

Abstract
Nitric oxide acts substantially in cellular signal transduction through stimulus-coupled S-nitrosylation of cysteine residues. The mechanisms that might subserve protein denitrosylation in cellular signaling remain uncharacterized. Our search for denitrosylase activities focused on caspase-3, an exemplar of stimulus-dependent denitrosylation, and identified thioredoxin and thioredoxin reductase in a biochemical screen. In resting human lymphocytes, thioredoxin-1 actively denitrosylated cytosolic caspase-3 and thereby maintained a low steady-state amount of S-nitrosylation. Upon stimulation of Fas, thioredoxin-2 mediated denitrosylation of mitochondria-associated caspase-3, a process required for caspase-3 activation, and promoted apoptosis. Inhibition of thioredoxin-thioredoxin reductases enabled identification of additional substrates subject to endogenous S-nitrosylation. Thus, specific enzymatic mechanisms may regulate basal and stimulus-induced denitrosylation in mammalian cells.
Cellular functions of nitric oxide (NO) are carried out in part through S-nitrosylation of Cys residues within a broad functional spectrum of proteins (1). Like protein phosphorylation, S-nitrosylation thus mediates or modulates transduction of myriad cellular signals. Although the NO synthases (NOSs) and S-nitrosoglutathione reductase (GSNOR) govern S-nitrosylation by NO or S-nitrosothiols (SNOs) (1), little is known about the nature of or even necessity for enzymatic mechanisms that may directly remove NO groups from Cys thiols. Indeed, stimulus-coupled denitrosylation has been shown to precisely modulate the function of proteins that are constitutively S-nitrosylated (2, 3), but the cellular effectors of denitrosylation remain undetermined.
Some caspases, members of a family of proteases that mediate apoptosis, are subject to inhibitory S-nitrosylation, and stimulation of death receptors of the tumor necrosis factor superfamily results in caspase denitrosylation (2, 4, 5). In particular, a subpopulation of caspase-3 (a major executioner caspase) that is associated with mitochondria is constitutively S-nitrosylated at the active site Cys, and engagement of the Fas receptor promotes denitrosylation (2, 6). However, the role of denitrosylation in transduction of the apoptotic signal has not been fully elucidated, and the molecular mechanism of denitrosylation is unknown.
Identification of thioredoxin as a caspase-3 denitrosylase
To assess protein denitrosylation, we developed an assay based on the reactivation of caspase that results from denitrosylation of the active site Cys (7) (fig. S1A). Recombinant caspase-3 was S-nitrosylated (SNO–caspase-3), and the inhibited enzyme was immobilized on nickel-coated plates. Bound SNO–caspase-3 was incubated with cellular extracts, and caspase-3 activity was then measured with a fluorogenic substrate. Addition of Jurkat cell cytosolic extract rapidly restored the activity of caspase-3 (Fig. 1A). Loss of the SNO moiety was verified by Hg-coupled photolysis-chemiluminescence, which measures the amount of NO displaced from Cys thiol by ultraviolet (UV) light (2) (Fig. 1A). This cytosolic denitrosylating activity was detected in numerous human cell types and in rat tissues (Fig. 1B). A subcellular fraction enriched in mitochondria also displayed denitrosylating activity (fig. S1B).
Fig. 1.
