

Abstract
Autophagy mediates the bulk turnover of cytoplasmic constituents in lysosomes. During embryonic development in animals, a dramatic degradation of yolk proteins and synthesis of zygotic proteins takes place, leading to intracellular remodeling and cellular differentiation. Zebrafish represents a unique system to study autophagy due in part to its rapid embryonic development relative to other vertebrates. The technical advantages of this organism make it uniquely suited to various studies including high-throughput drug screens. To study autophagy in zebrafish, we identified two zebrafish Atg8 homologs, lc3 and gabarap, and generated two transgenic zebrafish lines expressing GFP-tagged versions of the corresponding proteins. Similar to yeast Atg8 and mammalian LC3, zebrafish Lc3 undergoes post-translational modification starting at the pharyngula stage during embryonic development. We observed a high level of autophagy activity in zebrafish embryos, which can be further upregulated by the TOR inhibitor rapamycin or the calpain inhibitor calpeptin. In addition, zebrafish Gabarap accumulates within lysosomes upon autophagy induction. Thus, we established a convenient zebrafish tool to assay autophagic activity during embryogenesis in vivo.
Keywords: embryogenesis, lysosome, LysoTracker, protein targeting, stress
Introduction
Autophagy is a cellular degradative pathway that delivers cytoplasmic cargo to the lysosome. There are many forms of autophagy including macroautophagy, microautophagy and chaperone-mediated autophagy. The best studied of these processes is macroautophagy, which will be referred to as autophagy hereafter. During autophagy, a double-membrane vesicle called an autophagosome is formed around cargo. Autophagosomes subsequently fuse with a lysosome, allowing breakdown of the vesicle inner membrane; hydrolases then degrade the cargo, and the resulting macromolecules are released back into the cytosol for use as cellular nutrients. The study of autophagy is important because of its roles in human health and pathology including host immune defense, antigen presentation, tumor suppression, cardiovascular disease, gastrointestinal disorders, neurodegeneration and longevity.1-5
Mutant screens in S. cerevisiae have revealed approximately 31 genes required for autophagy (ATG genes) and many of these have mammalian homologs.6 Autophagy requires two highly conserved ubiquitin-like conjugation systems. The ubiquitin-like protein Atg12 is activated by an E1 enzyme (Atg7), subsequently transferred to an E2 enzyme (Atg10) and finally conjugated to its target Atg5. The second ubiquitin conjugation system promotes the C-terminal processing of the ubiquitin-like protein Atg8 and its eventual attachment to phosphatidylethanolamine (PE). Atg8 processing occurs by means of its initial cleavage by Atg4, activation by the E1 enzyme Atg7 and transfer to the E2 enzyme Atg3, which lipidates Atg8.7,8 Unlike most of the other known autophagy gene products, a population of Atg8 remains associated with the autophagosome and is degraded in the lysosome.9 This unique localization pattern of Atg8 has made it an excellent tool for monitoring the formation of autophagosomes in multiple organisms including S. cerevisiae,10,11 A. thaliana,12 D. melanogaster13 and mice,14 and also in cell culture.9
Humans have approximately seven Atg8 homologs, including microtubule-associated protein 1 light chain 3 (LC3) isoforms A, B and C, gamma-aminobutyric acid A receptor-associated protein (GABARAP) and its L3 isoform, Golgi-associated ATPase enhancer of 16 kDa (GATE-16) and Atg8-like protein (ATG8L),15 and the best studied of these is LC3. LC3 is conjugated to PE in response to starvation conditions or chemical treatments that upregulate autophagy. GABARAP shows many similarities with LC3 but its conjugation is only mildly affected by starvation, and under certain conditions conjugation may be activated independent of mTOR inactivation.16,17 GATE-16 is proposed to be involved in intra-Golgi transport and postmitotic Golgi reassembly.18 ATG8L is the least studied homolog, and the function of GATE-16 and ATG8L in autophagy is not well understood.
Whereas the study of autophagy in multiple organisms has proceeded at a rapid pace over the last decade,19 little autophagy research has been performed in zebrafish. This paucity is likely due to the current lack of tools to monitor autophagy in this organism. To expand in vivo research of autophagy in zebrafish we have created transgenic GFP-Lc3 and GFP-Gabarap fish.
Results
Zebrafish Lc3 is converted to form II at the pharyngula stage during embryogenesis
Yeast Atg8 and its mammalian homolog LC3 specifically label the growing phagophores and completed autophagosomes.9,23 GABARAP, a second mammalian homolog of Atg8, is another modifier in lipidation reactions mediated by Atg7, Atg3 and Atg4 in the same manner as Atg8 and LC3, and localizes to autophagosome membranes when overexpressed.16 In order to generate a marker protein for autophagosomes in zebrafish, we cloned cDNAs of two zebrafish homologs of Atg8. Both genes are predicted to encode a protein of 122 amino acids, which undergoes cleavage at the C terminus (Fig. 1A and B). Based on phylogenetic analysis and sequence homology, we named them Lc3 and Gabarap, with Lc3 more closely related to mammalian LC3 (91% identity) and Gabarap to mammalian GABARAP (94% identity) (Fig. 1C). Both proteins showed a high degree of homology to yeast Atg8 (Lc3 32%, and Gabarap 52% identity).
Figure 1.
