

Abstract
Parkinson’s disease is a debilitating movement disorder characterized by altered levels of α6β2* nicotinic acetylcholine receptors (nAChRs) localized on presynaptic striatal catecholaminergic neurons. α-Conotoxin MII (α-CTx MII) is a highly useful ligand to probe α6β2 nAChRs structure and function, but it does not discriminate among closely related α6* nAChR subtypes. Modification of the α-CTx MII primary sequence led to the identification of α-CTx MII[E11A], an analog with 500–5300 fold discrimination between α6* subtypes found in both human and non-human primates. α-CTx MII[E11A] binds most strongly (femtomolar dissociation constant) to the high affinity α6 nAChR, a subtype that is selectively lost in Parkinson’s disease. Here we present the three-dimensional solution structure for α-CTx MII[E11A] as determined by two-dimensional 1H NMR spectroscopy to 0.13 +/− 0.09 Ǻ backbone and 0.45 +/− 0.08 Ǻ heavy atom root mean square deviation from mean structure. Structural comparisons suggest that the increased hydrophobic area of α-CTx MII[E11A] relative to other members of the α-CTx family may be responsible for its exceptionally high affinity for α6α4β2* nAChR as well as discrimination between α6β2 and α3β2 containing nAChRs. This finding may enable the rational design of novel peptide analogs that demonstrate enhanced specificity for α6* nAChR subunit interfaces and provide a means to better understand nAChR structural determinants that modulate brain dopamine levels and the pathophysiology of Parkinson’s disease.
Keywords: NMR solution structure, conotoxin, Parkinson’s disease, nicotinic acetylcholine receptor
Introduction
Nicotinic acetylcholine receptors (nAChRs) are a diverse family of ligand-gated cation channels found primarily in neuronal and muscle cells in humans.1 Structurally, each nAChR exists as either a homopentamer of α subunits or a heteropentamer of α and β proteins. nAChRs can be formed by any grouping of α2-α6 subunits together with β2–β4 subunits; leading to a large assortment of different receptor subtype combinations.2,3 The α6 containing nAChRs are primarily expressed at the presynaptic endings of catecholaminergic neurons in the brain, where their activity modulates the release of dopamine (DA) and norepinephrine.4–8
α-CTx MII, isolated from the venom of Conus magus, is a potent antagonist of neuronal nAChRs.9 α-CTx MII is a 16 amino acid peptide with the sequence GCCSNPVCHLEHSNLC containing disulfide bonds connecting Cys2 to Cys8 and Cys3 to Cys16. α-CTx MII binds to the ligand binding domain (LBD) at the interface between the α 3 and β2 subunits of nAChRs and inhibits the receptor binding to acetylcholine (ACh) with an IC50 of 0.5 nM.9,10 At the time of it’s discovery in 1997, it was thought that the α3β2 subunit combination in nAChRs of presynaptic neurons in the brain was responsible for regulation of dopamine release into the synapse and transmission of cholinergic responses. More recently, it has been determined that nicotinic drugs acting on the closely related α6β2* nAChRs can alter dopamine release by DA neurons in the mid-brain and striatum.11 The reporting of α-CTx MII selectivity for α and β subunits has led to the discovery of an additional five native α-CTxs that bind both α3β2 and α6β2 nAChRs with variable potency and selectivity. α-CTx MII and α-CTx GIC bind to and inhibit α3β2 and α6β2 with approximately equal preference,9,12 while α-CTx OmIA shows ~20 fold preference for α3β2 over α6β2,13 α-CTx BuIA and α-CTx PIA show a ~20 fold and ~75 fold respective preference for α6β2 over α3β2.14,15 Each of these α-CTxs is an ACh antagonist with an IC50 of ~0.5–1 nM (with the exception of OmIA, IC50 ~11 nM). The single-site mutant, α-CTx MII[E11A], is a highly selective ligand for a α6α4β2* subtype of α6* nAChRs. In monkeys, α-CTx MII[E11A] binds putative α6α4β2* nAChRs with an affinity of 9.7 fM and α6 (non-α4)β2* nAChRs with an affinity of 51 pM. Thus, α-CTx MII[E11A] discriminates between these two closely related subtypes by 5300-fold.16
Treatment with the dopaminergic neurotoxin 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) creates irreversible damage that is widely used to mimic Parkinson’s disease. MPTP treatment of rodents and monkeys leads to selective loss of the α6α4β2* nAChRs in the central nervous system as identified by a decrease in bound α-CTx MII[E11A] in brain slice studies.16,17 Importantly, α6α4β2* nAChRs are also selectively lost in human Parkinson’s disease.18 Furthermore, striatal DA-ergic fiber cells have been shown to be heterogeneous based on nAChR subtype expression, containing two populations of DA-ergic fibers: low action potential threshold fibers versus high action potential threshold fibers for the release of DA. α-CTx MII[E11A] binds to the low threshold fibers whose action (DA release) is modulated by α6* nAChRs.16
Here we present the solution structure of α-CTx MII[E11A] and a structural comparison of α-CTx MII[E11A] to other α-CTxs that bind neuronal nAChRs. A structure to activity hypothesis is proposed based on primary sequence comparisons, mutagenesis data, and three dimensional (3D) structure features that are thought to contribute to α-CTx binding potency and specificity.
