

Abstract
During sleep, the mammalian CNS undergoes widespread, synchronized slow-wave activity (SWA) that directly varies with previous waking duration (Borbély, 1982; Dijk et al., 1990). When sleep is restricted, an enhanced SWA response follows in the next sleep period. The enhancement of SWA is associated with improved cognitive performance (Huber et al., 2004), but it is unclear either how the SWA is enhanced or whether SWA is needed to maintain normal cognitive performance. A conditional, CNS knock-out of the adenosine receptor, AdoA1R gene, shows selective attenuation of the SWA rebound response to restricted sleep, but sleep duration is not affected. During sleep restriction, wild phenotype animals express a rebound SWA response and maintain cognitive performance in a working memory task. However, the knock-out animals not only show a reduced rebound SWA response but they also fail to maintain normal cognitive function, although this function is normal when sleep is not restricted. Thus, AdoA1R activation is needed for normal rebound SWA, and when the SWA rebound is reduced, there is a failure to maintain working memory function, suggesting a functional role for SWA homeostasis.
Keywords: sleep, delta, adenosine, working memory, hippocampal function, memory, Cre-transgenic, metabolism
Introduction
The need for recovery of lost sleep, the homeostatic sleep response, is considered one of the universal characteristics of sleep (Mignot, 2008). One commonly used measure of this response is slow-wave activity (SWA) (Dijk et al., 1990). SWA is measured from the electroencephalogram and results from synchronized, neuronal membrane potential fluctuations of populations of cortical and thalamocortical neurons, with a dominant frequency component in the range of 0.5–4.5 Hz (Amzica and Steriade, 1998). Slow-wave activity is present across states but is predominate in the EEG during slow-wave sleep (SWS), also called non-rapid eye movement sleep, and in this sense, SWS may be considered permissive to the expression of SWA.
The magnitude of SWA is directly correlated with the time spent awake before the SWS episode (Franken et al., 2001) and may be locally increased in a use-dependent manner (Huber et al., 2004; Vyazovskiy et al., 2004). Thus, SWA is considered a marker for and is thought to be functionally involved in sleep/waking homeostasis (Daan et al., 1984).
Adenosine has been hypothesized to be involved in sleep/waking homeostasis by modulation of SWA. Converging evidence suggest that this SWA modulation results from A1 adenosine receptor (AdoA1R) activation. Adenosine, acting on the AdoA1R, has been shown to decrease cholinergic neuronal activity and to facilitate, at the single-cell level, slow oscillations (Pape, 1992; Rainnie et al., 1994; Benington et al., 1995; Porkka-Heiskanen et al., 1997). Pharmacological blockade of adenosine A1 and A2 receptors by caffeine results in increased arousal (and decreased sleep) in mammals (Fredholm et al., 2005). In rodents, the increased arousal effect may, in large part, be secondary to increased locomotor activity (Lindskog et al., 2002), mediated by the A2AdoR signaling system in the striatum that is, in itself, arousing. However, an AdoA1R-mediated action cannot be ruled out despite the evidence that the stimulatory action of caffeine is less marked acting by AdoA1Rs (Huang et al., 2005). Furthermore, the SWA-promoting actions of increased endogenous or exogenous adenosine in the cholinergic nuclei (Porkka-Heiskanen et al., 1997; Portas et al., 1997) seems most likely to be mediated by AdoA1Rs because AdoA2aRs have no electrophysiological activity in the cholinergic nuclei, in contrast to AdoA1Rs (Arrigoni et al., 2001). Thus, the physiological role of AdoA1Rs to increase SWA remains to be resolved.
We have used conditional AdoA1R knock-out mice, using a calcium/calmodulin-dependent protein kinase II (CaMKII)-promoting Cre transgene, to selectively reduce rebound SWA in response to moderate sleep restriction, as a means of assessing mechanisms responsible for this classic homeostatic sleep response. The conditional knock-out approach was justified over either the constitutive knock-out or the adenoassociated virus (AAV)-mediated Cre induction approaches for the following reasons. First, the constitutive deletion of the AdoA1R gene showed a minimal reduction of SWA in mice during the early part of the circadian day (at lights on) when sleep pressure is most likely to be the greatest (Stenberg et al., 2003), raising the possibility of developmental compensatory effects. These potential effects would be less of a factor with a conditional gene knock-out (Tsien et al., 1996). Second, although localized increases in adenosine are sufficient to increase SWA (Porkka-Heiskanen et al., 1997; Portas et al., 1997), this may require activation of presynaptic AdoA1R (Arrigoni et al., 2001, 2005; Brambilla et al., 2005). A localized AAV-Cre mediated AdoA1R gene deletion will only affect the neurons within the region of transfection, leaving presynaptic AdoA1Rs on terminals originating from neurons outside the transfected region unaffected.
Evidence is provided here suggesting that the normal rebound increase in SWA in response to sleep restriction is modulated by AdoA1Rs and that this modulation is associated with normal working memory performance under sleep restriction conditions.
Materials and Methods
Conditional gene deletion of AdoA1R.
A targeted gene insertion, using a C57BL/6 mouse and embryonic stem cells from 129SvJ strain, was made with loxP sites flanking the AdoA1R gene (Scammell et al., 2003). AdoA1R gene deletion was induced by crossing these mice with another strain (T50 line) containing a transgene expressing the recombinase, Cre, under the control of a CaMKII promoter (Tsien et al., 1996). The genetic background of the Cre cutter line was also C57BL/6. The AdoA1R−/− line was backcrossed a minimum of three times before use of offspring for experimentation. The promoter provides regional and developmental specificity of the Cre expression and, as a result, of the AdoA1R gene deletion.
During development from postnatal day 10 (P10) to P60, the expression pattern of the Cre recombinase becomes less restricted (Luikart et al., 2005; Monteggia et al., 2007) to include forebrain, diencephalon, and brainstem of the CNS as indicated by β-galactosidase expression in ROSA26 reporter mice (Tsien et al., 1996) crossed with mice containing the CaMKII-Cre transgene (Fig. 1).
Figure 1.

A conditional deletion of the AdoA1R gene. Cre expression in 6- to 8-week-old mice under the control of a CaMKII promoter induces recombination of floxed genes in both the forebrain and the hindbrain. First column, The reporter mouse ROSA26 was used to demonstrate Cre expression and therefore the location and spread of AdoA1R knock-out. Sections from an offspring of a ROSA26 reporter mouse crossed with a CaMKII-Cre mouse were stained with XGal (βGal stain) and neutral red (counterstain). ROSA26 mice express βGal wherever the transgene (in this case Cre) is expressed. The XGal stain is dark blue and apparent in both the cortex and the hindbrain tegmentum, and to a lesser extent in the thalamus. Second column, Autoradiographic label of mRNA message for AdoA1R is from comparable sections in Cre-negative AdoA1Rf/f mice. Third column, Cre-positive AdoA1R−/− mice probed for mRNA message for AdoA1R. AdoA1R message is reduced in regions that both are Cre+ (first column) and show AdoA1R message (compare second and third columns). Together, these results suggest that AdoA1R expression is reduced in most areas of the brain in the AdoA1R−/− mice.