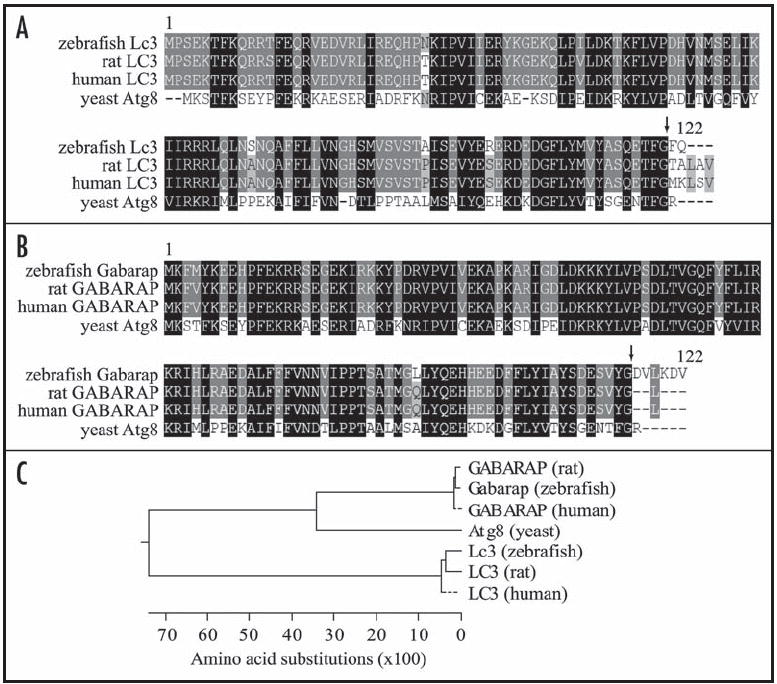
Alignment of zebrafish Lc3 (A) or Gabarap (B) with yeast and mammalian homologs. Protein sequences from S. cerevisiae (yeast), D. rerio (zebrafish), R. norvegicus (rat) and H. sapiens (human) were aligned using the ClustalW program. Amino acid identities, and high and low similarities are highlighted in black, dark gray and light gray, respectively. Arrows indicate the potential cleavage and lipidation site. (C) Phylogenetic tree of Atg8 homologs in yeast, zebrafish and mammals.
To determine when autophagy might be induced in embryos during development, we first analyzed the temporal expression pattern of Lc3 and Gabarap by RT-PCR. lc3 and gabarap transcripts were detected at the early cleavage stage (1–2 cell, 0 hours post-fertilization (hpf)) (Fig. 2A), indicating that both lc3 and gabarap mRNA are maternally deposited, since transcription of zebrafish zygotic DNA does not start until 3 hpf.24 To investigate whether the presence of lc3 transcripts corresponded to expression of the protein and if conjugation of Lc3 occurred, we examined the appearance of Lc3 with an antimammalian LC3 antibody that cross-reacts with the zebrafish homolog. We were able to detect a 16 kDa band that presumably corresponds to the Lc3-I form and a 14 kDa band that is equivalent to Lc3-II (Fig. 2B). In contrast to mouse embryos, in which LC3 conversion is observed even in metaphase II oocytes,25 the conversion of Lc3-I to Lc3-II was evident at 48 hpf but not at 24 hpf. In addition, the total expression level of Lc3 increased in 48 hpf embryos compared to 24 hpf. The earliest time point at which we observed conversion to the Lc3-II form was at 32 hpf (data not shown), suggesting that autophagy is upregulated in zebrafish embryos at the pharyngula period. One possible explanation for the delay in autophagy induction is that other proteins essential for autophagy are not yet produced. In support of this hypothesis we found that although lc3 and beclin 1 are maternally deposited, some other predicted autophagy-related genes in zebrafish start transcription at, or after, approximately 24 hpf (Fig. 2C). In particular, mRNA transcripts for the zebrafish homologs of ulk1 and atg9 were not detected at 0 hpf, indicating that they were not maternally deposited, whereas beclin 1 transcripts were detected at 0 hpf similar to lc3.
Figure 2.
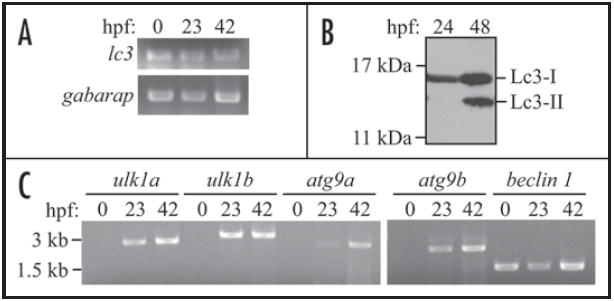
The zebrafish LC3 homolog Lc3 undergoes post-translational modification during embryonic development. (A) lc3 and gabarap mRNAs are maternally deposited in zebrafish embryos. RT-PCR was performed with RNA isolated from 0, 23 and 42 hpf wild-type embryos using gene-specific primers. (B) Lc3-I converts to Lc3-II after 24 hpf. Protein extracts were isolated from 24 and 48 hpf wild-type embryos, analyzed by SDS-PAGE and detected using anti-LC3 antibody. (C) Some autophagy-related genes start transcription at or after 23 hpf. RT-PCR was performed with RNA isolated from 0, 23, and 42 hpf wild-type embryos using specific primers to the zebrafish ulk1a, ulk1b, atg9a, atg9b and beclin 1 genes.