Materials and Methods
NMR Spectroscopy
An NMR sample of α-CTx MII[E11A] was initially prepared at a concentration of 4.2 mM in 90% H2O and 10% D2O (%v/v). Properly folded synthetic peptide was generously donated by the laboratory of Dr. J. Michael McIntosh, University of Utah. High resolution 1D 1H, DQF-COSY, TOCSY and NOESY spectra were acquired at 11.75 T on a Varian Inova spectrometer at a temperature of 15°C. Time course deuterium exchange experiments were performed on a freeze dried sample that was re-suspended in deuterium oxide followed by data reacquisition in 100% D2O. Subsequent high resolution 1D 1H, DQF-COSY, TOCSY, NOESY, 1H/13C HSQC, and 1H/15N HSQC NMR spectra were performed on 0.8 mM α-CTx MII[E11A] in 95% H2O and 5% D2O at a spectrometer field strength of 14.1 T in a Bruker Avance III spectrometer utilizing a TXI cryogenically cooled probe. Two-dimensional 1H NMR experiments and interpretation of spectra were based on established methods.19,20 Proton DQF-COSY,21 NOESY,22 and TOCSY23 spectra were acquired with the transmitter set to 4.76 ppm and a spectral window of 4500 Hz. Spectra in H2O were acquired with the transmitter set at 4.8 ppm and a spectral window of 6500 Hz. Transmitter presaturation was applied at the solvent frequency to suppress the H2O resonance. A series of NOESY spectra were acquired with mixing times of 150, 250, and 350 ms. TOCSY spectra were acquired with mixing times of 80 ms. Slowly exchanging NH protons were detected by acquiring a series of 1D spectra of the fully protonated peptide immediately following dissolution in 100% D2O. Spectra were processed using Bruker TopSpin (v. 2.1), MestReC, or Varian VNMR software.
Restraint Set Generation
The 3JNH-α values for Cys2, Ser4, Leu10, Ala11, His12 and Cys16 were extracted from a high resolution 1D 1H NMR spectrum. Backbone phi dihedral angle restraints were set to −120 +/− 40–or 3JNH-α greater than 8.0 Hz and to −65 +/− 25° for a 3JNH-α less than 5 Hz. Inter- and intraproton distance ranges were derived from NOESY spectra recorded at 15°C. A visual analysis of peak intensity was used to assign NOE intensity as strong, medium, or weak. As a distance reference, the intensity of the geminal protons was considered strong and the distance constraint was set to 1.8 Å. Restraints were set to 1.8–2.7, 1.8–3.3, and 1.8–5.0 Å for strong, medium, and weak intensity signals in the NOESY spectra. Only cross peaks that indicated reasonable correlation between the different mixing time NOESY spectra were included as constraints for molecular dynamics calculations.24
Structure Calculations
One thousand initial structures generated from random starting conformations were input into the CYANA (V. 2.1) software.25 The distance geometry and gradient minimization calculations were performed to find the conformations which best modeled the 91 upper and 17 lower distance limits, 6 phi angle, 4 hydrogen bonds, and 2 disulfide bonds consistent with the NMR data and the molecular backbone, respectively. A pseudo atom correction of 0.5 Å was added to the upper bounds of restraints for methyl and methylene groups. The final set of 20 structures demonstrated an overall root-mean-square deviation among backbone atoms of 0.13 +/− 0.09 Å.