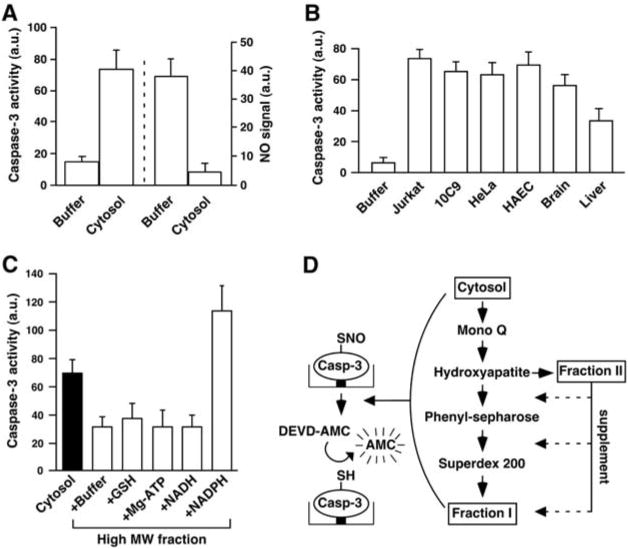
Characterization of an enzymatic activity that denitrosylates SNO–caspase-3. Data in (A) to (C) are presented as mean ± SEM; n ≥ 3. (A) Reactivation of SNO–caspase-3 protease activity by cell cytosol. Immobilized SNO–caspase-3 (∼100 nM) was incubated for 30 min with a cytosolic extract prepared from Jurkat cells. (Left) Caspase activity was determined by using Z-DEVD-7-amino-4-methylcoumarin (DEVD-AMC). (Right) SNO content was assayed by using Hg-coupled photolysis-chemiluminescence. a.u., arbitrary unit. (B) Caspase activity after 30-min incubation of SNO–caspase-3 with cytosolic fractions (100 μg of protein) prepared from human cells or rat tissues. HAEC, primary human aortic endothelial cells. (C) SNO–caspase-3 was incubated with the cytosolic fraction from Jurkat cells or with cytosolic fractions after size-exclusion chromatography through Sephadex G-25 [high molecular weight (MW) fraction] and supplemented with GSH (0.5 mM), ATP (10 μM), NADH (10 μM), or NADPH (10 μM). (D) The procedure used to partially purify a SNO–caspase-3 denitrosylase (fig. S1, A and E, and table S1).
Cytosolic denitrosylating activity was present in a fraction enriched for large molecular size (>10 kD) and was diminished after exposure to heat or trypsin (fig. S1C). Activity was also reduced after fractionation by size-exclusion chromatography (Sephadex G-25), indicating a possible requirement for a small cofactor, and activity was restored and potentiated by reduced nicotinamide adenine dinucleotide phosphate (NADPH) but not nicotinamide dinucleotide (NADH), glutathione (GSH), or adenosine triphosphate (ATP) (Fig. 1C). Thus, the cytosolic denitrosylating activity exhibits properties of an NADPH-dependent oxidoreductase.
We derived, by a four-step chromatographic purification from Jurkat cells, a highly active fraction (designated fraction I) (Fig. 1D and table S1), whose activity was dependent on a second fraction added in limiting amounts (1:10) (designated fraction II) (Fig. 1D and fig. S1D). Fraction I contained eight proteins (fig. S1E), which were identified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (table S2). Of these, only thioredoxin-1 (Trx1) could be ascribed a redox-related function. Recombinant Trx reductase (TrxR) could substitute for fraction II, fully reconstituting the denitrosylating activity of fraction I (fig. S1D).
Depletion of Trx1 from HeLa cells with small interfering RNA (siRNA) correlated with a loss of SNO–caspase-3 denitrosylating activity in vitro (Fig. 2A and fig. S2A), and denitrosylating activity was restored by adding back recombinant Trx1 but not an active site mutant Trx [Cys32 → Ser32, Trx1(C32S)] (fig. S2A). In contrast, siRNA-mediated depletion of an additional member of the Trx family, Trx-related protein 14 (TRP14) (8), had no effect on denitrosylating activity (fig. S2B). Similarly, immunodepletion of Trx1 but not TRP14 abolished denitrosylating activity (Fig. 2B). A reconstituted Trx system [10 nM Trx and TrxR (Trx-TrxR) and including NADPH] efficiently denitrosylated an excess of SNO–caspase-3 (Fig. 2C). Denitrosylation by Trx1 in the absence of TrxR1 was ineffective but was restored when concentrations of Trx1, but not Trx1(C32S), approached or exceeded that of SNO–caspase-3 (Fig. 2D and fig. S2C), suggestive of single-turnover denitrosylation coupled to Trx1 oxidation.