Lc3-I to Lc3-II conversion is enhanced in the presence of lysosomal inhibitors
Having established that the Lc3 protein is expressed and that it undergoes post-translational modification similar to its mammalian and yeast homologs during zebrafish embryonic development, we decided to study the extent of autophagy during embryogenesis by analyzing Lc3-II conversion. To evaluate the basal level of autophagy, we utilized two lysosomal protease inhibitors, pepstatin A, an inhibitor of cathepsins D and E, and E64d, an inhibitor of cathepsins B, H and L.26 At the concentration and time used in this analysis, these inhibitors imposed no readily observable effects on embryo viability when applied in the embryo water (data not shown). We treated two days postfertilization (dpf) embryos with the above drugs for 24 hours, and observed the accumulation of Lc3-II (Fig. 3). Treatment with lysosomal protease inhibitors alone resulted in a substantial increase in the Lc3-II to Lc3-I ratio, suggesting that constitutive autophagy occurred at a high basal level during normal embryogenesis; since Lc3-II is turned over in the lysosome, stabilization of the protein when lysosomal degradation is blocked is an indication of autophagic flux.27
Figure 3.
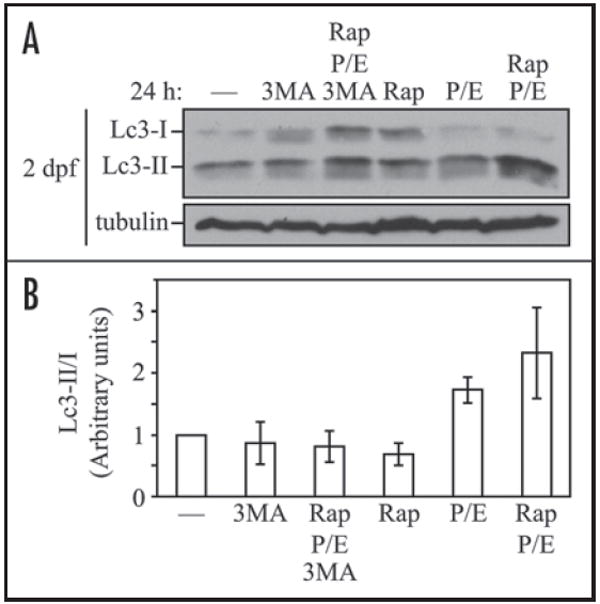
Lc3-II accumulates in response to rapamycin and lysosomal inhibitor treatment. (A) GFP-Lc3 embryos at 2 dpf were treated with the indicated chemicals or the solvent DMSO (—) for 24 h. Protein extracts were analyzed by SDS-PAGE and detected using anti-LC3, or anti-tubulin antibody as a loading control. (B) The Lc3-II to Lc3-I ratio in (A) was quantified using ImageJ software (http://rsb.info.nih.gov/ij/). The value for DMSO-treatment was set to 1.0 and other values were normalized. Error bars represent the standard deviation (s.d.) of three independent experiments. Rap, rapamycin; 3MA, 3-methyladenine; P/E, pepstatin A and E64d.
Starvation is a known inducer of autophagy; however, because the embryonic yolk is a stable nutrient supply during embryogenesis, we tested rapamycin, a TOR inhibitor that is widely used to induce autophagy in yeast and mammalian cells by mimicking starvation conditions.28 With a final concentration of 1 μM rapamycin in embryo water, which has been previously shown to inhibit the TOR signaling pathway in zebrafish embryos and larvae but not cause cell death,29 we observed no detectable change in the Lc3-II/Lc3-I ratio after 24 hours of treatment (Fig. 3A and B); this may be explained by the possibility that rapamycin treatment enhanced the overall autophagic flux, and Lc3-II was rapidly degraded in lysosomes under these conditions.27 Therefore, we simultaneously treated embryos with rapamycin and lysosomal protease inhibitors, and in this case observed an increase in Lc3-II levels. To determine if the change in Lc3-II level was dependent on autophagy machinery, we used 3-methyladenine (3-MA), which inhibits the class III phosphatidylinositol 3-kinase (PtdIns3K) and autophagosome formation. 3-MA treatment blocked the increase in the Lc3-II/Lc3-I ratio seen with rapamycin in the presence of lysosomal protease inhibitors, confirming that the Lc3-II upregulation was a consequence of autophagy (Fig. 3).
Generation of GFP-Lc3 and GFP-Gabarap transgenic zebrafish
To extend our in vivo analysis of autophagy in live embryos, we generated transgenic zebrafish lines expressing an N-terminal green fluorescent protein (GFP)-fused Lc3 (GFP-Lc3) under the control of the constitutive CMV (human cytomegalovirus) promoter (Fig. 4A). The transgenic line showed no developmental or adulthood abnormalities. GFP-Lc3 transgene expression was visualized from the early cleavage stage and throughout adulthood (Fig. 4B). The expression of GFP-Lc3 was observed in various tissues, with the highest expression in muscle, lens and spinal cord. We also generated a GFP-Gabarap transgenic line (Fig. 4), to study whether Gabarap participates in autophagy in vivo. The GFP-Gabarap transgene showed a similar expression pattern as GFP-Lc3.
Figure 4.