Results
Assignment of Resonances
The complete 1H resonance assignment for α-CTx MII[E11A] was achieved using the method of Wüthrich.19 A combination of DQF-COSY, NOESY, and TOCSY spectra acquired at 15°C in 95% H2O and 5% D2O were used to eliminate any ambiguities in assignment. 1H NMR spectra were observed with good chemical shift dispersion in the amide region ranging from 9.18 to 7.45 ppm. Fourteen of the 16 amino acid spin systems for α-CTx MII[E11A] were identified in the “fingerprint” region of a 80 ms mixing time TOCSY spectrum. Pro6 lacks a resonance in the “fingerprint” region of the spectrum and was identified later. Gly1 was never assigned in the TOCSY spectrum. All spin systems with the exception of Gly1 and Pro6 were confirmed in the “fingerprint” region of a DQF-COSY spectrum and assigned to Cys2, Cys3, Ser4, Asn5, Val7, Cys8, His9, Leu10, Ala11, His12, Ser13, Asn14, Leu15 and Cys16. Verification of α- and β-proton resonances and the location of the Pro6 spin system were extracted from TOCSY and DQF-COSY spectra acquired in 100% D2O. A complete and thorough mapping of all amino acid spin systems was accomplished in this manner.
The assignment of spin systems to amino acids in the primary sequence of the peptide went as follows. The methyl region between 0.79 and 1.06 ppm in the TOCSY spectrum acquired in 95% H2O provided information needed to identify Val7, Leu10 and Leu15. Diagnostic resonances were also seen for His9, His12 and Pro6. The imidizole ring protons of His9 and His12 were found in the TOCSY and 1H/13C HSQC spectra between 6.99 and 8.71 ppm. The spin system assigned to Pro6 originated at 4.26 ppm in the TOCSY spectrum. The δ-protons at 4.07 and 3.93 ppm were helpful for assigning resonances for Pro6 because its α-proton at 4.26 ppm was within 0.01 ppm of the α-protons of His9 and Ala11. A complete trace of the Pro6 spin system was the only way to distinguish the amino acid from those with overlapping spin systems. The remaining amino acids, Cys2, Cys3, Ser4, Asn5, Cys8, His12, Ser13, Asn14, and Cys16 all share the same AMX pattern and were sequentially assigned from the 250 ms mixing time NOESY spectrum by way of a “NOESY walk”.
Sequential Assignments
The NOESY data were sufficient to observe proton-proton NOE’s between the amide proton of one residue and the α-proton of proceeding residue for Cys3, Ser4, Asn5, Cys8, His9, Leu10, Ala11, His12, Ser13, Asn14, Leu15, and Cys16. These residues were assigned based on sequential information combined with Hβ i to HNi+1 cross peaks from the 250 ms NOESY spectrum.
Off-diagonal resonances were observed for the amide and α-protons of Cys2 at 8.89 ppm and 4.52 ppm to the β-protons of Cys3 at 3.24 and 2.84 ppm. There was a weak NOE interaction between the amide proton of Cys2 at 8.89 ppm and the β-proton of Cys8 at 3.29 ppm that helped to identify Cys8 and verify the assignment of Cys2. Likewise, a weak NOE was observed between the amide proton of Cys3 at 8.69 and the β-protons of Cys16 at 2.63 and 2.23 ppm. A summary of restraints entered into CYANA and the results of the molecular dynamics calculations are provided in Table 1.
Table 1.
Restraints and structural statistics for the final 20 structures generated by the CYANA software based on NMR derived constraints for α-CTx MII[E11A].
