus, our data suggest that in the context of active inflammation in asthma patients, blocking dominant stimulatory effects of IL-4 and IL-13 via STAT6 on disease-associated Th2 cells would likely result in a net beneficial effect. The results of our experiments also suggest that inhibition of IL-4/IL-13 effects in vivo combined with adoptive transfer of activated Tregs could result in even greater clinical efficacy.
Acknowledgments
The authors thank Amgen, Inc. (Thousand Oaks, CA) for providing the plasmid expressing caSTAT6, Drs. W. Paul and J. Zhu for the retroviral construct expressing caSTAT6 and for critical reading of the manuscript and M. Fei for assistance with cell isolation from lung tissue.
This work was supported by NIH grants RO1 AI 48927, P50HL84932 and RO1 HL77430 (to A.R.), RO1 HL60207 (to P.R.), T32 CA82084 (to O. F.), and F30 ES014776 (to B.P.) from the National Institutes of Health.
References
- 1.Thomson AW. The cytokine handbook. Acadmic Press; San Diego, CA: 2003. [Google Scholar]
- 2.Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev. 2003:CD001186. doi: 10.1002/14651858.CD001186. [DOI] [PubMed] [Google Scholar]
- 3.Janeway C. Immunobiology : the immune system in health and disease. Garland Science; New York: 2005. [Google Scholar]
- 4.Paul WE. Fundamental immunology. Lippincott Williams & Wilkins; Philadelphia: 2003. [Google Scholar]
- 5.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 6.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DA, Doherty PC, Grosveld G, Paul WE, Ihle JN. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 7.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 8.Noben-Trauth N, Shultz LD, Brombacher F, Urban JF, Jr, Gu H, Paul WE. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc Natl Acad Sci U S A. 1997;94:10838–10843. doi: 10.1073/pnas.94.20.10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kruse S, Japha T, Tedner M, Sparholt SH, Forster J, Kuehr J, Deichmann KA. The polymorphisms S503P and Q576R in the interleukin-4 receptor alpha gene are associated with atopy and influence the signal transduction. Immunology. 1999;96:365–371. doi: 10.1046/j.1365-2567.1999.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hershey GK, Friedrich MF, Esswein LA, Thomas ML, Chatila TA. The association of atopy with a gain-of-function mutation in the alpha subunit of the interleukin-4 receptor. N Engl J Med. 1997;337:1720–1725. doi: 10.1056/NEJM199712113372403. [DOI] [PubMed] [Google Scholar]
- 11.Ostroukhova M, Seguin-Devaux C, Oriss TB, Dixon-McCarthy B, Yang L, Ameredes BT, Corcoran TE, Ray A. Tolerance induced by inhaled antigen involves CD4(+) T cells expressing membrane-bound TGF-beta and FOXP3. J Clin Invest. 2004;114:28–38. doi: 10.1172/JCI20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CH, Seguin-Devaux C, Burke NA, Oriss TB, Watkins SC, Clipstone N, Ray A. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J Exp Med. 2003;197:1689–1699. doi: 10.1084/jem.20021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, Teper A, Gaillard M, Heinrich J, Krensky AM, Rosenfeld RG, Lewis DB. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol. 2006;177:2770–2774. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 15.Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172:6519–6523. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- 16.Thornton AM, Piccirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol. 2004;34:366–376. doi: 10.1002/eji.200324455. [DOI] [PubMed] [Google Scholar]
- 17.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. 2004;172:5213–5221. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 18.Bensinger SJ, Walsh PT, Zhang J, Carroll M, Parsons R, Rathmell JC, Thompson CB, Burchill MA, Farrar MA, Turka LA. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J Immunol. 2004;172:5287–5296. doi: 10.4049/jimmunol.172.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–2027. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- 20.Ostroukhova M, Qi Z, Oriss TB, Dixon-McCarthy B, Ray P, Ray A. Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-beta. J Clin Invest. 2006;116:996–1004. doi: 10.1172/JCI26490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pillemer BB, Xu H, Oriss TB, Qi Z, Ray A. Deficient SOCS3 expression in CD4(+)CD25(+)FoxP3(+) regulatory T cells and SOCS3-mediated suppression of Treg function. Eur J Immunol. 2007;37:2082–2089. doi: 10.1002/eji.200737193. [DOI] [PubMed] [Google Scholar]
- 22.Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. 1997;272:21597–21603. doi: 10.1074/jbc.272.34.21597. [DOI] [PubMed] [Google Scholar]
- 23.Zhu J, Guo L, Watson CJ, Hu-Li J, Paul WE. Stat6 is necessary and sufficient for IL-4’s role in Th2 differentiation and cell expansion. J Immunol. 2001;166:7276–7281. doi: 10.4049/jimmunol.166.12.7276. [DOI] [PubMed] [Google Scholar]
- 24.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends in molecular medicine. 2007;13:108–116. doi: 10.1016/j.molmed.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nature reviews. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao D, Malmstrom V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol. 2003;33:215–223. doi: 10.1002/immu.200390024. [DOI] [PubMed] [Google Scholar]