The expression pattern we observed has been previously reported (Monteggia et al., 2007), using similar histological techniques as used in our study. Cre expression was not observed in putative interneurons or in the inhibitory nucleus reticularis of the thalamus. At high-power magnification, β-galactosidase staining was clearly apparent in the nuclei of all the thalamocortical neurons (data not shown), despite its apparent absence of expression in the thalamus in low-power micrographs (Fig. 1, left column). β-Galactosidase expression was concentrated in the nuclei of cells in the ROSA26 mouse, and this appeared especially to be the case for the thalamic neurons.
We used standard autoradiographic techniques to image 35S-labeled mRNA probe for the AdoA1R transcript as described previously (Scammell et al., 2003). The homozygous “floxed” AdoA1R strain (AdoA1Rf/f) has a wild-type expression pattern for AdoA1R message (Fig. 1, middle column). The AdoA1R−/− strain (homozygous for the floxed allele, AdoA1Rf, and positive for the CaMKII-Cre transgene) shows a loss of AdoA1R expression, and this loss was restricted to regions that were positive for both Cre and AdoA1R expression (Fig. 1, right column).
Quantitative densitometry indicates that the AdoA1R message is reduced in the AdoA1R−/− strain by >80% in sleep-relevant areas that include the following: (1) in the brainstem, the dorsal pontine tegmentum; (2) in the diencephalon, the thalamus, with the exception of the inhibitory nucleus reticularis, and the hypothalamus; (3) in the forebrain, the parietal neocortex. Using the identical AdoA1Rf/f strain, we have previously shown that, wherever Cre is expressed, there is an associated loss of AdoA1R-dependent electrophysiological responses (Scammell et al., 2003; Arrigoni et al., 2005).
Housing and surgical procedures.
Adult male AdoA1Rf/f and AdoA1R−/− mice aged 9–16 weeks were housed singly in standard mouse cages with access to food and water ad libitum. Cages were located in a climate-controlled environment with a 12 h light/dark schedule with lights on at 8:00 A.M. To determine sleep/waking activity, EEG and EMG electrodes were surgically implanted. Briefly, mice were anesthetized with isoflurane and placed in a Kopf stereotax. The scalp was sheared, cleaned, and incised. Four holes were drilled for EEG electrodes, which consisted of a piece of coated wire capped by a gold amphenol pin on one side (Plastics One) and a small screw on the other. Two electrodes were placed bilaterally over the frontal area [anteroposterior (AP), +1.1 mm; mediolateral (ML), ±1.45 mm], and two electrodes were placed bilaterally over the parietal area (AP, −3.5 mm; ML, ±1.45 mm). Two panel EMG electrodes (Plastics One) were placed bilaterally into the nuchal musculature. The electrode caps were threaded into a six-pin pedestal (Plastics One) and affixed to the skull with dental acrylic (Thermo Fisher Scientific). After surgery, the base of the implant was coated with antibiotic to prevent infection and the animals were given buprinex (0.1 ml, i.p.) for pain relief. Animals were given 10 d to recover from surgery before any experimental manipulations. All experimental procedures were approved by the North Texas Veterans Administration Institutional Animal Care and Use Committees.
EEG/EMG activity recording procedures and sleep restriction.
After 10 d of recovery from surgical procedures, cages were placed in custom wooden boxes. A lightweight cable (Plastics One) was secured to the implant and to a commutator (Plastics One) located on the ceiling of the box. The commutator allows for relatively free movement and prevents the cable from tangling. Animals were given at least 5 d to acclimate to the cable. Next, animals were moved to cages that were bottomless and suspended ∼0.125 inch above a treadmill (Fig. 2). While on the treadmill, the lightweight cable was attached to a counterbalanced lever arm (Instech Laboratories) that allowed for relatively free movement and prevented tangling. Animals were given 2 d to acclimate to the treadmill environment before baseline recording. Baseline sleep/waking activity was measured for 24 h, followed by 48 h of sleep restriction. For sleep restriction, the treadmill was engaged for 48 more hours with a cycle of 4 h on (TM on) and 2 h off (TM off) for a total of eight complete 6 h cycles. This treadmill was remotely controlled by a computer programmed to implement the 6 h cycles. The treadmill speed was set at 3 cm/s because this was the slowest speed that reliably eliminated sleep (as assessed using EEG and EMG sleep scoring criteria) throughout the 48 h restricted sleep period. When the treadmill was moving, urine and feces were swept under the cage, and the feces dropped onto a trash bag under the belt. The treadmill was cleaned between sets of animals. A camera was placed over the treadmill and connected to a monitor in a separate room to allow remote observation.
Figure 2.

Sleep restriction apparatus. To sleep restrict mice, a 6 h cycle composed of 4 h of enforced waking and 2 h of uncontrolled behavioral state (4 h on and 2 h off) was used. Mice were placed in a Plexiglas cage with no bottom, ¼ inch above a treadmill that moved at a speed of 3 cm/s. Food and water tubes were placed in each cage.
A Grass model 15 data acquisition system (Astro-Med) was used to record EEG/EMG activity. An electrode board (Astro-Med) was used to reference the left frontal lead to the left parietal lead, the right frontal lead to the right parietal lead, and the left nuchal lead to the right nuchal lead for two channels of EEG and one channel of EMG. EEG and EMG signals were fed from the electrode board to an amplifier and then to a PC. Signals were sampled at 128 Hz, filtered between 0.3 and 100, and amplified. After recording was complete, data were copied to CD and scored off-line using Rodent Sleep Scorer (Astro-Med). Epochs were scored in 10–15 s and assigned into one of three sleep/waking states (see Fig. 3). Waking consists of low-amplitude, high-frequency EEG and high EMG activity; SWS consists of high-amplitude, low-frequency EEG with little EMG modulation; and REM consists of low-amplitude, desynchronized EEG with occasional muscle twitches on a background of low EMG activity. Rodent Sleep Scorer (Astro-Med) also performed a fast Fourier transform analysis with a Hamming window function for each epoch. Delta power from 3.0 to 4.5 Hz was defined as slow-wave activity and averaged across all epochs and across all SWS sleep epochs for statistical comparisons across conditions. This delta power range was chosen based on the findings of Maret et al. (2005) of an altered SWA power frequency band distribution observed in mice with a mutated retinoic acid receptor β gene during SWS primarily in the 3.0–4.5 Hz range.
Figure 3.

The AdoA1R-inducible gene deletion did not significantly affect the relative time spent in waking or SWS but does affect SWA across states and within SWS. A, Percentage time in waking and SWS (sleep time) is shown for AdoA1R−/− and AdoA1Rf/f mice analyzed for four 2 h periods (equivalent to TM-off periods of sleep restriction days) under baseline conditions. B, SWA for all states (total) and during waking and (C on right) during SWS was analyzed for the same four 2 h periods during baseline conditions. C, The loss of the AdoA1R gene resulted in significantly less average SWA power during SWS and in an attenuated increase in SWA power from waking to the SWS state. This attenuation is attributable primarily to decreased SWA during SWS in the AdoA1R−/− mutants. Error bars indicate SEM.
Working memory; eight-arm radial maze testing.