| Parameter | Value |
|---|---|
| Number of NOE distance constraints | 92 |
| Intra-residue | 16 |
| Sequential | 27 |
| Medium range | 32 |
| Long range | 17 |
| Dihedral angle restraints | 6 |
| Hydrogen bond constraints | 4 |
| Ramachandran plot regions (%) | |
| Most favored region | 51.9 |
| Additionally allowed region | 40.4 |
| Generously allowed region | 7.7 |
| Disallowed region | 0.0 |
| RMSDs from the average structure (Å) | |
| Backbone atoms | 0.13 ± 0.09 |
| Heavy atoms | 0.42 ± 0.08 |
Discussion
Structural description of α-CTx MII[E11A]
α-CTx MII[E11A] folds into a highly compact globular structure consisting of a central amphipathic α-helix between residues 6–13 that is flanked by β-turns at the N- and C-termini. Deuterium exchange studies were used to identify four hydrogen bonds that stabilize the α-helix of α-CTx MII[E11A]. This α-helix is held in place by i to i+4 interactions between the carbonyl oxygen atoms of Pro6, Val7, Cys8, and His9, and the amide protons on Leu10, Ala11, His12, and Ser13, respectively. At the N-terminus, residues 2–5 are involved in a type II β-turn, stabilized by an i to i+3 hydrogen bond between the amide proton of Asn5 and the carbonyl oxygen of Cys2. The C-terminal region exhibits a type III β-turn between Ser13 and Cys16; this turn may be stabilized by an i to i+4 hydrogen bond between the amide proton of Cys16 and the carbonyl oxygen of Ser13, although the amide proton of Cys16 was not observed to persist for more than 20 minutes following dissolution in D2O. The exterior surface of the α-helix has a hydrophobic surface created by Pro6, Val7, Leu10, and Ala11 on one side, and a hydrophilic face consisting of Cys8, His9, and Ser13 on the opposite side of the helix. Since the α-helix has been determined to be significant for peptide activity, the potential for the cis isomer of the proline (Pro6) that initiates the helix was explored. The presence of two strong intensity NOE cross peaks in the 350 ms mixing time NOESY spectrum is indicative of the proximity of the Asn5 αH to Pro6 δH’s and is consistent with a structure containing the trans isomer of the Pro6 residue. 26,27 Additional inspection of the 80 ms TOCSY spectrum indicated a single spin system for Pro6 originating at 4.26 ppm; again consistent with the trans isomer and the absence of the cis isomer in the overall structure. Surprisingly, the hydrophobic residues of the α-helix are more exposed to the solvent than the charged/hydrophilic residues, with the exception of His12. This residue, which has been shown to be very significant for biological activity, is oriented toward the surface of the molecule forming its own prominent electropositive surface feature.28
Structural Comparison between α-CTx MII[E11A] and wildtype α-CTx MII
As expected, the structure of α-CTx MII[E11A] is very similar to that of native α-CTx MII (Figure 1). However, there are desirable differences in the electrostatic topography of the peptide that may explain the observed enhancement in binding selectivity for the α6β2 subunit interface as compared to α3β2. As reported by both Shon et al. and Hill et al., the α-helix of α-CTx MII originates at Pro6 and terminates at His12, while in α-CTx MII[E11A] the α-helix is observed to extend to Ser13.29,30 The increased length of the helix may be attributed to the single point mutation at Glu11. This observation is consistent with the increase in α-helical content as measured by circular dichroism (CD) reported by Everhart et al.31 According to their analysis, α-CTx MII consists of 31.2±0.5% α-helix versus 44.9±1.3% for α-CTx MII[E11A]. The change in α-helix does not result in a dramatic change in the overall three dimensional (3D) structure, and a change in the backbone helix itself would not be expected to be the ultimate reason for the observed changes in specificity between α-CTx MII and the [E11A] analog.
Figure 1.

A structure comparison of α-CTxs MII (top row) and E11A (bottom row). From left to right, an overlay of the 20 backbone atoms for the final 20 structures (for MII, Shon et al. structure) with the N- and C-termini labeled, a tube structure with Pro6 labeled as the point of initiation of the α-helix and residues in positions 11 and 12 labeled, and lastly, the surface of each peptide colored by atom charge where red is negative, blue is positive, and white is neutral for the peptides at a pH of 3.