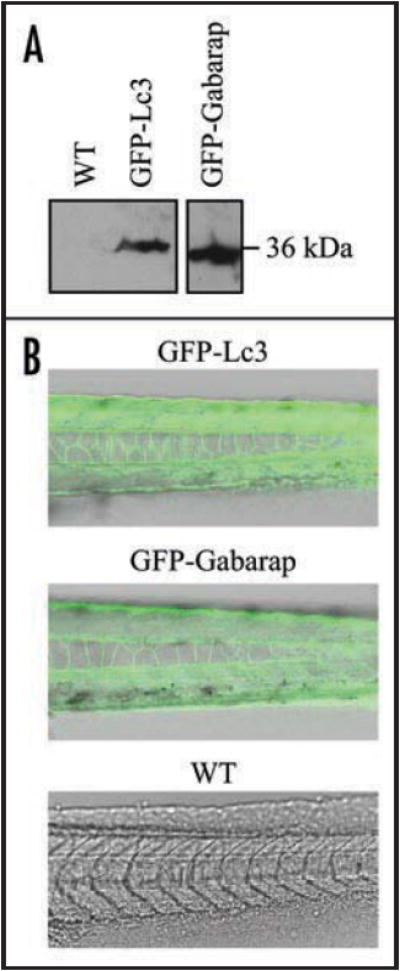
Generation of GFP-Lc3 and GFP-Gabarap transgenic zebrafish. (A) GFP-Lc3 and GFP-Gabarap transgene expression was confirmed by western blot of protein lysates from GFP-Lc3, GFP-Gabarap or nontransgenic wild-type (WT) embryos using anti-GFP antibody. (B) GFP-Lc3, GFP-Gabarap or nontagged wild-type embryos were observed by confocal fluorescence microscopy and merged with bright-field images.
Basal autophagy in zebrafish embryos can be further upregulated by rapamycin or calpeptin
Using the GFP-Lc3 line, we examined autophagic responses in zebrafish embryos. We found that LysoTracker Red, which accumulates in acidic organelles in living cells, could successfully stain lysosomes in live embryos when supplied in water (Fig. 5A). Embryos at the hatching period contained a number of lysosomes, which were further induced upon rapamycin treatment. Even though basal levels of autophagy were easily detected by western blot of Lc3 (Fig. 3), we observed very few GFP-Lc3 punctate dots (indicative of autophagosomes) prior to drug treatment, whereas following rapamycin treatment the GFP-Lc3 puncta increased; however, these puncta did not display extensive colocalization with LysoTracker Red. When lysosomal protease inhibitors were also added, the total number of puncta increased as did the extent of colocalization. This result suggested that although formation of GFP-Lc3 puncta could be induced by rapamycin alone, the chimeric protein was degraded within the lysosome except in the presence of lysosomal protease inhibitors, again indicating the occurrence of autophagic flux. The colocalization of GFP-Lc3 puncta with lysosomes was also dramatically increased when embryos were treated with pepstatin A and E64d alone, supporting the idea that basal autophagic flux is high in embryos. In addition, 3-MA suppressed the accumulation of Lc3-positive puncta in the presence of lysosomal inhibitors and rapamycin, whereas lysosomes in embryos seemed not affected by 3-MA (Fig. 5A). We further quantified the colocalization of Lc3 puncta with lysosomes (Fig. 5B), and the results suggest that formation of GFP-Lc3 puncta correlates with conversion of endogenous Lc3-I to Lc3-II (Fig. 3).
Figure 5.
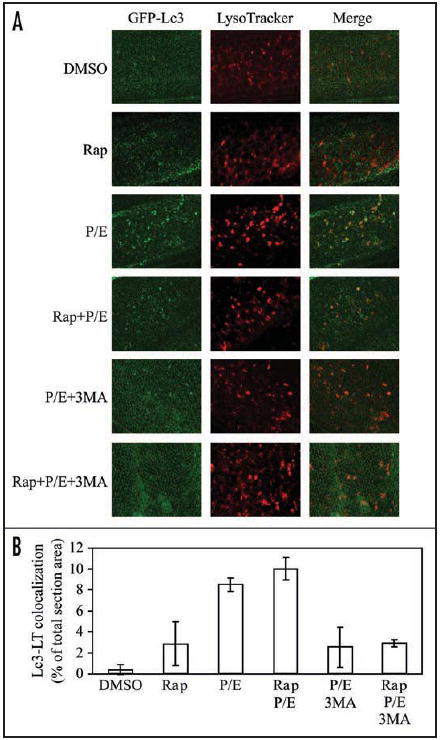
GFP-Lc3 puncta formation and lysosome colocalization are induced by rapamycin and lysosomal inhibitors. (A) GFP-Lc3 embryos at 3 dpf were treated with the indicated chemicals or DMSO for 24 h, and then stained with LysoTracker Red for 1 h before imaging by confocal fluorescence microscopy. (B) Colocalization of GFP-Lc3 puncta with LysoTracker Red (LT) in (A) was quantified using Leica Simulator SP5 software. Rap, rapamycin; 3MA, 3-methyladenine; P/E, pepstatin A and E64d.