A standard eight-arm radial maze protocol was used to assess working memory. The mice used for cognitive testing were not tethered to the recording system, but were otherwise treated in a similar manner to the recorded animals. All animals were weighed daily, maintained at or above 85% of their ad libitum feeding weight, and were housed on the treadmill (not moving) from training onward. The radial maze is constructed of clear plastic and placed on a table surface with visual cues from the room readily visible.
The task involved obtaining rewards that were hidden from sight in small cups at the end of each arm. Over 2 weeks of training (two trials per day), all the animals learned to visit each of the eight arms only once and rarely made mistakes (average, less than two mistakes per trial). These observations are similar to other assessments of hippocampal-independent working memory (Winocur, 1982) in mice using a similar radial maze protocol (Schmitt et al., 2003). Control trials in trained animals using unbaited cups confirmed that olfactory cues obtained from the reward itself were not needed to locate the correct cup.
Mice had two training sessions per day, a morning session at 8:00 A.M. and an afternoon session at 2:00 P.M. At the beginning of each training trial, eight arms were baited with a small piece of chocolate (Hershey's Hugs; The Hershey Company). Mice were placed into the central hub of the maze and allowed to enter any arm. A revisit error was scored if a mouse traversed more than one-third of an arm from which they had already retrieved the chocolate. Mice were removed from the maze after successfully retrieving all eight chocolate pieces or after a maximum of 10 min. Training continued until animals reached asymptotic performance for 3 d, after which animals began 2 d of testing, followed by 3 d of recall. Testing procedures were the same as training, with the exception that the treadmill was set to the 6 h cycle (4 h on; 2 h off). Recall was the same as training (TM off).
Episodic memory; paired associates testing.
The protocol for the paired associated task was the same as what we used previously (Rajji et al., 2006). Briefly, mice were first shaped for 10 d to dig into a sand-filled cup for a chocolate piece buried in the sand. Animals who failed to retrieve the chocolate rewards on day 10 were excluded from the study. After shaping, animals were given two combinations of odor (X or Y; mixed in sand) and contexts (A or B) and trained to associate one odor (X) with reward in one context (room A) and the other odor (Y) with reward in the other context (room B). Animals were given eight trials (four for each of two contexts) per day across 11 d. Similar to the eight-arm radial maze procedure, animals were housed on the treadmill (not moving) through training and the TM 6 h cycle began for testing. During testing, animals were given new odor–context cues to learn. Animals were sleep restricted for 6 d and, beginning on day 2, were trained for 5 d (using the same protocol as that used for control conditions with new contexts and scent cues) to acquire new paired associates during the sleep restriction.
During testing on both tasks, animals were moved from the treadmill to the task apparatus and placed back on the treadmill after task completion. All trials occurred during the treadmill-on phase.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism (GraphPad Software). Group comparisons of sleep time and SWA were made using a Mann–Whitney U test. Data are expressed as mean ± SEM. For baseline (undisturbed time) sleep/waking time analyses, percentage time in waking, SWS, and REM sleep was calculated from four 2 h blocks (matched to the TM-off times: 12:00–2:00 P.M., 6:00–8:00 P.M., 12:00–2:00 A.M., 6:00–8:00 A.M.). Animals showing >90% waking in any baseline 2 hour block were eliminated from additional analysis. Time in SWS over the 24 h baseline period was calculated for a subset of mice for comparisons of daily SWS time. For SWA analyses, SWA was averaged in 1 h bins for baseline and both sleep restriction days. For the eight-arm radial maze, the number of revisit errors and latency to complete the task were averaged for two sessions, one beginning after 18 h of sleep restriction and the other 24 h later. Comparisons were made between the average performance on the last two training days, sleep restriction days, and recovery day, along with weight on each of these days. A Wilcoxon matched-pairs test was used to compare error rate between baseline and probe conditions within each group. A Mann–Whitney U test was used to compare error rate, latency, and weight between groups. For the paired associates task, percentage correct on the new context trials during testing was calculated. A two-way ANOVA was used to compare genotype (AdoA1Rf/f and AdoA1R−/−) and time (trial 1 and trial 5).
Results
Spontaneous waking, SWS, and slow-wave activity
The expression of baseline sleep/waking states were not different between genotypes (Fig. 3). The AdoA1R−/− mice had a similar percentage time in waking (n = 5; 47.2 ± 9.2%) (Fig. 3 A), SWS (43.9 ± 7.9%) (Fig. 3 A), and REM sleep (8.2 ± 2.5%) (data not shown) compared with AdoA1Rf/f mice (n = 5; waking mean, 50 ± 8.4%; SWS mean, 41.4 ± 6.4%; REM mean, 8.6 ± 2.4%). As previously reported for AdoA1R constitutive gene deletion mutants (Stenberg et al., 2003), we did not detect a significant phenotype with respect to the percentage of total time spent in any one of the three sleep/waking states.
The loss of AdoA1R expression resulted in a decrease of the average SWA power (frequency range of 3.0–4.5 Hz) over the baseline recording period (AdoA1Rf/f SWA, 59.3 ± 2.2 μV2/epoch; AdoA1R−/− SWA, 49.8 ± 1.1 μV2/epoch; p < 0.001) (Fig. 3 B). This genotype difference in SWA was amplified during SWS, the state in which SWA predominates (AdoA1Rf/f SWS-SWA, 83.6 ± 2.8 μV2/epoch; AdoA1R−/− SWS-SWA, 69.5 ± 2.3 μV2/epoch; p < 0.0005) (Fig. 3 C) and was absent during waking (AdoA1Rf/f waking–SWA, 40.8 ± 12.5 μV2/epoch; AdoA1R−/− waking–SWA, 37.2 ± 8.3 μV2/epoch) (Fig. 3 B). The ability to increase SWA from waking to SWS was significantly attenuated by the loss of AdoA1R expression (AdoA1Rf/f, increased SWA by 110 ± 6.22%; AdoA1R−/−, 82.1 ± 7.96%; p = 0.01) (Fig. 3 C). Thus, even under baseline sleep conditions, SWA expression was attenuated during SWS in animals with disrupted AdoA1R expression.
An altered SWA power frequency band distribution was observed in mice with a mutated retinoic acid receptor β gene during SWS primarily in the 3.0–4.5 Hz range (Maret et al., 2005). The frequency-specific contribution to the power of EEG signal during SWS was assessed for AdoA1R−/− mutants and AdoA1Rf/f mice. No change in the power distribution in the SWA frequency range of 0.5–15.0 Hz between the two groups was apparent (Fig. 4).
Figure 4.

SWS EEG power distribution by genotype. Power distribution (calculated as a percentage of total EEG power for each 1.0 Hz bin) in the frequency range from 0.5 to 15 Hz during SWS did not significantly differ between AdoA1Rf/f and AdoA1R−/− mice (n = 5 per genotype). Thus, although AdoA1R−/− animals show decreased SWA in the 3.0–4.5 Hz band, this frequency range (3.0–4.0, 4.0–5.0 Hz) makes up the same relative amount of the total power during SWS. Error bars indicate SEM.