- 28.Marshall NA, Christie LE, Munro LR, Culligan DJ, Johnston PW, Barker RN, Vickers MA. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood. 2004;103:1755–1762. doi: 10.1182/blood-2003-07-2594. [DOI] [PubMed] [Google Scholar]
- 29.Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, Kambouchner M, Valeyre D, Chapelon-Abric C, Debre P, Piette JC, Gorochov G. The immune paradox of sarcoidosis and regulatory T cells. J Exp Med. 2006;203:359–370. doi: 10.1084/jem.20050648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor JJ, Mohrs M, Pearce EJ. Regulatory T cell responses develop in parallel to Th responses and control the magnitude and phenotype of the Th effector population. J Immunol. 2006;176:5839–5847. doi: 10.4049/jimmunol.176.10.5839. [DOI] [PubMed] [Google Scholar]
- 31.Ward SM, Fox BC, Brown PJ, Worthington J, Fox SB, Chapman RW, Fleming KA, Banham AH, Klenerman P. Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J Hepatol. 2007;47:316–324. doi: 10.1016/j.jhep.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. 2008;29:114–126. doi: 10.1016/j.immuni.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Yu A, Malek TR. Selective availability of IL-2 is a major determinant controlling the production of CD4+CD25+Foxp3+ T regulatory cells. J Immunol. 2006;177:5115–5121. doi: 10.4049/jimmunol.177.8.5115. [DOI] [PubMed] [Google Scholar]
- 35.Turka LA, Walsh PT. IL-2 signaling and CD4+ CD25+ Foxp3+ regulatory T cells. Front Biosci. 2008;13:1440–1446. doi: 10.2741/2773. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee DK, Dhodapkar MV, Matayeva E, Steinman RM, Dhodapkar KM. Expansion of FOXP3high regulatory T cells by human dendritic cells (DCs) in vitro and after injection of cytokine-matured DCs in myeloma patients. Blood. 2006;108:2655–2661. doi: 10.1182/blood-2006-03-011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takaki H, Ichiyama K, Koga K, Chinen T, Takaesu G, Sugiyama Y, Kato S, Yoshimura A, Kobayashi T. STAT6 Inhibits TGF-beta1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem. 2008;283:14955–14962. doi: 10.1074/jbc.M801123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pace L, Pioli C, Doria G. IL-4 modulation of CD4+CD25+ T regulatory cell-mediated suppression. J Immunol. 2005;174:7645–7653. doi: 10.4049/jimmunol.174.12.7645. [DOI] [PubMed] [Google Scholar]
- 39.Pace L, Rizzo S, Palombi C, Brombacher F, Doria G. Cutting edge: IL-4-induced protection of CD4+CD25- Th cells from CD4+CD25+ regulatory T cell-mediated suppression. J Immunol. 2006;176:3900–3904. doi: 10.4049/jimmunol.176.7.3900. [DOI] [PubMed] [Google Scholar]
- 40.Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MG, Jr, Glasheen E, Lane WS, Pierce JH, White MF. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377:173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 41.Vella A, Teague TK, Ihle J, Kappler J, Marrack P. Interleukin 4 (IL-4) or IL-7 prevents the death of resting T cells: stat6 is probably not required for the effect of IL-4. J Exp Med. 1997;186:325–330. doi: 10.1084/jem.186.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zamorano J, Wang HY, Wang LM, Pierce JH, Keegan AD. IL-4 protects cells from apoptosis via the insulin receptor substrate pathway and a second independent signaling pathway. J Immunol. 1996;157:4926–4934. [PubMed] [Google Scholar]
- 43.Daniel C, Salvekar A, Schindler U. A gain-of-function mutation in STAT6. J Biol Chem. 2000;275:14255–14259. doi: 10.1074/jbc.c000129200. [DOI] [PubMed] [Google Scholar]
- 44.Lee DU, Rao A. Molecular analysis of a locus control region in the T helper cytokine gene cluster: a target for STAT6 but not GATA-3. Proc Natl Acad Sci USA. 101:16010–16015. doi: 10.1073/pnas.0407031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-b signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 47.Zhang DH, Yang L, Cohn L, Parkyn L, Homer R, Ray P, Ray A. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity. 1999;11:473–482. doi: 10.1016/s1074-7613(00)80122-3. [DOI] [PubMed] [Google Scholar]
- 48.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 49.Mohrs M, Lacy DA, Locksley RM. Stat signals release activated naive Th cells from an anergic checkpoint. J Immunol. 2003;170:1870–1876. doi: 10.4049/jimmunol.170.4.1870. [DOI] [PubMed] [Google Scholar]
- 50.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O’Shea JJ, Shevach EM. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mantel PY, Kuipers H, Boyman O, Rhyner C, Ouaked N, Ruckert B, Karagiannidis C, Lambrecht BN, Hendriks RW, Crameri R, Akdis CA, Blaser K, Schmidt-Weber CB. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol. 2007;5:e329. doi: 10.1371/journal.pbio.0050329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 53.Kubo T, Hatton RD, Oliver J, Liu X, Elson CO, Weaver CT. Regulatory T cell suppression and anergy are differentially regulated by proinflammatory cytokines produced by TLR-activated dendritic cells. J Immunol. 2004;173:7249–7258. doi: 10.4049/jimmunol.173.12.7249. [DOI] [PubMed] [Google Scholar]