A recent study revealed a TOR-independent pathway that can enhance autophagy.30 In this case, adenylyl cyclase produces cyclic AMP, which increases intracellular Ca2+ levels and calpain activity. This further results in cleavage and activation of the small G protein Gsα, which in turn activates adenylyl cyclase causing a loop that inhibits autophagy. Various chemicals have been tested to target different steps in this pathway to induce autophagy, including 2’5’-ddA (adenylyl cyclase inhibitor), verapamil (L-type Ca2+ channel antagonist), calpeptin (calpain inhibitor) and clonidine (Gi protein activator). We decided to examine the effects of these chemical inducers on autophagy in zebrafish embryos. The calpain inhibitor calpeptin appeared to be very potent for inducing formation of Lc3-positive puncta and autolysosomes, as judged by the colocalization of GFP-Lc3 and LysoTracker Red (Fig. 6A and B). Conversion of endogenous Lc3 was also upregulated by calpeptin (Fig. 6C). The other treatments had less dramatic (2’5’-ddA and verapamil) or essentially no effect (clonidine) on autophagy at the concentrations and times used in this analysis. We also confirmed the conversion of the GFP-Lc3 fusion protein by examining embryos treated with rapamycin or calpeptin, although GFP-Lc3 was overexpressed and largely present as the unconjugated form (Fig. 6D). However, the level of GFP-Lc3-II was dramatically increased when rapamycin or calpeptin were added in the presence of lysosomal protease inhibitors (Fig. S1), in agreement with our previous results examining Lc3-II levels (Fig. 3). Finally, compared with the no treatment control, the concentrations and time used in the chemical treatments did not induce apparent apoptotic cell death, as assessed by staining with the vital dye acridine orange (Fig. S2). Thus, we established two pharmacological strategies to effectively upregulate autophagy in zebrafish embryos, that is, targeting either TOR or a TOR-independent autophagy pathway by rapamycin or calpeptin, respectively.
Figure 6.
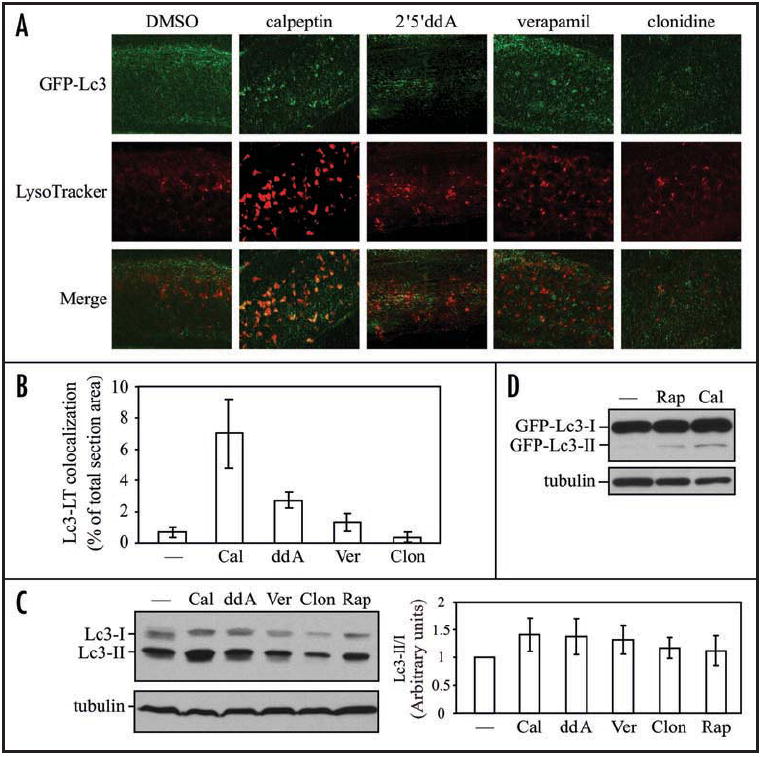
Autophagy is upregulated in response to chemical inducers functioning independently of the TOR pathway. (A) GFP-Lc3 embryos at 3 dpf were treated with the indicated drugs or DMSO for 24 h, and then stained with LysoTracker Red for 1 h before imaging by confocal fluorescence microscopy. (B) Colocalization of GFP-Lc3 dots with LysoTracker Red (LT) in (A) was quantified using Leica Simulator SP5 software. (C and D) Wild-type (C) or GFP-Lc3 (D) embryos at 2 dpf were treated with the indicated chemicals or the solvent DMSO (—) for 24 h. Protein extracts were analyzed by SDS-PAGE and detected by western blot using anti-LC3, or anti-tubulin antibody as a loading control. (C, right) The relative increase of the Lc3-II/Lc3-I ratio induced by chemicals was quantified by ImageJ software (http://rsb.info.nih.gov/ij/) and represented as mean ± s.d. of three independent experiments. The value of Lc3-II/Lc3-I from the DMSO-treated sample was set to 1.0 and other values were normalized. Rap, rapamycin; Cal, calpeptin; ddA, 2’5’-dideoxyadenosine; Ver, verapamil; clon, clonidine.
Gabarap accumulates within lysosomes under autophagy-inducing conditions
Whether another mammalian Atg8 homolog, GABARAP, is involved in autophagy is controversial. One paper shows that overexpressed mammalian GABARAP is subject to lipidation by the same machinery as LC3 and localizes to autophagosome membranes under starvation conditions,16 whereas another group reports that puncta formation and lysosomal turnover of endogenous GABARAP is not activated during starvation-induced autophagy.17 Notably, different cell culture lines were used in these reports.