Fig. 2.
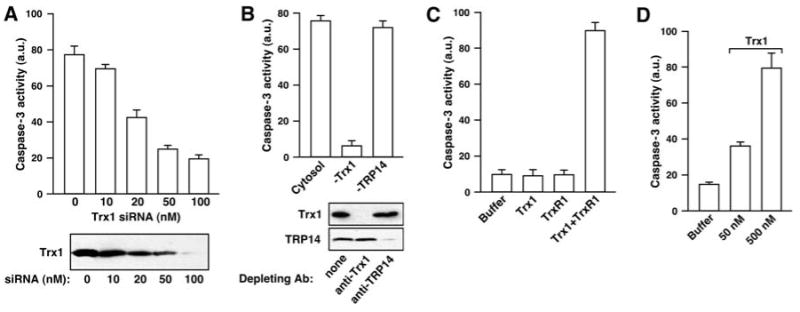
The Trx system is a major SNO–caspase-3 denitrosylating activity. Data are presented as mean ± SEM; n = 3. (A) Caspase-3 activity was determined (with Z-DEVD-AMC) after a 30-min incubation of SNO–caspase-3 (∼100 nM) with a cytosolic fraction prepared from untreated HeLa cells or from cells that were transfected for 3 days with siRNA for Trx1. (B) Caspase-3 activity was determined after a 30-min incubation of SNO–caspase-3 with HeLa cytosolic extract or cytosol that had been depleted of Trx1 or TRP14 by using specific antibodies against Trx1 or TRP14. (C) Caspase-3 activity was determined after a 30-min incubation of SNO–caspase-3 (∼100 nM) with NADPH (100 μM) and recombinant human Trx1 (10 nM) and/or recombinant rat TrxR1 (10 nM). (D) Caspase-3 activity after a 30-min incubation with recombinant Escherichia coli Trx1.
Dynamic regulation of cellular protein S-nitrosylation by Trx-TrxR
Vicinal dithiols, as present in Trx (CXXC, where C represents Cys and X is another amino acid), exhibit a denitrosylating activity in vitro (9), and various redox enzymes, including dithiol proteins, can catalyze denitrosylation reactions ex vivo (10–13). Trx may also break down SNOs within cells exposed to high concentrations of a nitrosylating agent (14, 15), as might occur during a nitrosative stress. However, in the context of signaling by endogenously derived NO, Trx has been proposed to promote S-nitrosylation of proteins (including caspase-3) (16, 17). Thus, in sum, it is not known whether Trx might have a physiological role in mediating either basal or stimulus-induced denitrosylation in vivo.
The extent of S-nitrosylation reflects the equilibrium between addition and removal of NO groups. We used RNA interference to assess whether Trx governs basal S-nitrosylation of caspase-3 in human B lymphocytes (10C9 cells), where most caspase-3 is cytosolic and in the reduced form (i.e., only the minor population of caspase-3 associated with the mitochondria is constitutively S-nitrosylated) (6). Two nonoverlapping siRNAs efficiently decreased the amount of cellular Trx1, and amounts of endogenous SNO–caspase-3 [assessed by the biotin switch technique, in which S-nitrosylated cysteines are selectively biotinylated (18, 19)] were increased in proportion to Trx1 depletion (Fig. 3A). siRNA-mediated depletion of iNOS or NOS inhibition with NG-monomethyl-l-arginine (L-NMMA) prevented the increase in SNO–caspase-3 (fig. S3, A and B). Depletion of TrxR1 in 10C9 cells (Fig. 3A and fig. S3C) and in human embryonic kidney (HEK) cells stably expressing a second NOS isoform, nNOS (HEK-nNOS) (fig. S3D), also resulted in increased SNO–caspase-3, even more efficiently than did depletion of Trx1. Thus, TrxR1 activity may be rate-limiting for denitrosylation of cytosolic SNO–caspase-3 in vivo. Inhibition of Trx or TrxR might alter the cellular redox balance (e.g., decrease intracellular GSH levels) and thus affect the abundance of SNO-proteins independently of specific denitrosylating activity (19). However, knockdown of Trx1 or TrxR1 did not alter levels of acid soluble thiols (which consist primarily of GSH) (fig. S3E).