There appear to be differences in the nature of the C-terminal β-loop between Ser13 and Cys16 of α-CTx MII and α-CTx MII[E11A]. The C-terminal turn of α-CTx MII is described as a distorted 310-helix, whereas the C-terminal portion of α-CTx MII[E11A] more closely resembles a type III β-turn.30 There are also differences in the orientation of several amino acid side chains. This is probably due to the relatively few NOE constraints observed in this region of the peptide, and does not significantly affect the results of this study. The helical portion of α4/7 conotoxins (named according to the number of amino acids in each disulfide loop) is generally accepted to be functionally critical in binding.12–15
In α-CTx MII there is a hydrophobic patch consisting of residues Pro6, Val7, and Leu10 on the exposed face of the α-helix. The size of this hydrophobic area is larger in α-CTx MII[E11A] with the addition of the nonpolar Ala11 side chain. The significance of the hydrophobic regions of α-CTx MII toward binding selectivity was first demonstrated by consecutive substitution of each peptide amino acid with alanine.28 In that study the [V7A] and [L10A] mutations showed a relative shift in binding towards α3β2 versus α6β2 nAChRs. In α-CTx MII[V7A] and α-CTx MII[L10A] the hydrophobicity of the amphipathic α-helix was effectively decreased by substitution of the isopropyl or isobutyl side chains, respectively, for a methyl substituent. Conversely, alanine substitution at His9 and Asn14 increased the hydrophobic character of the α-helix, which sharpened selectivity of the peptide mutants for α6β2 versus α3β2 nAChRs. The ratios of [H9A] and [N14A] IC50 for α3β2 to α6β2 were 74.7 and 24.2, respectively, indicating a significant enhancement of binding preference for the α6β2 subunit interface. To put this in perspective, α-CTx MII[E11A] has an IC50 α3β2 to IC50 α6β2 ratio of 54.5, yet a 500-fold discrimination index for human and 5300-fold for monkey α6α4β2* versus α6β2* nAChRs.16
α-CTx MII [E11A] has the same secondary structure as the native peptide albeit with an extension of the α-helix, and a distinct difference in electrostatic topography surrounding His12. In previous studies, His12 and Asn5 were found to be important determinants of α-CTx MII potency, as was the protonation state of His12,28,31 The difference in selectivity of α-CTx MII and α-CTx MII[E11A] may be attributed to a change in the position, orientation, or pKa of His12. The distance between the Asn5 and His12 side chains appears to be greater in α-CTx MII[E11A] than α-CTx MII, but caution must be taken in structural evaluation based on side chain atom position, as the orientation of these atoms is dynamic.
Structural Comparison between α-CTx MII[E11A] and other select α-CTxs
The NMR solution structures for α-CTxs and the X-ray crystal structures that are currently available have shown that despite differences in primary sequence there is a highly conserved secondary structural motif. It appears that α-CTx peptide binding specificity and potency is dependent on the amino acid side chains which the backbone serves to present to the receptor on the conserved scaffold. Table 2 shows the sequences for the α-CTxs that demonstrate binding to α3β2 and α6β2 containing nAChR. The recently reported novel α-CTx TxIA was also included due to its similarity to other α-CTxs and because of its binding affinity to an acetylcholine binding protein from Aplysia californica (Ac-AChBP). The Ac-AChBP serves as a model for nAChRs because it has highly conserved LBD residues, and biochemical, pharmacological, and functional attributes consistent with indigenous nAChRs in the human body.32,33 Co-crystallization of α-CTx TxIA and Ac-AChBP demonstrate that Arg5, Asn11, and Asn12 are key side chain residues on the peptide ligand that stabilize binding to the receptor by electrostatic and hydrogen bonding interactions.32 As well, disulfide stacking between Cys2–Cys8 of the peptide ligand and adjacent disulfides in the receptor appear to stabilize binding in the AChBP receptor model.32
Table 2.
Primary sequence comparison of α-CTxs that bind to both α3β2 and α6β2 subtype nAChRs. The scaffold disulfide bond motif common to these conotoxins is shown for PIA. Included are the IC50 values for α3β2 and α6β2 subtype nAChR and the PDB code for each conotoxin.
| α-CTx | Primary Sequence | IC50 α3β2nM | IC50 α6β2nM | Ratio IC50 α3β2 to α6β2 | Reference | PDB |
|---|---|---|---|---|---|---|
| PIA | RDPCCSNPVCTVHNPQIC | 74.20 | 0.95 | 78.1 | [15] | 1ZLC |
| MII[E11A] | GCCSNPVCHLAHSNLC | 8.72 | 0.16 | 54.5 | [26] | -- |
| BuIA | GCCSTPPC– – –AVLYC | 5.72 | 0.26 | 22.0 | [14] | 2I28 |
| MII | GCCSNPVCHLEHSNLC | 2.20 | 0.39 | 5.6 | [9] | 1M2C |
| GIC | GCCSHPACAGNNQHIC | 1.10 | ~3 | 0.4 | [12] | 1UL2 |
| OmIA | GCCSHPACNVNNPHICG | 11.00 | 201 | 0.0547 | [13] | 2GCZ |
| TxIA | GCCSRPPCIANNPDLC | 3.50 | N/A | N/A | [32] | U2Z6 |
Previous conotoxins have been identified that bind both α6β2 and α3β2 nAChRs; these include α-CTxs MII, PIA, GIC, BuIA and OmIA. Interestingly, α-CTxs MII[E11A], BuIA and PIA show an increased size of the hydrophobic face of the peptide as compared to α-CTxs MII, OmIA and GIC. The size of the hydrophobic region of the amphipathic α-helix is hypothesized to be important for selective binding of peptide to α6β2 versus α3β2 containing nAChRs. Surface representations for these toxins and for the newly determined α-CTx MII[E11A] are shown in Figure 2. As mentioned previously, His12 and Asn5 were found to be important determinants of α-CTx MII potency.31 The electronic and steric character of a histidine is similar to that of an asparagine at neutral pH, and it is worth noting that histidine and asparagine are interchangeably found in α-CTxs (Table 2, Figure 2). Either histidine or asparagine is found in homologous locations to the His12 and Asn5 positions of α-CTx MII[E11A] in each of the α-CTxs displayed in Figure 2, with the exception of BuIA. α-CTx BuIA has an amidated C-terminus which occupies the same site as His12 in [E11A], which may serve as an asparagine side chain homolog for binding. Figure 2 also shows that each of the specified α-CTxs has these residues positioned with the same orientation to create a similar electrostatic surface for receptor interaction. The positive and negative surface charges associated with the peptides in addition to the two disulfide bonds (shown in yellow, Figure 2) provide variation to account for the observed binding differences from one peptide to another.