Restricted sleep response: acute 4 h sleep deprivation
The reduced SWA in the AdoA1R−/− mice may have resulted from aberrant homeostatic control in relation to waking duration. However, the AdoA1R−/− mice had spontaneous waking periods of similar length as the waking periods of AdoA1Rf/f mice (average duration of waking for AdoA1Rf/f, 2.3 ± 0.4 min; AdoA1R−/−, 2.1 ± 0.1 min). They nevertheless expressed significantly less SWA during the ensuing SWS periods (Fig. 3 C). The magnitude of the SWA power is closely correlated with waking duration (Dijk et al., 1990; Franken et al., 2001). If these correlations are causally related, then our observations of the genotype-dependent difference in SWA are consistent with an altered, less effective SWA-inducing feedback.
To test for a genotype-dependent change in the relationship between a controlled waking duration (i.e., forced waking) and SWA, both genotypes of mice (n = 7 per genotype) were maintained awake on a slowly moving treadmill for a 4 h period, followed by a 2 h undisturbed period (treadmill was turned off). Figure 5 A shows an example of raw EEG signals from both groups during baseline and after enforced waking conditions. Both genotypes responded to the enforced waking with increased SWA during the 2 h undisturbed period, but the magnitude of the SWA during SWS was significantly greater for the AdoA1Rf/f genotype compared with the AdoA1R−/− group (AdoA1Rf/f, 69 ± 0.6 μV2/epoch; AdoA1R−/−, 59.3 ± 3.0 μV2/epoch; p = 0.01) (Fig. 5 B). However, there was no difference in percentage time in SWS between genotypes (AdoA1Rf/f, 45.5 ± 4%; AdoA1R−/−, 50.2 ± 5.8%). This indicates an adenosine-mediated role in the rebound SWA response to prolonged waking acts via the AdoA1R.
Figure 5.
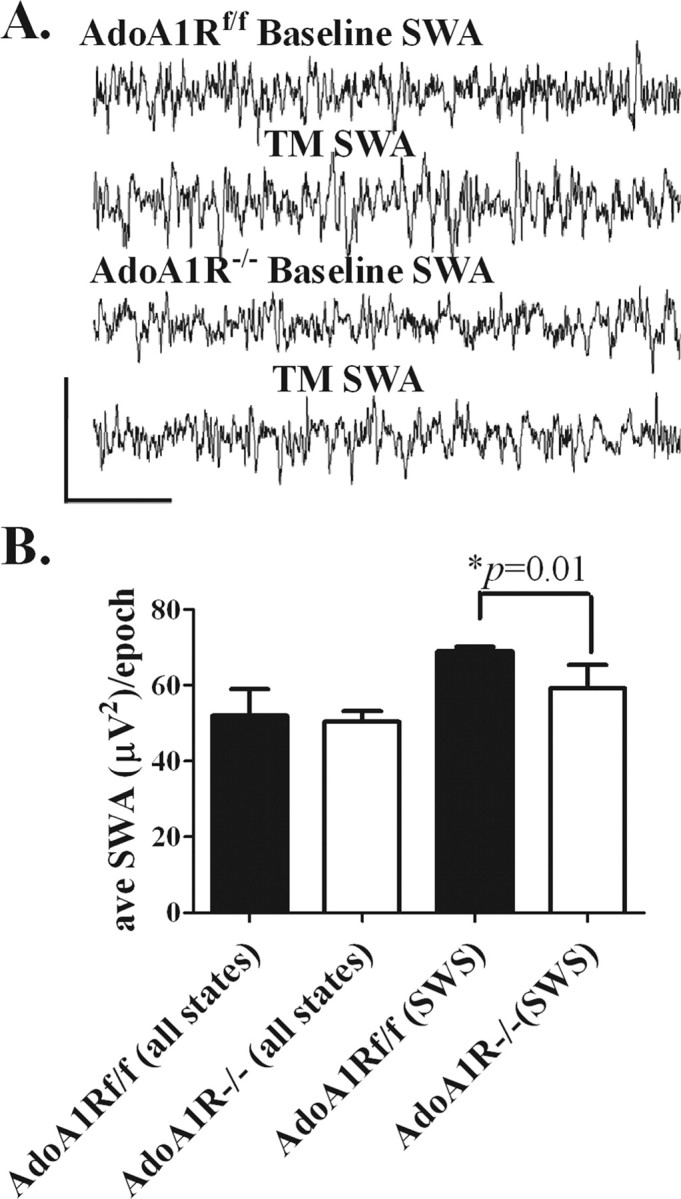
SWA response to 4 h of sleep deprivation. The magnitude of SWA during SWS, assessed for 2 h after 4 h of sleep deprivation, was less in AdoA1R−/− mice. A, The top two lines show an EEG trace from an AdoA1Rf/f mouse during SWS under uncontrolled sleep (baseline) and after sleep deprivation (TM) conditions. The bottom two lines show a similar set of EEG traces from an AdoA1R−/− mouse. Calibration: 300 μV, 2.2 s. B, When SWA was calculated for all states, there was no difference between genotypes (left). However, AdoA1R−/− showed significantly less SWS-specific SWA (right). The difference in SWA between genotypes during SWS can be seen in the raw data. Error bars indicate SEM.
The restricted sleep response: chronic sleep restriction, sleep duration, and slow-wave activity
In mice, 4 h of sleep deprivation did not result in any significant change in SWS time, but SWA during SWS was greatly enhanced. To test whether chronic sleep restriction in mice can be functionally compensated by a rebound SWA increase in an AdoA1R-dependent manner, SWS time and SWA were assessed in both genotypes under conditions of chronic sleep restriction.
The opportunity to sleep was restricted to a 2 h period per 6 h cycle by enforcing waking for 4 h with a slowly moving treadmill. The 6 h cycle (4 h TM on and 2 h TM off) was repeated for 48 h (eight complete cycles). Notably, there was no change in SWS time during spontaneous sleep periods (baseline) compared with the same time during TM off for either AdoA1Rf/f mice (n = 5; 44.9 ± 2.3% baseline; 49.2 ± 3.2% restricted sleep TM off) or for AdoA1R−/− mice (n = 5; 44.6 ± 2.2% baseline; 49.9 ± 4.6% restricted sleep TM off), nor was there any difference in SWS time between genotypes. Mice, of either genotype, spent 10.1 h per d in SWS under baseline conditions, whereas under the sleep restriction paradigm only 3.5 h per day was spent in SWS, a 65% reduction in SWS time.
In contrast to SWS time, SWA recorded during the 2 h TM-off phase, compared with the baseline sleep day (recorded at the same circadian time), showed an increase over baseline magnitude for both genotypes. SWA was significantly lower for the AdoA1R−/− mice on every cycle during the TM-off phases (AdoA1R−/− average, 51.3 ± 2.1 μV2/epoch; average AdoA1Rf/f, 70.8 ± 2.4 μV2/epoch; p < 0.001) (Fig. 6 A,C). Thus, during chronic restricted sleep, SWA, regardless of state, was attenuated in an AdoA1R-dependent manner with no change in SWS time.
Figure 6.