In order to address this discrepancy in vivo, we analyzed the localization of Gabarap under autophagy-inducing conditions using the GFP-Gabarap transgenic embryos. We treated 3 dpf embryos with rapamycin and lysosomal inhibitors for 24 h, and stained them with LysoTracker Red. Rapamycin triggered GFP-fused Gabarap puncta formation and translocation to lysosomes, which were enhanced in the presence of lysosomal protease inhibitors (Fig. 7A and B). However, puncta formation of Gabarap was not as significant as that of Lc3, although colocalization with LysoTracker Red was similar, indicating that Gabarap may localize to a lesser degree than Lc3 on autophagosomal membranes (Figs. 5B versus 7B). In addition, western blotting showed that the GFP-Gabarap fusion protein possessed two forms, and the conversion from form I to II was also induced by calpeptin (Fig. 7C). We found that there was evidence for a low level of autophagic flux based on treatment with lysosomal protease inhibitors, suggesting that the overall rate of turnover was quite low. Therefore, in zebrafish embryos, Gabarap was likely turned over constitutively, but at a low level, within lysosomes, and during autophagy induction its lysosomal accumulation increased.
Figure 7.
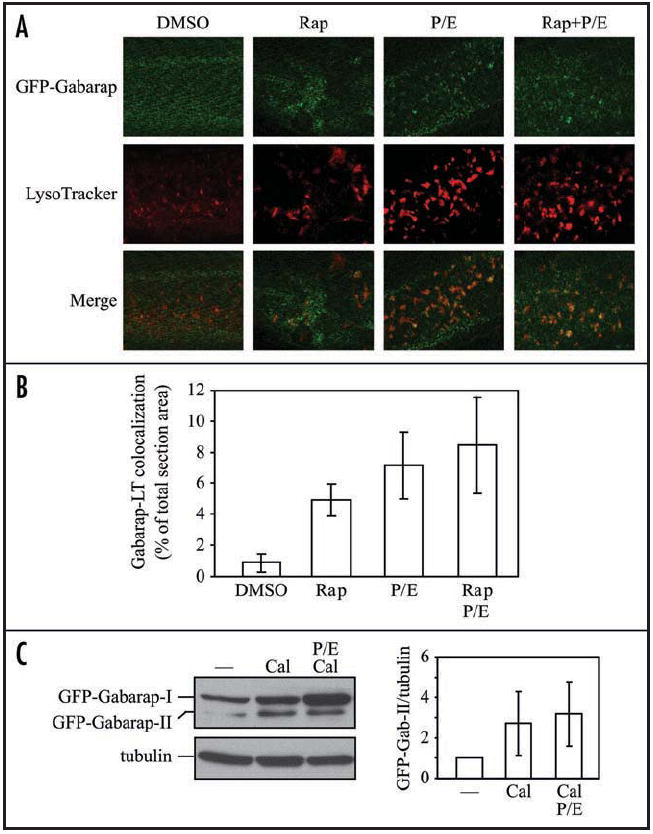
Gabarap accumulates within lysosomes after treatment with rapamycin and lysosomal inhibitors. (A) GFP-Gabarap embryos at 3 dpf were treated with the indicated chemicals or DMSO for 24 h, and then stained with LysoTracker Red for 1 h before imaging by confocal fluorescence microscopy. (B) Colocalization of GFP-Gabarap puncta with LysoTracker Red (LT) in (A) was quantified using Leica Simulator SP5 software. (C, left) GFP-Gabarap embryos at 2 dpf were treated with the indicated chemicals or the solvent DMSO (—) for 24 h. Protein extracts were analyzed by SDS-PAGE and detected using anti-GABARAP, or anti-tubulin antibody as a loading control. (C, right) The relative increase of the GFP-Gabarap-II/tubulin ratio induced by chemicals was quantified by ImageJ software (http://rsb.info.nih.gov/ij/). The value of GFP-Gabarap-II versus tubulin from the DMSO-treated sample was set to 1.0 and other values were normalized. Error bars represent the s.d. of three independent experiments. Rap, rapamycin; Cal, calpeptin; P/E, pepstatin A and E64d.
Discussion
Due to their optical clarity and short development time, zebrafish are ideal for studying development. Through a BLAST search, we found that homologs of multiple autophagy-related genes, including ulk1/atg1, atg2, atg3, atg4, atg5, beclin 1/atg6, atg7, lc3/atg8, atg9, atg12 and atg16 are present in the zebrafish genome, which suggests that this organism may be useful as a model system for studying the function and mechanism of autophagy. It has recently been reported that autophagy is required in mice for preimplantation embryonic development; autophagosomes are observed in GFP-LC3 mouse embryos at the one- to four-cell stage, and autophagy-defective embryos fail to develop beyond the four- and eight-cell stage.25 In contrast to zebrafish development that proceeds quickly after fertilization, mammalian development progresses relatively slowly. It is unknown if autophagy is required during the one- to four-cell stage in nonmammalian vertebrates such as fish, amphibians, or birds; however, it is unlikely that zebrafish undergo autophagy during the 1–4 cell stages as ulk1a, ulk1b, atg9a and atg9b (genes that are presumably required for autophagy as they are in other organisms) were not expressed in 0 hpf embryos (Fig. 2C). However, by 24 hpf ulk1a, ulk1b, atg9b and to a lesser extent atg9a transcripts were easily detected. PE-conjugated Lc3 was not seen in 24 hpf embryos, but Lc3-II was clearly observed by 48 hpf. The appearance of transcripts for genes necessary for autophagy by 24 hpf, the subsequent appearance of PE-conjugated Lc3 by 48 hpf, and the high basal level of autophagy by the 48 hpf time point opens the possibility that autophagy may play a role in zebrafish development during later developmental stages.