Figure 2.
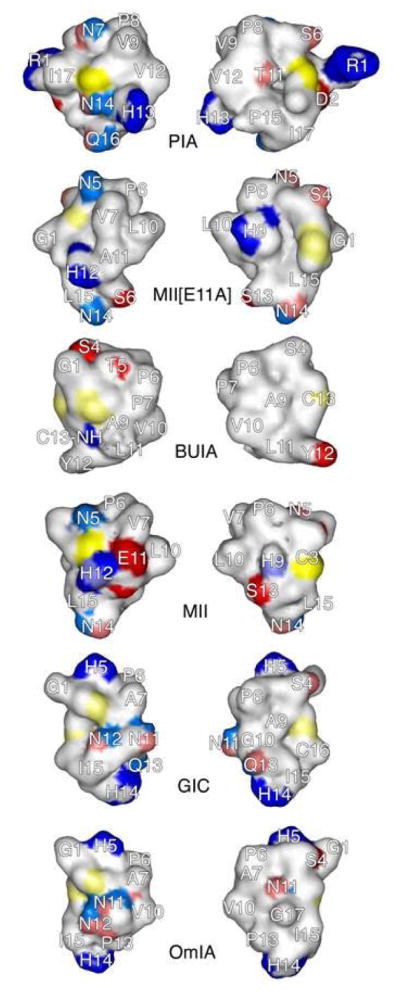
Surface comparison of α-CTxs PIA, MII[E11A], BuIA, MII, GIC, OmIA from top to bottom, respectively. Blue regions represent electropositive surfaces, red regions are electronegative surfaces, and white/faintly colored regions are hydrophobic/nonpolar surfaces. Sulfur atoms of cystine residues are yellow. Left panel: front view of peptide surface. Right panel: backside view of peptide surface.
The crystal structures currently available to serve as binding models for the α-CTxs, contain ImI, PnIA, and Tx1A bound in the ligand binding domain of the AChBP from Lymnea stagnalis, Botulinus subtiles, or Aplysia californica. These X-ray crystal structures of the AChBP with bound α-CTxs ImI, PnIA, and Tx1A all indicate that the Cys2 to Cys8 cystine bridge of the peptide is involved via disulfide bond stacking with two adjacent cysteine residues in the receptor.33–35 Figure 2 shows that in each of these conotoxins, the Cys2 to Cys8 disulfide bridge lies directly between either an asparagine or a histidine in the same position as Asn5 and His12 in α-CTx MII. In addition, these toxins show a prominent and contiguous hydrophobic region of varying size on the right side of the “front view” and the left side of the “back view” of each structure corresponding to hydrophobic residues in the α-helical portion of these peptides. This hydrophobic region is consistent with the observation that hydrophobic interaction is a predominant factor for ligand-binding to nAChRs.36–38 It appears that the hydrophilic residues at positions 5 and 12 (either Asn or His) in combination with the α-helix and disulfide bonds that dictate the peptide scaffold are significant contributors to binding specificity.