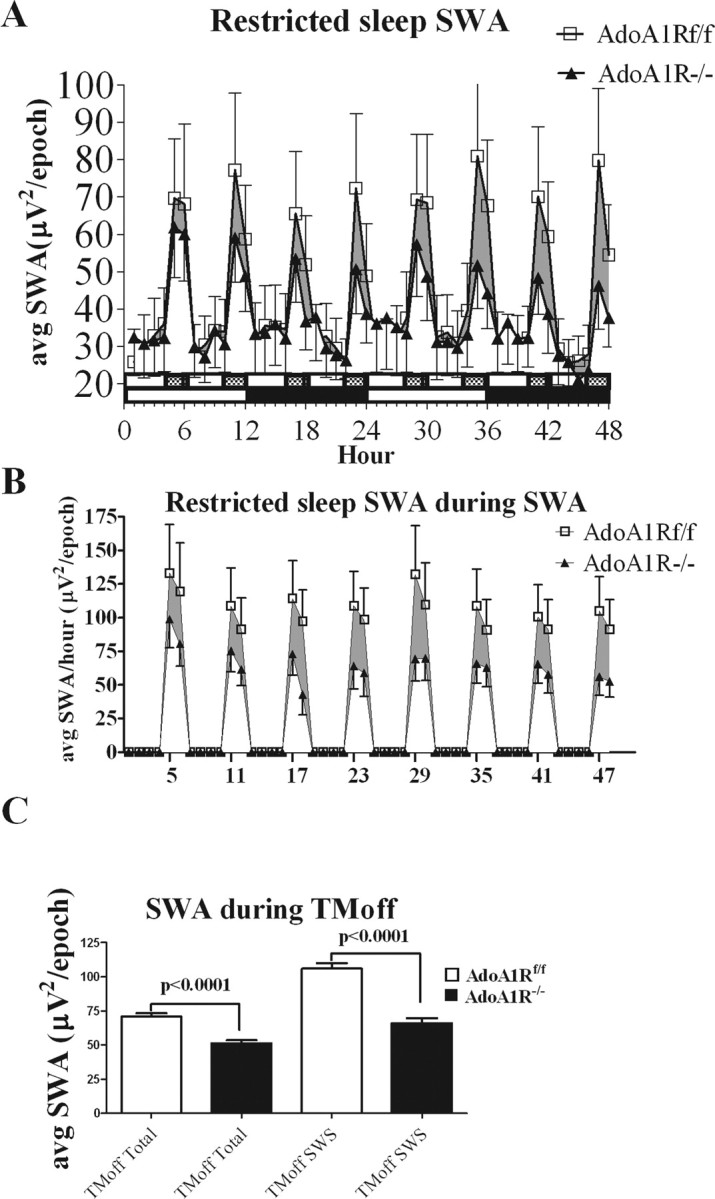
SWA during chronic sleep restriction. The enhancement of synchronized SWA after each 4 h enforced-waking period is markedly attenuated in AdoA1R−/− mice compared with the AdoA1Rf/f littermates. A, A graph is shown of the SWA power/10 s epoch (y-axis) averaged over each hour of the 48 h spent of the treadmill (x-axis) from data pooled from AdoA1R−/− (▴; n = 5) and AdoA1Rf/f (□; n = 5) mice. The slow-moving treadmill was on for 4 h (indicated by open 4 h horizontal bar) and off for 2 h (filled 2 h bar) for repeating 6 h cycles. Mice were maintained on a 12 h light (open 12 h horizontal bar)/12 h dark (filled 12 h bar) schedule. Eight cycles were completed in the 48 h experiment, and during the 2 h treadmill-off phase, the SWA enhancement by the AdoA1R−/− mice was consistently attenuated (shaded region indicates the power difference) compared with the AdoA1Rf/f genotype. B, Mice were scored for behavioral state, and average SWA power only during SWS was plotted for each of the 48 h of the experiment on the treadmill. C, The averaged SWA power/epoch for all of the treadmill-off phases showed a significant difference between genotypes, indicating a reduced SWA response to the 4 h waking period (left two bars). The largest difference in SWA power was observed during SWS during the TM off phase (right two bars). Error bars indicate SEM.
Because SWA predominates during SWS, SWA power recorded during SWS during sleep restriction was compared across genotype. The largest SWA difference between genotypes was observed under these conditions (AdoA1Rf/f, 106.2 ± 3.4 μV2/epoch; AdoA1R−/−, 65.9 ± 3.2 μV2/epoch), showing ∼30 μV2/epoch difference (p < 0.0001) (Fig. 6 B,C). The percentage change in SWA from the TM-on phase when the animals were awake to SWS during the TM-off phase (calculated as [SWS_SWA TM off–SWA TM on]/SWA TM on) was, for AdoA1Rf/f, 254 ± 11%; for AdoA1R−/−, 139 ± 12.3%; significant at p < 0.0002 (Fig. 7). This suggests the AdoA1R is needed for the full expression of the rebound SWA response during sleep restriction.
Figure 7.

Percentage change in SWA within and between conditions. Left bars (white), During the 48 h restricted-sleep condition, AdoA1R−/− showed a reduced percentage increase in SWA from enforced waking (during the TM-on phase) to SWS (during the TM-off phase). Right bars (gray), The increase in SWA from waking to SWS state in the restricted-sleep condition is greater than the increase in SWA from waking to SWS in baseline condition and this is quantified as percentage change in SWA (y-axis). This additional increase during restricted sleep is significantly greater for the AdoA1Rf/f mice compared with the AdoA1R−/− mice. Error bars indicate SEM.
These observations suggest that the AdoA1R may be particularly important to the increase in SWA from waking to SWS when sleep time is restricted, compared with our baseline unrestricted conditions. Indeed, AdoA1Rf/f animals showed a significant increase in SWS SWA after forced waking compared with baseline SWS SWA (baseline, 83.6 ± 2.8 μV2/epoch; TM off, 106.2 ± 4.8 μV2/epoch; p < 0.001) (Fig. 7), whereas there was no significant increase in SWS SWA in AdoA1R−/− animals (baseline, 69.5 ± 2.3 μV2/epoch; TM off, 65.9 ± 3.9 μV2/epoch).
We compared the increase in SWA activity from waking to SWS under baseline conditions (Fig. 3 C) to the increase in SWA from waking to SWS under sleep-restricted conditions (Fig. 7, left pair of bars). Notably, AdoA1Rf/f mice were able to further increase SWA from waking to SWS in sleep-restricted conditions compared with baseline conditions. This additional increase was significantly greater than that observed with mice with disrupted AdoA1R activity (AdoA1Rf/f, 42.3 ± 12.8%; AdoA1R−/−, 9.81 ± 7.37%; p = 0.005) (Fig. 7, right pair of bars). This change was calculated as follows: [increase of SWA_restricted − increase SWA_baseline]/increase of SWA_baseline. The findings are consistent with a ceiling effect on SWA during SWS in AdoA1R−/− mice, suggesting that additional enhancement of SWA in sleep-restricted conditions requires the AdoA1R.
Finally, we compared the percentage change in SWA during SWS from baseline to sleep restriction under acute (one 6 h cycle) (Fig. 5) and chronic (eight 6 h cycles) (Fig. 6) sleep restriction. There was no difference in percentage change between acute and chronic sleep restriction in either group (data not shown).