In recent years autophagy has been shown to play multiple important roles in human health and pathology.4,5,31 Therefore, it is likely that chemical screens for compounds that regulate autophagy, and subsequent dissection of the mechanisms involved, will become an important area of research. Indeed a screen using a panel of 253 compounds was recently performed to identify drugs that induce autophagy.30 As a secondary screen these researchers used a zebrafish Huntington disease model to determine if these drugs caused clearance of aggregate-prone proteins, but they did not directly show autophagy induction in zebrafish.30 Here, we have shown for the first time that aqueous administration of rapamycin, calpeptin and 2’5’-ddA, and to a lesser extent verapamil, induce autophagy (Figs. 3 and 6). Our findings not only provide the first instance of monitoring chemical upregulation of autophagy in zebrafish, but also corroborate the hypothesis that these drugs induce autophagy in this system. Because drug administration can be achieved simply by aqueous exposure, zebrafish would be an ideal organism for primary or secondary in vivo small molecule screens for compounds that regulate autophagy.32,33
The optical transparency of zebrafish combined with their external development make them an ideal organism for in vivo microscopy analysis, especially during the developmental period. We therefore have created transgenic GFP-Lc3 and GFP-Gabarap zebrafish lines. Both GFP-Lc3 and GFP-Gabarap accumulate in lysosomes in response to drug treatment, suggesting these GFP-tagged Atg8 homologs are incorporated into autophagosomes as found in other organisms. These fish will provide an essential tool for the study of autophagy by allowing monitoring of autophagosome formation in this model system as has been done in other organisms.
Materials and Methods
Zebrafish strains and maintenance
The AB and local strains of wild-type fish were used in this study. Embryos were collected from pairwise matings of adults and raised at 28.5°C. Larvae were staged according to morphological features previously described.20
Cloning and RT-PCR
Total RNA was extracted from zebrafish embryos using Trizol (Invitrogen, 15596026). Reverse transcription was performed using oligo-dT and SuperScript II (Invitrogen, 18064014), and PCR reactions using the Phusion High Fidelity DNA polymerase (New England Biolabs, F530L). Zebrafish lc3, gabarap, ulk1a, ulk1b, atg9a, atg9b and beclin 1 were identified by a BLAST search of the zebrafish genomic database with mammalian homologs of autophagy genes. RT-PCR primers were designed based on the BLAST search results and the products were sequenced to confirm the homology. Primer sequences are available upon request.
Generation of transgenic zebrafish
gabarap was cloned into the pEGFP-C1 vector (Clontech, 6084-1) using EcoRI and KpnI. The fragment containing the CMV promoter, GFP-Gabarap and the polyA sequence was then released using AseI and MluI, blunted by the Klenow enzyme and ligated into the pminiTol2 vector linearized by EcoRV, resulting in pminiTol2-GFP-Gabarap. For construction of pminiTol2-GFP-Lc3, the gabarap gene was replaced by lc3 introduced into the vector using XhoI and XmaI.
pT3TS-Tol2 was linearized by XbaI and transcribed with T3 RNA polymerase using the Ambion mMESSAGE mMACHINE® kit (Ambion, AM1348) to produce Tol2 transposase mRNA. Approximately 25 ng of plasmid DNA (pminiTol2-GFP-Lc3 or pminiTol2-GFP-Gabarap) and 100 ng of Tol2 mRNA were coinjected into newly fertilized embryos at the one-cell stage to produce transgenic fish. Injected embryos were raised to adulthood and out-crossed to wild-type fish to identify transgenic founders.
Immunoblotting
Embryos were dechorionated, deyolked and homogenized in SDS loading buffer. After being boiled at 95°C for 5 min, embryo lysates were resolved on 10% or 15% gels and probed by western blot with anti-LC3 (Novus Biologicals, NB100-2331), anti-GABARAP21 or anti-tubulin (Sigma, T6793) antibodies. The specificity of the LC3 antibody NB100-2331 to Lc3 was confirmed by detecting GFP-Lc3 with both NB100-2331 and an anti-GFP antibody.
Drug treatment
Drug treatment was performed in 24-well plates. The following chemicals dissolved in DMSO were added to the embryo water at the final concentrations indicated in parentheses: rapamycin (1 μM), calpeptin (50 μM), 2’5’-dideoxyadenosine (2’5’-ddA; 100 μM), verapamil (3 μM), clonidine (3 μM), pepstatin A (10 μg/ml) and E64d (5 μg/ml). 3-methyladenine was dissolved in water as a 0.33 M stock and used at a final concentration of 10 mM.
LysoTracker assay
Embryos were incubated for 1 h in water with 10 μM LysoTracker Red (Invitrogen, L7528), and rinsed several times with fresh water before imaging by confocal microscopy.
Acridine orange staining assay
Postfertilization embryos at 2 days were subject to 24 h-chemical treatment and stained with acridine orange as previously reported.22 Four embryos from each treatment were collected, washed three times with fresh water and imaged on an Olympus IX71 fluorescence microscope using the GFP filter at a magnification of 20X.