Another interesting observation from this study is that the hydrophilic locus occupied by a serine following the second cysteine in the primary sequence of each toxin is ubiquitous. α-CTxs MII, OmIA and GIC have relatively large hydrophilic domains that are much larger than those in the other toxins, while the reduced size of this hydrophilic domain to those found in α-CTxs PIA, BuIA and MII[E11A] may be responsible for reduced selectivity toward binding tothe α3β2 subunit interface. The back side of the toxins shows a common hydrophilic region on the left formed by three hydrophilic (polar or negatively charged) residues (Figure 2). Thus, the presence of such common hydrophobic and hydrophilic domains, although different in their exact sizes, is likely to represent the structural basis for binding selectivity towards α3β2 or α6β2 nAChR subtypes. These structural comparisons of α-CTxs suggest that binding affinity may be caused by the hydrophobic region of the peptide while subunit binding selectivity may be attributed to the hydrophilic patches.
Acknowledgments
This work was supported by St. Luke’s Mountain States Tumor Medical Research Institute (6PR3381000154), Research Corporation Cottrel College Scholars Program (6PR3381000172), NIH Grant # P20 RR016454 from the INBRE Program of the National Center for Research Resources, and the National Science Foundation CRIF-MU program (0639251). The authors wish to thank Drs. Clemens Anklin and Donna Baldasseri at Bruker Biospin, Inc. for helpful discussions and NMR data acquisition. In addition we would like to thank Mr. Sean Ruettgers and Mr. Reed Jacob at Boise State University who provided technical assistance and made artistic contributions to this work.
List of abbreviations
- NMR
nuclear magnetic resonance
- nAChR
nicotinic acetylcholine receptor
- AChBP
acetylcholine binding protein
- DA
dopamine
- CTx
conotoxin
- DQF-COSY
double quantum filtered-correlation spectroscopy
- NOESY
nuclear Overhauser effect spectroscopy
- TOCSY
total correlation spectroscopy
- HSQC
heteronuclear single quantum coherence spectroscopy
- LBD
ligand binding domain
- MPTP
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
Footnotes
Indicates the possible presence of additional subunits
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA. Trends Neurosci. 2004;27:329. doi: 10.1016/j.tins.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Lindstrom J. Ion Channels. 1996;4:377. doi: 10.1007/978-1-4899-1775-1_10. [DOI] [PubMed] [Google Scholar]
- 3.Dani JA, Bertrand D. Annu Rev Pharmacol Toxicol. 2007;47:699. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 4.Le Novere N, Zoli M, Changeux JP. Eur J Neurosci. 1996;8:2428. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 5.Azam L, Winzer-Serhan UH, Chen Y, Leslie FM. J Comp Neurol. 2002;444:260. doi: 10.1002/cne.10138. [DOI] [PubMed] [Google Scholar]
- 6.Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Mol Pharmacol. 2004;65:1526. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- 7.Azam L, McIntosh JM. J Pharmacol Exp Ther. 2005;312:231. doi: 10.1124/jpet.104.071456. [DOI] [PubMed] [Google Scholar]
- 8.Azam L, McIntosh JM. Mol Pharmacol. 2006;70:967. doi: 10.1124/mol.106.024513. [DOI] [PubMed] [Google Scholar]
- 9.Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. J Biol Chem. 1996;271:7522. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- 10.Harvey SC, McIntosh JM, Cartier GE, Maddox FN, Luetje CW. Mol Pharmacol. 1997;51:336. doi: 10.1124/mol.51.2.336. [DOI] [PubMed] [Google Scholar]
- 11.Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, Bupp S, Heintz N, McIntosh JM, Bencherif M, Marks MJ, Lester HA. Neuron. 2008;60:123. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McIntosh JM, Dowell C, Watkins M, Garrett JE, Yoshikami D, Olivera BM. J Biol Chem. 2002;277:33610. doi: 10.1074/jbc.M205102200. [DOI] [PubMed] [Google Scholar]