These data suggest the AdoA1R is necessary for the compensatory SWS-specific SWA rebound that follows sleep restriction. This may indicate the possibility of functional implications for the compensatory SWA increase.
Working memory performance and sleep restriction
A recent study suggests that an increase in SWA during SWS follows new learning, and improved performance is correlated with this increase in SWA (Huber et al., 2004). Because the AdoA1R−/− mice show a selective deficit in the SWA response to restricted sleep, we examined their performance on a working memory task in comparison with AdoA1Rf/f mice, before, during, and after 48 h of sleep restriction. Ten animals per genotype were examined on a working memory task. These animals were not connected to the EEG/EMG recording system.
There was no difference in working memory performance between genotypes before testing (AdoA1Rf/f, 1.6 ± 0.3 revisit errors/trial; AdoA1R−/−, 1.6 ± 0.3 revisit errors/trial) (Fig. 8 A). The two genotypes were then sleep restricted on the treadmill with the 4 h TM-on/2 h TM-off protocol for three full cycles (18 h). At the beginning of the fourth cycle, they were tested on the eight-arm maze working memory task (during the time when waking was enforced). They were then returned to the sleep restriction protocol in time to begin the 2 h TM-off phase of the fourth cycle (hours 18–24). Twenty-four hours later, at the beginning of the eighth cycle, the mice were tested again for the working memory task, and then allowed to sleep ad libitum for another 24 h when they were tested again for recovery (Fig. 8 A). During the sleep restriction, the AdoA1R−/− mice showed a significant increase in the number of revisit errors (4.1 ± 1.3 errors; p < 0.05), whereas the AdoA1Rf/f mice performed at a level of accuracy that was no different from baseline (1.5 ± 0.2 errors). The difference in revisit error rate was also significant between the two groups (p < 0.05) (Fig. 8 A). The significant worsening in the working memory behavior of the AdoA1R−/− mice was associated with an increase in the SEM, indicating that not all of the knock-out mice were affected to the same degree. Nevertheless, all of the AdoA1Rf/f mice tested either improved in performance or did not change, whereas only 4 of 10 AdoA1R−/− mice did not worsen in performance. This difference in performance is not attributable to a difference in sensory/motor ability or motivation as assessed by latency to complete the task and by weights of the animals (Fig. 8 B,C). Thus, the ability to functionally compensate for restricted sleep in a cognitive performance task requires the AdoA1R gene and is directly associated with enhancement of SWA.
Figure 8.

SWA-dependent deficit in working memory performance. Sleep restriction reduces working memory capacity in animals with conditional loss of AdoA1R gene expression, but it has no effect on those with intact AdoA1R expression. A, Pooled data for each genotype during baseline and sleep restriction (TM) conditions showed a performance deficit (increase in revisit errors) for AdoA1R−/− animals, but no deficit for AdoA1Rf/f animals. The deficit was reversed after a 1 d return to baseline sleeping conditions (recovery). B, C, There was no difference in the weight (B) or the time to task completion (C) between groups, suggesting that there was no difference in motivation or in sensory/motor ability to perform the task. Error bars indicate SEM.
Episodic memory and sleep restriction
Next, we tested whether hippocampus-dependent learning is affected by the inducible AdoA1R deletion using a task that involves hippocampal-dependent acquisition of paired associates.
AdoA1Rf/f and AdoA1R−/− animals (n = 4 per genotype) acquired this task under baseline conditions (Fig. 9) with no difference between genotypes (data not shown). Both genotypes were sleep restricted for 6 d and, beginning on day 2, were trained for 5 d (using the same protocol as that used for control conditions, except with new contexts and scent cues) to acquire new paired associates during the sleep restriction. Again, no difference was observed between groups or within groups with respect to learning under baseline conditions compared with learning during sleep restriction. A two-way ANOVA testing the effect of genotype/condition on trial day response was not significant (Fig. 9), although there was an effect of trial day on acquisition (p < 0.005) (Fig. 9). The hippocampal-dependent task was thus distinguished from the working memory task by the lack of significant effect compared with the significant effect of the AdoA1R gene on working memory task performance when sleep was restricted.
Figure 9.

Sleep restriction did not affect acquisition of a hippocampal-dependent appetitive, paired associates learning task used to assess episodic memory. Mice learned that one of two odor cues was rewarded in one context, and that in another context the reward assignment was reversed. After training, acquisition of a new odor/context pair was then tested during 5 d of the 4 h TM-on, 2 h TM-off sleep restriction condition. There was no difference in performance (percentage of correct trials) between groups (white/black bars); however, both genotypes showed significant improvement across days (gray cross-hatched bars). Error bars indicate SEM.
Discussion
In summary, these findings suggest that AdoA1 receptors mediate a feedback response to restricted sleep comprised of a compensatory enhancement of synchronized SWA. The loss of AdoA1R expression results in a selective attenuation of this homeostatic SWA response. The attenuation of SWA is associated with compromised working memory-dependent performance, indicative of a functional role for AdoA1R-dependent SWA homeostasis in maintaining this cognitive performance when sleep is restricted.
Several studies indicate that the expression of SWA is under genetic control (Franken et al., 1998, 2001; Wisor et al., 2002; Maret et al., 2005), but the specific molecular, neurochemical, and physiological mechanisms responsible for this control remain mostly uncharacterized. At the cellular level, AdoA1Rs mediate an increase in GIRK channel currents and a decrease in hyperpolarization activated currents, both of which facilitate the oscillations that underlie SWA (Pape, 1992). In addition, activation of AdoA1Rs on presynaptic terminals reduces synaptic glutamate release, thereby reducing excitatory drive (Brambilla et al., 2005) and allowing electrophysiological relaxation of neurons toward a more hyperpolarized membrane potential needed for the emergence of endogenously generated SWA oscillations (McCormick and Pape, 1990). Application of exogenous adenosine and AdoA1R agonists either systemically or locally to the diencephalon increases SWS (Radulovacki, 1985) and SWA recorded from the surface EEG (Benington and Heller, 1995; Benington et al., 1995).
An indirect AdoA1R-mediated mechanism to increase SWS and SWA in response to waking may result from a local increase in extracellular adenosine in cholinergic arousal centers. Increased glutamate-mediated excitatory input to neurons of these centers, associated with prolonged waking, increases AdoA1R activation (Rainnie et al., 1994; Arrigoni et al., 2001; Brambilla et al., 2005). This activation reduces arousal center activity, acting at both presynaptic and postsynaptic inhibitory AdoA1Rs (Rainnie et al., 1994; Arrigoni et al., 2001), and accordingly decreases cholinergic tone in the target sites of these centers, including the thalamocortical system. The decrease in cholinergic tone facilitates SWA as observed when adenosine reuptake is blocked (increasing endogenous adenosine concentration) in the cholinergic basal forebrain (Porkka-Heiskanen et al., 1997). Thus, AdoA1R-mediated actions can potentially facilitate the increase in SWA by two complementary mechanisms during prolonged waking (Rainnie et al., 1994; Porkka-Heiskanen et al., 1997; Portas et al., 1997; Arrigoni et al., 2001). However, the requirement for AdoA1R-mediated increase in homeostatic SWA has not previously been demonstrated.