Confocal fluorescence microscopy
Embryos were transferred into 0.003% (w/v) 1-phenyl-2-thiourea prior to 24 hours postfertilization to prevent pigmentation. They were mounted live in water containing 0.16 mg/ml tricaine (Sigma, A5040) for imaging. Images were taken using the Leica SP5 confocal system under a 40X water immersion lens.
Quantitative analyses of colocalization of GFP-Lc3 or GFP-Gabarap with LysoTracker Red signals were performed with images using the Leica Simulator SP5 software. Since GFP-Lc3 and GFP-Gabarap showed both a uniform cytosolic signal and more intense spots, threshold values were set to reduce the cytosolic signal and identify the more intense dots. The same threshold value was applied for all samples in an indicated experiment. The amount of colocalization between LysoTracker Red signals and GFP-Lc3 or GFP-Gabarap dots was quantified in three independent visual fields from three independent embryos. The Y-axis represents the percentage of the colocalization area per visual field. All values are represented as mean ± standard deviation (s.d.).
Supplementary Material
Acknowledgments
We thank Drs. Takashi Ueno and Eiki Kominami (Juntendo University, Japan) for the anti-GABARAP antibody, Drs. David Ginsburg and Friedhelm Hildebrandt for providing zebrafish facilities, Dr. Gregg Sobocinski for technical assistance with the confocal microscope, Dr. Jordan Shavit for technical assistance with zebrafish, and members of the Klionsky lab for helpful discussions and comments. We also owe our great thanks to Dr. Stephen Ekker (Mayo Clinic), for kindly providing us the pminiTol2 and pT3TS-Tol2 plasmids. This work was supported by a Rackham Predoctoral Fellowship from the University of Michigan to C.H., and National Institutes of Health Public Health Service grant GM53396 to D.J.K.
Footnotes
Note Supplementary materials can be found at: www.landesbioscience.com/supplement/HeAUTO5-4-Sup.pdf
References
- 1.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 3.Martinet W, Knaapen MW, Kockx MM, De Meyer GR. Autophagy in cardiovascular disease. Trends Mol Med. 2007;13:482–91. doi: 10.1016/j.molmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6:1837–49. doi: 10.4161/cc.6.15.4511. [DOI] [PubMed] [Google Scholar]
- 6.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 7.Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008;9:859–64. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–92. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 9.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Huang W-P, Klionsky DJ. Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p and the autophagy conjugation complex. J Cell Biol. 2001;152:51–64. doi: 10.1083/jcb.152.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- 12.Yoshimoto K, Hanaoka H, Sato S, Kato T, Tabata S, Noda T, et al. Processing of ATG8s, ubiquitin-like proteins, and their deconjugation by ATG4s are essential for plant autophagy. Plant Cell. 2004;16:2967–83. doi: 10.1105/tpc.104.025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rusten TE, Lindmo K, Juhász G, Sass M, Seglen PO, Brech A, et al. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7:179–92. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP and Apg8L. J Biol Chem. 2003;278:51841–50. doi: 10.1074/jbc.M308762200. [DOI] [PubMed] [Google Scholar]
- 16.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–12. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 17.Tanida I, Wakabayashi M, Kanematsu T, Minematsu-Ikeguchi N, Sou YS, Hirata M, et al. Lysosomal turnover of GABARAP-phospholipid conjugate is activated during differentiation of C2C12 cells to myotubes without inactivation of the mTor kinase-signaling pathway. Autophagy. 2006;2:264–71. doi: 10.4161/auto.2871. [DOI] [PubMed] [Google Scholar]
- 18.Sagiv Y, Legesse-Miller A, Porat A, Elazar Z. GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO J. 2000;19:1494–504. doi: 10.1093/emboj/19.7.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 20.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 21.Tanida I, Komatsu M, Ueno T, Kominami E. GATE-16 and GABARAP are authentic modifiers mediated by Apg7 and Apg3. Biochem Biophys Res Commun. 2003;300:637–44. doi: 10.1016/s0006-291x(02)02907-8. [DOI] [PubMed] [Google Scholar]
- 22.Tucker B, Lardelli M. A rapid apoptosis assay measuring relative acridine orange fluorescence in zebrafish embryos. Zebrafish. 2007;4:113–6. doi: 10.1089/zeb.2007.0508. [DOI] [PubMed] [Google Scholar]
- 23.Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–46. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelegri F. Maternal factors in zebrafish development. Dev Dyn. 2003;228:535–54. doi: 10.1002/dvdy.10390. [DOI] [PubMed] [Google Scholar]
- 25.Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117–20. doi: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- 26.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 27.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 29.Makky K, Tekiela J, Mayer AN. Target of rapamycin (TOR) signaling controls epithelial morphogenesis in the vertebrate intestine. Dev Biol. 2007;303:501–13. doi: 10.1016/j.ydbio.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinet W, Knaapen MW, Kockx MM, De Meyer GR. Autophagy in cardiovascular disease. Trends Mol Med. 2007;13:482–91. doi: 10.1016/j.molmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Berger J, Currie P. The role of zebrafish in chemical genetics. Curr Med Chem. 2007;14:2413–20. doi: 10.2174/092986707781745532. [DOI] [PubMed] [Google Scholar]
- 33.Redfern WS, Waldron G, Winter MJ, Butler P, Holbrook M, Wallis R, et al. Zebrafish assays as early safety pharmacology screens: Paradigm shift or red herring? J Pharm Toxic Meth. 2008;58:110–7. doi: 10.1016/j.vascn.2008.05.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.