Fig. 3.
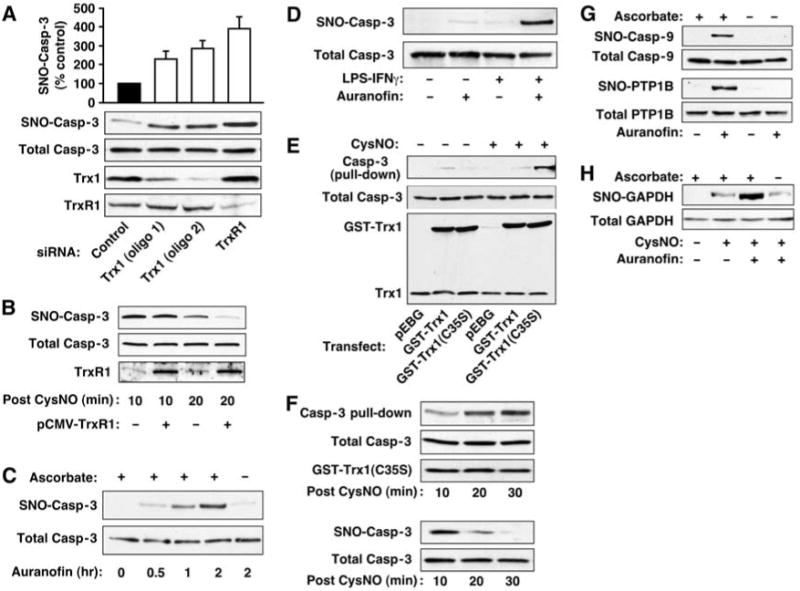
Trx1-TrxR1 mediates protein denitrosylation in vivo. Results are representative of three experiments. (A) 10C9 cells were transfected for 3 days with siRNA specific for Trx1 or TrxR1 or with control RNA. SNO–caspase-3 was assayed by biotin switch. The histogram summarizes results (mean ± SEM) of three experiments. (B) HEK cells were transfected for 24 hours with TrxR1 before treatment with CysNO (200 μM). Whole-cell extracts were prepared 10 or 20 min after CysNO exposure, and SNO–caspase-3 was assayed by biotin switch. (C) 10C9 cells were treated with auranofin (2 μM) for the times indicated, and SNO–caspase-3 was assayed by biotin switch. To verify biotin labeling specificity for SNO, the effect of omitting ascorbate in the assay is shown. (D) RAW264.7 cells were treated with auranofin (1 μM, 1 hour) or with LPS (1 μg/ml)–IFNγ (10 ng/ml) for 16 hours, or LPS-IFNγ plus auranofin (added for the last hour), and SNO–caspase-3 was assayed by biotin switch. (E) SNO-dependent interaction of Trx1 and caspase-3. HEK cells were co-transfected with caspase-3 and either GST-tagged wild-type Trx1, GST-tagged Trx1(C35S), or empty GST vector before treatment with CysNO (500 μM; 10 min). Affinity purification (pull-down) from lysates was with GSH-agarose. (F) HEK cells were treated as in (E) and lysed at different times after CysNO exposure. Pull-down of proteins was as in (E) (top). SNO–caspase-3 was assessed by biotin switch (bottom). (G and H) 10C9 cells were treated with auranofin (2 μM) or vehicle [dimethyl sulfoxide (DMSO)] for 2 hours, and S-nitrosylation of endogenous caspase-9, PTP1B, and GAPDH was assessed by biotin switch. For GAPDH (H), CysNO (500 μM; 20 min) was added after auranofin treatment.