Adenosine is the endogenous ligand for the AdoA1R and its extracellular concentration increases whenever the ratio of metabolite availability to metabolite demand decreases (McIlwain and Poll, 1986; Greene and Haas, 1991; Dunwiddie and Masino, 2001). An AdoA1R-mediated inhibitory tone, present under physiological conditions, is positively modulated with a slow time constant in response to maintained increases in excitatory synaptic glutamate release as occurs with maintained waking (Brambilla et al., 2005). Because extracellular adenosine concentration reflects the intracellular concentration that is maintained in equilibrium with ATP/ADP ratio by adenosine kinase (Muchmore et al., 2006) in both glia and neurons (Studer et al., 2006), the AdoA1R inhibitory tone reflects the intracellular metabolic state of nervous tissue. Accordingly, intracellular metabolic state can modulate the functionally relevant enhancement of synchronized SWA during the SWS state. Although the loss of AdoA1Rs in the conditional gene deletion used in this study are exclusively neuronal (Tsien et al., 1996), the source of the adenosine may include both neurons and glia (Pascual et al., 2005; Studer et al., 2006) and reflect the metabolic state both kinds of neural tissue.
The data presented here suggest that, under physiological conditions during sleep restriction, AdoA1R activation is needed for the full expression of a homeostatic increase of SWA. Nevertheless, SWA still predominates during SWS in the absence of AdoA1R activation because it was clearly observed in AdoA1R−/− mice. Furthermore, in a mutant mouse with constitutive deletion of the AdoA1R gene, SWA was not altered in spontaneously sleeping mice and mice sleep deprived by gentle handling (Stenberg et al., 2003). These findings are most consistent with a modulatory role for AdoA1R activation in SWA expression.
Additionally, the working memory deficits seen in the AdoA1R−/− mice under sleep restriction may be attributable to factors other than the decreased SWA, such as stress or changes in other frequency bands. It is unlikely that a stress effect would only appear in the knock-out animals because all mice had the same amount of forced waking at the same belt speed. We cannot rule out differences in other frequency bands in the AdoA1R−/− animals; however, the greatest changes in frequency during SWS after sleep restriction are in the SWA frequency band (Borbély, 1982). Thus, it is reasonable to conclude that the working memory effects seen by knocking out the AdoA1 receptor are most likely attributable to the concurrent changes in SWA.
The effect of the inducible deletion of the AdoA1R gene in CNS was remarkably selective for an attenuated rebound response in SWA to sleep restriction. SWA was only slightly attenuated in baseline conditions, and the percentage of time spent in SWS in either baseline or during sleep restriction was not affected by the gene deletion. This selectivity offered an experimental opportunity to dissociate changes in SWA from changes in SWS time because AdoA1R−/− mice had an attenuated homeostatic SWA response but an unchanged SWS time in our sleep restriction paradigm. The ability to further increase SWA expressed during SWS under baseline conditions to the larger amplitude of SWA expressed during SWS under the sleep restriction conditions was marked in the AdoA1Rf/f mice and almost completely absent in the AdoA1R−/− mice (a >40% compared with <10% additional increase). This suggests an AdoA1R-dependent compensatory increase in SWA.
AdoA1R−/− mice could perform a working memory cognitive task normally under baseline conditions, but performance was compromised under conditions of sleep restriction. This deficit in performance was associated with the deficit in the compensatory SWA response, and the deficit was recovered when sleep was no longer restricted. Thus, loss of AdoA1R function compromises working memory performance but only under conditions involving the reduction of rebound SWA, consistent with a role for rebound SWA in cognitive performance.
Notably, attenuated SWA did not affect a hippocampal-dependent, paired associates, learning task involving acquisition and next day improvement in performance. This task was used for several reasons. First, the paradigm is sensitive to loss of only 10–30% of CA3 NMDA receptors (Rajji et al., 2006), indicating its sensitivity to intact hippocampal, NMDA receptor function including NMDA receptor-dependent synaptic plasticity. Second, it does not require working memory to perform, because all the needed cues are always present. Third, attentional processes may be less critical for the performance of this task for the same reason (i.e., all cues are always present, a condition that contrasts with the eight-arm working memory task). Our observations suggest that the neural processes involved in hippocampal-dependent learning and episodic memory (Rajji et al., 2006) are less sensitive or even insensitive to the AdoA1R loss of function and the attenuated SWA compared with those required for normal working memory capacity. It is conceivable that any negative effect of the disrupted rebound SWA response was opposed by the loss of AdoA1R inhibitory tone in the hippocampus, although these same speculated opposing effects ought to apply to the neural mechanisms involved in performance of the working memory task as well, and this was not the case. Synaptic plasticity responsible for the acquisition, retention, and consolidation involved in the paired associate task was less sensitive than the neural mechanism(s) needed to perform the eight-arm maze working memory task.
With respect to the mechanisms responsible for performance of the working memory task, the observed reversibility of the sleep restriction-induced deficit in AdoA1R−/− mice suggests that synaptic plasticity and/or consolidation processes were not involved. A similar kind of selective loss of function of working memory compared with reference memory has been described as resulting from the deletion of the GluR1 subunit of the AMPA receptor (Schmitt et al., 2003) consistent with a greater sensitivity of working memory to perturbations of glutamatergic function rather than mechanisms directly related to synaptic plasticity. A major difference in neuronal function with respect to working memory performance compared with episodic memory is that the former requires sustained, selective, circuit activity of the prefrontal cortical circuits (Constantinidis et al., 2002; Lee and Kesner, 2003; Wang et al., 2004). The loss of AdoA1R function and the associated rebound SWA may be one of the factors that selectively compromise the more metabolically demanding activities such as those involved in sustained circuit activity under conditions that produce rebound SWA-like restricted sleep and thus affect working memory performance.
The nature of sleep function remains enigmatic (Greene and Siegel, 2004), especially because the molecular mechanisms controlling a homeostatic increase in SWA are not well understood. It has been suggested that sleep function is involved in the consolidation of learning (Walker and Stickgold, 2004; Stickgold and Walker, 2005) or in the global homeostasis of synaptic plasticity of learning and memory (Vyazovskiy et al., 2008), based on observations of performance with or without intervening episodes of sleep. Nevertheless, whether these sleep dependent changes are attributable to a direct modulation of synaptic plasticity or to an indirect outcome from some other process that impinges on synaptic plasticity remains to be established. Indeed, there is considerable evidence consistent with a role in metabolism, feeding, and sleep based on known modulators of sleep that involve metabolism and feeding, including NPAS2 (Rutter et al., 2001; Dudley et al., 2003), clock (Naylor et al., 2000; Turek et al., 2005), leptin (Laposky et al., 2006), orexin/hypocretin (Chemelli et al., 1999; Lin et al., 1999; Willie et al., 2001) and adenosine (Brambilla et al., 2005).
Together, our findings suggest that metabolically associated feedback signals of waking, involving AdoA1R activation, influence homeostatic sleep functions in the CNS. The homeostatic sleep function requires rebound synchronized SWA, as suggested by the observation that loss of AdoA1R function and the selective attenuation of this homeostatic sleep response reduces working memory capacity.