- 13.Talley TT, Olivera BM, Han KH, Christensen SB, Dowell C, Tsigelny I, Ho KY, Taylor P, McIntosh JM. J of Biol Chem. 2006;281:24678. doi: 10.1074/jbc.M602969200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azam L, Dowell C, Watkins M, Stitzel JA, Olivera BM, McIntosh JM. J Biol Chem. 2005;280:80. doi: 10.1074/jbc.M406281200. [DOI] [PubMed] [Google Scholar]
- 15.Dowell C, Olivera BM, Garrett JE, Staheli ST, Watkins M, Kuryatov A, Yoshikami D, Lindstrom JM, McIntosh JM. J Neurosci. 2003;23:8445. doi: 10.1523/JNEUROSCI.23-24-08445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bordia T, Grady SR, McIntosh JM, Quik M. Mol Pharmacol. 2007;72:52. doi: 10.1124/mol.107.035998. [DOI] [PubMed] [Google Scholar]
- 17.Meyer EL, Yoshikami D, McIntosh JM. J Neurochem. 2008;105:1761. doi: 10.1111/j.1471-4159.2008.05266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quik M, Bordia T, O’Leary K. Biochem Pharmacol. 2007;74:1224. doi: 10.1016/j.bcp.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wüthrich K. NMR of Proteins and Nucleic Acids. Wiley; New York: 1986. [Google Scholar]
- 20.Bax A. Annu Rev Biochem. 1989;58:223. doi: 10.1146/annurev.bi.58.070189.001255. [DOI] [PubMed] [Google Scholar]
- 21.Rance M, Sørensen OW, Bodenhausen G, Wagner G, Ernst RR, Wüthrich K. Biochem Biophys Res Commun. 1983;117:479. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 22.Jeener J, Meier BH, Bachmann P, Ernst RR. J Chem Phys. 1979;71:4546. [Google Scholar]
- 23.Braunschweiler L, Ernst RR. J Magn Reson. 1983;53:521. [Google Scholar]
- 24.Keepers JW, James TL. J Magn Reson. 1984;57:404. [Google Scholar]
- 25.Güntert P, Mumenthaler C, Wüthrich K. J Mol Biol. 1997;273:283. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 26.Kaerner A, Rabenstein DL. Stability and structure-forming properties of the two disulfide bonds of α-conotoxin GI. Biochemistry. 1999;38:5459–5470. doi: 10.1021/bi9826658. [DOI] [PubMed] [Google Scholar]
- 27.Larive CK, Guerra L, Rabenstein DL. Cis/trans conformational equilibrium across cysteine6-proline peptide bond of oxytocin, arginine, vasopressin, and lysine vasopressin. J Am Chem Soc. 1992;114:7331–7337. [Google Scholar]
- 28.McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Mol Pharmacol. 2004;65:944. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- 29.Hill JM, Oomen CJ, Miranda LP, Bingham J-P, Alewood PF, Craik DJ. Biochemistry. 1998;37:15621. doi: 10.1021/bi981535w. [DOI] [PubMed] [Google Scholar]
- 30.Shon K-J, Koerber SC, Rivier JE, Olivera BM, McIntosh JM. Biochemistry. 1997;36:15693. doi: 10.1021/bi971443r. [DOI] [PubMed] [Google Scholar]
- 31.Everhart D, Cartier GE, Malhorta A, Gomes A, McIntosh JM, Luetje CW. Biochemistry. 2004;43:2732. doi: 10.1021/bi036180h. [DOI] [PubMed] [Google Scholar]
- 32.Tomizawa M, Maltby D, Medzihradszky KF, Zhang N, Durkin KA, Presley J, Talley TT, Taylor P, Burlingame AL, Casida JE. Biochemistry. 2007;46:8798. doi: 10.1021/bi700667v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Celie PH, Kasheverov IE, Mordvintsev DY, Hogg RC, van Nierop P, van Elk R, van Rossum-Fikkert SE, Zhmak MN, Bertrand D, Tsetlin V, Sixma TK, Smit AB. Nat Struct Mol Biol. 2005;12:582. doi: 10.1038/nsmb951. [DOI] [PubMed] [Google Scholar]
- 34.Dutertre S, Ulens C, Buttner R, Fish A, Hopping G, Alewood P, Schroeder C, Nicke A, Smit A, Sixma T, Lewis R. EMBO J. 2007;26:3858. doi: 10.1038/sj.emboj.7601785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansen SB, Talley TT, Radic Z, Taylor P. J Biol Chem. 2004;279:24197. doi: 10.1074/jbc.M402452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bren N, Sine SM. J Biol Chem. 2000;275:12692. doi: 10.1074/jbc.275.17.12692. [DOI] [PubMed] [Google Scholar]
- 37.Quiram PA, McIntosh JM, Sine SM. J Biol Chem. 2000;275:4889. doi: 10.1074/jbc.275.7.4889. [DOI] [PubMed] [Google Scholar]
- 38.Bourne Y, Talley TT, Hansen SB, Taylor P, Marchot P. EMBO J. 2005;24:1512. doi: 10.1038/sj.emboj.7600620. [DOI] [PMC free article] [PubMed] [Google Scholar]