Equivalent amounts of SNO–caspase-3 accumulated in HEK cells overexpressing either TrxR1 or empty vector shortly after treatment with S-nitrosocysteine (CysNO), a cell-permeable S-nitrosylating compound (Fig. 3B; 10 min). However, after 20 min, the amount of SNO–caspase-3 was lower in TrxR1-transfected cells (Fig. 3B). Therefore, augmenting TrxR1 enhances SNO–caspase-3 denitrosylation.
We examined in intact cells the effects on denitrosylation of 1-chloro-2,4-dinitrobenzene (DNCB) or S-triethylphosphine-gold(I)-2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (auranofin), two rapidly acting and potent but structurally dissimilar inhibitors of TrxR (20, 21). Treatment of 10C9 cells with either DNCB or auranofin increased the amount of SNO–caspase-3 in cells exposed to CysNO (fig. S3F). Remarkably, in the absence of an exogenous NO donor, cells treated with auranofin also accumulated SNO–caspase-3 (Fig. 3C and fig. S3G). We validated the identification of endogenous SNO in assays with auranofin by showing the dependence of signals on ascorbate (18, 19) as well as by the elimination of signals after UV irradiation, which liberates NO from SNO-proteins (19) (Fig. 3C and fig. S3H), and we observed that auranofin did not alter the abundance of acid-soluble thiols (fig. S3E). Thus, constitutive Trx1 activity apparently controls the steady-state amount of caspase-3 S-nitrosylation, a role similar to that of phosphatases in controlling basal phosphorylation.
To explore the role of Trx in reversing stimulus-coupled caspase-3 S-nitrosylation, we treated RAW264.7 macrophages with lipopolysaccharide (LPS) plus interferon-γ (IFNγ), which induces iNOS. Whereas LPS-IFNγ or auranofin alone weakly elicited S-nitrosylation of caspase-3, robust S-nitrosylation was seen when the two treatments were combined (Fig. 3D). Thus, S-nitrosylation of caspase-3 appears to reflect dynamic turnover of the NO group, mediated by the opposing activities of NOS and Trx-TrxR.
The reduction of substrate disulfides by Trx involves a CXXC motif (Cys32 and Cys35 in human Trx1) (22) (fig. S4A). A C35S mutant Trx can be used to trap substrates in a mixed disulfide (23). We developed an analogous SNO-based trapping strategy to examine the interaction of Trx with SNO–caspase-3. We transfected HEK cells with caspase-3 and Trx1(C35S) fused to glutathione S-transferase (GST). After treatment with CysNO, caspase-3 co-precipitated with Trx1(C35S) but not with wild-type Trx1 (Fig. 3E). The amount of caspase-3 trapped by Trx1(C35S) increased from 10 to 30 min after treatment with CysNO, during which time denitrosylation occurred (Fig. 3F). Additionally, in HEK cells transiently expressing iNOS, Trx(C35S) but not wild-type Trx formed mixed disulfides with multiple proteins in an NO-dependent manner (fig. S4D). Thus, Trx1 appears to interact with SNO–caspase-3 in vivo and to denitrosylate via a mixed-disulfide intermediate (fig. S4B); however, a mechanism involving transnitrosylation (14) is not excluded (fig. S4C).
Trx might serve as a denitrosylase for a broad spectrum of proteins. To identify endogenously S-nitrosylated substrates of Trx under basal conditions, we inhibited Trx in 10C9 cells with auranofin and identified caspase-9 and protein tyrosine phosphatase 1B (PTP1B) as substrates for Trx-mediated denitrosylation (Fig. 3G). The S-nitrosylated form of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), whose proapoptotic function is induced by NO (24), was not detected in untreated cells and did not appreciably accumulate 2 hours after exposure to auranofin. However, treatment with CysNO, representing a nitrosative stress, resulted in accumulation of SNO-GAPDH, and the amounts of SNO-GAPDH were further increased markedly by auranofin (Fig. 3H). Similarly, inhibition of TrxR or Trx increased the abundance of additional SNO-proteins after exposure to CysNO (fig. S5, A and B) as well as slowed their denitrosylation (fig. S5, C and D). Further, in HEK-nNOS cells, depletion of TrxR1 with siRNA increased the amounts of whole-cell SNO (Fig. S5E). Collectively, these studies suggest that Trx-TrxR acts as a multisubstrate denitrosylase.
Regulation of caspase-3 denitrosylation and apoptosis by mitochondrial Trx-TrxR
We then asked whether the Trx system also mediates stimulus (Fas)-induced denitrosylation, first described for mitochondria-associated SNO–procaspase-3 in 10C9 cells (2, 6). Both siRNA-mediated depletion of TrxR2, the isoform of TrxR that is localized selectively to mitochondria (25), and auranofin [which effectively inhibits TrxR2 (26)] abrogated Fas-dependent denitrosylation of mitochondrial caspase-3 (Fig. 4, A and B, and fig. S6A). Thus, Fas-induced denitrosylation of mitochondrial SNO–caspase-3 appears to be mediated by Trx2-TrxR2, a conclusion supported further by the finding that Fas stimulation resulted in an increase in mitochondrial Trx2 [152% ± 12% (mean ± SEM) relative to control, n = 4] (fig. S6B). These results suggest the possibility that Trx2-mediated denitrosylation [acting in concert with cleavage by initiator caspase(s)] may promote full activation of caspase-3 and thereby facilitate apoptosis.
Fig. 4.
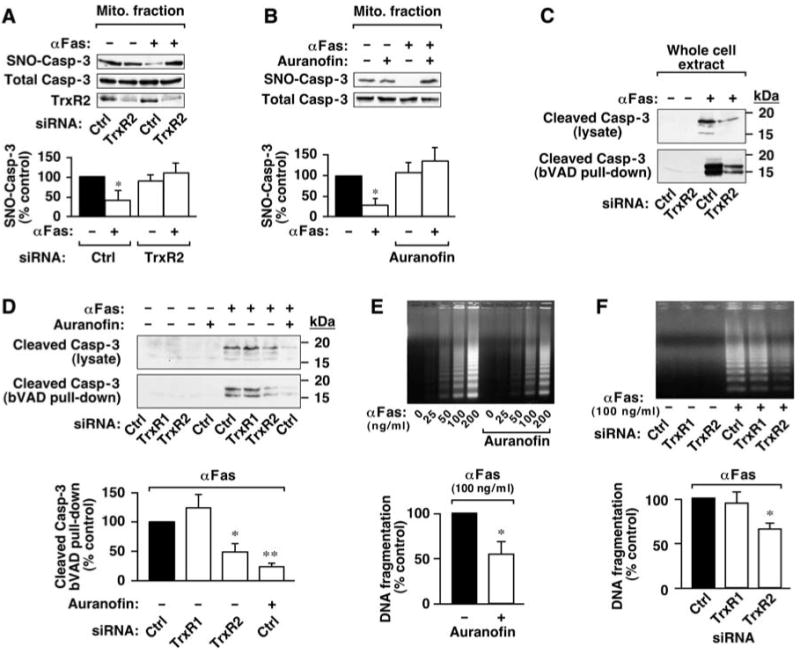
The mitochondrial Trx system mediates Fas-induced denitrosylation of mitochondria-associated SNO–caspase-3 and promotes apoptotic signaling. (A) 10C9 cells were transfected for 3 days with siRNA for TrxR2 or with control RNA before exposure to CH11 monoclonal antibody against Fas (αFas; 50 ng/ml) for 2 hours. The amount of SNO–caspase-3 in a subcellular fraction enriched for mitochondria was evaluated by biotin switch. Results are mean ± SEM of four experiments. *P < 0.05 by analysis of variance (ANOVA). (B) 10C9 cells were left untreated or treated with auranofin for 1 hour, followed by Fas receptor stimulation and evaluation of mitochondrial SNO–caspase-3 as in (A). Results are the mean ± SEM of three experiments. *P < 0.05 by ANOVA. (C) 10C9 cells treated as in (A) were lysed in the presence of bVAD-FMK (10 μM), an affinity ligand for active caspase. Cleaved caspase-3 (<20 kD) was assessed by immunoblotting with antibodies against caspase-3 in lysates and after purification of active, bVAD-FMK–bound caspases with streptavidin-agarose (pull-down). Results are representative of three experiments. (D) 10C9 cells were transfected with siRNA specific for TrxR1 or TrxR2 or with control RNA. Cells were left untreated or treated with auranofin (1 μM; 1 hour) followed by treatment with anti-Fas (100 ng/ml; 2 hours). Caspase-3 cleavage and activation were assessed as in (C). Results (Fas-activated samples) are mean ± SEM of three experiments. *P < 0.05, **P < 0.01 by ANOVA. (E and F) 10C9 cells were treated with auranofin or transfected with siRNA specific for TrxR1 or TrxR2 as in (A) to (D). DNA fragmentation was assessed 6 hours after treatment with anti-Fas. Results (Fas-activated samples) are mean ± SEM of four experiments. *P < 0.05 by ANOVA.
We further examined this possibility by assessing the effects of mitochondrial TrxR2 knockdown or inhibition on two molecular events that characterize the execution phase of Fas- and caspase-3-mediated apoptosis: caspase-3 activation and DNA fragmentation (27). In Fas-stimulated 10C9 cells, both depletion of TrxR2 with siRNA and acute inhibition with auranofin reduced both the amount of cleaved, active caspase-3 [captured with biotin-Val-Ala-Asp(OMe) fluoromethyl ketone (bVAD-FMK)] (Fig. 4, C and D, and figs. S6C and S7A) and caspase-3-like activity [cleavage of the tetrapeptide Asp-Glu-Val-Asp (DEVD)] (fig. S6D). In support of these data, activation of caspase-8 (which lies upstream of cytosolic caspase-3) was also diminished by TrxR2 inhibition (fig. S7A). In contrast, depletion of TrxR1 had no appreciable effect on caspase activity (Fig. 4D and fig. S6C). Furthermore, treatment of 10C9 cells with auranofin and knockdown of TrxR2 (but not TrxR1) with siRNA decreased DNA fragmentation by 45%±14 (n = 4) (Fig. 4E) and 34% ± 6 (Fig. 4F and fig. S7B), respectively. Although the precise sequence of events subserving transmission of the NO-regulated apoptotic signal from mitochondrial to cytosolic compartments remains to be elucidated fully (further discussed in fig. S7C), our findings suggest that denitrosylation of mitochondria-associated SNO–caspase-3 by Trx2-TrxR2 promotes Fas-induced apoptosis. More generally, inhibition of Trx2-TrxR2 provides a means to selectively manipulate mitochondria-associated caspase.
Concluding remarks
Many SNO-proteins are present constitutively in low amounts, and both S-nitrosylation and denitrosylation have been observed after stimulation of multiple classes of receptors (1). The present findings provide evidence for a specific enzymatic mechanism of protein denitrosylation, acting in distinct cellular compartments to regulate basal and stimulus-coupled protein denitrosylation, and thus complement the recent description of S-nitrosylation and -denitrosylation governed by GSNOR (11). Identification of mitochondria- and cytosol-specific denitrosylases not only elucidates the mechanism by which Fas induces denitrosylation of mitochondria-associated caspases selectively but also indicates a function for Trx2 in amplifying apoptotic signals.
Previous studies have raised the idea that Trx1 may promote S-nitrosylation of proteins through mechanisms involving nonactive site thiols Cys69 or Cys73 (16, 17), but these residues are not present in Trx2. Our finding that protein S-nitrosylation is augmented by genetic or pharmacologic inhibition of either Trx1-TrxR1 or Trx2-TrxR2 suggests that a major function of the Trx system (involving active-site Cys32) is protein denitrosylation. Most commonly, the Trx system is characterized as playing a highly conserved role in protection from cellular oxidative stress. The present findings indicate that SNO-protein denitrosylation (and thus regulation of NO-based signaling) represents an additional function of the Trx system in metazoan organisms, in which several classes of SNO-regulating enzymes—NO synthases, GSNO reductases, and Trx-TrxRs—are expressed ubiquitously across cell types.
Supplementary Material
Acknowledgments
The authors thank B. Li and A. Hausladen for technical assistance. M.B. was supported by a Ruth Kirschstein National Research Service Award fellowship from the NIH.
Footnotes
Supporting Online Material: www.sciencemag.org/cgi/content/full/320/5879/1050/DC1
References and Notes
- 1.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Nat Rev Mol Cell Biol. 2005;6:150. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 2.Mannick JB, et al. Science. 1999;284:651. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 3.Erwin PA, Lin AJ, Golan DE, Michel T. J Biol Chem. 2005;280:19888. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 4.Hoffmann J, Haendeler J, Zeiher AM, Dimmeler S. J Biol Chem. 2001;276:41383. doi: 10.1074/jbc.M107566200. [DOI] [PubMed] [Google Scholar]
- 5.Kim JE, Tannenbaum SR. J Biol Chem. 2004;279:9758. doi: 10.1074/jbc.M312722200. [DOI] [PubMed] [Google Scholar]
- 6.Mannick JB, et al. J Cell Biol. 2001;154:1111. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Materials and methods are available on Science Online.
- 8.Jeong W, Yoon HW, Lee SR, Rhee SG. J Biol Chem. 2004;279:3142. doi: 10.1074/jbc.M307932200. [DOI] [PubMed] [Google Scholar]
- 9.Arnelle DR, Stamler JS. Arch Biochem Biophys. 1995;318:279. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 10.Nikitovic D, Holmgren A. J Biol Chem. 1996;271:19180. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 11.Liu L, et al. Nature. 2001;410:490. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 12.Ravi K, Brennan LA, Levic S, Ross PA, Black SM. Proc Natl Acad Sci U S A. 2004;101:2619. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sliskovic I, Raturi A, Mutus B. J Biol Chem. 2005;280:8733. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- 14.Stoyanovsky DA, et al. J Am Chem Soc. 2005;127:15815. doi: 10.1021/ja0529135. [DOI] [PubMed] [Google Scholar]
- 15.Sengupta R, et al. Biochemistry. 2007;46:8472. doi: 10.1021/bi700449x. [DOI] [PubMed] [Google Scholar]
- 16.Haendeler J, et al. Nat Cell Biol. 2002;4:743. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell DA, Morton SU, Fernhoff NB, Marletta MA. Proc Natl Acad Sci U S A. 2007;104:11609. doi: 10.1073/pnas.0704898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Nat Cell Biol. 2001;3:193. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 19.Forrester MT, Foster MW, Stamler JS. J Biol Chem. 2007;282:13977. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 20.Arner ES, Bjornstedt M, Holmgren A. J Biol Chem. 1995;270:3479. doi: 10.1074/jbc.270.8.3479. [DOI] [PubMed] [Google Scholar]
- 21.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. J Biol Chem. 1998;273:20096. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 22.Lillig CH, Holmgren A. Antioxid Redox Signal. 2007;9:25. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 23.Verdoucq L, Vignols F, Jacquot JP, Chartier Y, Meyer Y. J Biol Chem. 1999;274:19714. doi: 10.1074/jbc.274.28.19714. [DOI] [PubMed] [Google Scholar]
- 24.Hara MR, et al. Nat Cell Biol. 2005;7:665. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 25.Lee SR, et al. J Biol Chem. 1999;274:4722. doi: 10.1074/jbc.274.8.4722. [DOI] [PubMed] [Google Scholar]
- 26.Pia Rigobello M, et al. J Inorg Biochem. 2004;98:1634. doi: 10.1016/j.jinorgbio.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 27.Nagata S. Annu Rev Genet. 1999;33:29. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.