Footnotes
This work was supported by the Department of Veterans Affairs and National Institutes of Health Grant R01 MH 06777. We gratefully acknowledge the scientific direction and oversight of Dr. Margaret Thompson in the generation of the AdoA1Rf/f mice, and the technical assistance of To Thai, Lehong Nguyen, and Dr. Gerald Marks.
References
- Amzica F, Steriade M. Electrophysiological correlates of sleep delta waves. Electroencephalogr Clin Neurophysiol. 1998;107:69–83. doi: 10.1016/s0013-4694(98)00051-0. [DOI] [PubMed] [Google Scholar]
- Arrigoni E, Rainnie DG, McCarley RW, Greene RW. Adenosine-mediated presynaptic modulation of glutamatergic transmission in the laterodorsal tegmentum. J Neurosci. 2001;21:1076–1085. doi: 10.1523/JNEUROSCI.21-03-01076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni E, Crocker AJ, Saper CB, Greene RW, Scammell TE. Deletion of presynaptic adenosine A1 receptors impairs the recovery of synaptic transmission after hypoxia. Neuroscience. 2005;132:575–580. doi: 10.1016/j.neuroscience.2004.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- Benington JH, Kodali SK, Heller HC. Stimulation of A1 adenosine receptors mimics the electroencephalographic effects of sleep deprivation. Brain Res. 1995;692:79–85. doi: 10.1016/0006-8993(95)00590-m. [DOI] [PubMed] [Google Scholar]
- Borbély AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- Brambilla D, Chapman D, Greene R. Adenosine mediation of presynaptic feedback inhibition of glutamate release. Neuron. 2005;46:275–283. doi: 10.1016/j.neuron.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Constantinidis C, Williams GV, Goldman-Rakic PS. A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci. 2002;5:175–180. doi: 10.1038/nn799. [DOI] [PubMed] [Google Scholar]
- Daan S, Beersma DG, Borbély AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246:R161–R183. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- Dijk DJ, Brunner DP, Beersma DG, Borbély AA. Electroencephalogram power density and slow wave sleep as a function of prior waking and circadian phase. Sleep. 1990;13:430–440. doi: 10.1093/sleep/13.5.430. [DOI] [PubMed] [Google Scholar]
- Dudley CA, Erbel-Sieler C, Estill SJ, Reick M, Franken P, Pitts S, McKnight SL. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science. 2003;301:379–383. doi: 10.1126/science.1082795. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M. Genetic variation in EEG activity during sleep in inbred mice. Am J Physiol. 1998;275:R1127–R1137. doi: 10.1152/ajpregu.1998.275.4.R1127. [DOI] [PubMed] [Google Scholar]
- Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–2621. doi: 10.1523/JNEUROSCI.21-08-02610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- Greene R, Siegel J. Sleep: a functional enigma. Neuromolecular Med. 2004;5:59–68. doi: 10.1385/NMM:5:1:059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene RW, Haas HL. The electrophysiology of adenosine in the mammalian central nervous system. Prog Neurobiol. 1991;36:329–341. doi: 10.1016/0301-0082(91)90005-l. [DOI] [PubMed] [Google Scholar]
- Huang ZL, Qu WM, Eguchi N, Chen JF, Schwarzschild MA, Fredholm BB, Urade Y, Hayaishi O. Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nat Neurosci. 2005;8:858–859. doi: 10.1038/nn1491. [DOI] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- Laposky AD, Shelton J, Bass J, Dugovic C, Perrino N, Turek FW. Altered sleep regulation in leptin-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R894–R903. doi: 10.1152/ajpregu.00304.2005. [DOI] [PubMed] [Google Scholar]
- Lee I, Kesner RP. Time-dependent relationship between the dorsal hippocampus and the prefrontal cortex in spatial memory. J Neurosci. 2003;23:1517–1523. doi: 10.1523/JNEUROSCI.23-04-01517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Lindskog M, Svenningsson P, Pozzi L, Kim Y, Fienberg AA, Bibb JA, Fredholm BB, Nairn AC, Greengard P, Fisone G. Involvement of DARPP-32 phosphorylation in the stimulant action of caffeine. Nature. 2002;418:774–778. doi: 10.1038/nature00817. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret S, Franken P, Dauvilliers Y, Ghyselinck NB, Chambon P, Tafti M. Retinoic acid signaling affects cortical synchrony during sleep. Science. 2005;310:111–113. doi: 10.1126/science.1117623. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol. 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain H, Poll JD. Adenosine in cerebral homeostatic role: appraisal through actions of homocysteine, colchicine, and dipyridamole. J Neurobiol. 1986;17:39–49. doi: 10.1002/neu.480170105. [DOI] [PubMed] [Google Scholar]
- Mignot E. Why we sleep: the temporal organization of recovery. PLoS Biol. 2008;6:e106. doi: 10.1371/journal.pbio.0060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61:187–197. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Smith RA, Stewart AO, Cowart MD, Gomtsyan A, Matulenko MA, Yu H, Severin JM, Bhagwat SS, Lee CH, Kowaluk EA, Jarvis MF, Jakob CL. Crystal structures of human adenosine kinase inhibitor complexes reveal two distinct binding modes. J Med Chem. 2006;49:6726–6731. doi: 10.1021/jm060189a. [DOI] [PubMed] [Google Scholar]
- Naylor E, Bergmann BM, Krauski K, Zee PC, Takahashi JS, Vitaterna MH, Turek FW. The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci. 2000;20:8138–8143. doi: 10.1523/JNEUROSCI.20-21-08138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC. Adenosine promotes burst activity in guinea-pig geniculocortical neurones through two different ionic mechanisms. J Physiol. 1992;447:729–753. doi: 10.1113/jphysiol.1992.sp019026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portas CM, Thakkar M, Rainnie DG, Greene RW, McCarley RW. Role of adenosine in behavioral state modulation: a microdialysis study in the freely moving cat. Neuroscience. 1997;79:225–235. doi: 10.1016/s0306-4522(96)00640-9. [DOI] [PubMed] [Google Scholar]
- Radulovacki M. Role of adenosine in sleep in rats. Rev Clin Basic Pharm. 1985;5:327–339. [PubMed] [Google Scholar]
- Rainnie DG, Grunze HC, McCarley RW, Greene RW. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science. 1994;263:689–692. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajji T, Chapman D, Eichenbaum H, Greene R. The role of CA3 hippocampal NMDA receptors in paired associate learning. J Neurosci. 2006;26:908–915. doi: 10.1523/JNEUROSCI.4194-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter J, Reick M, Wu LC, McKnight SL. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science. 2001;293:510–514. doi: 10.1126/science.1060698. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Arrigoni E, Thompson MA, Ronan PJ, Saper CB, Greene RW. Focal deletion of the adenosine A1 receptor in adult mice using an adeno-associated viral vector. J Neurosci. 2003;23:5762–5770. doi: 10.1523/JNEUROSCI.23-13-05762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt WB, Deacon RM, Seeburg PH, Rawlins JN, Bannerman DM. A within-subjects, within-task demonstration of intact spatial reference memory and impaired spatial working memory in glutamate receptor-A-deficient mice. J Neurosci. 2003;23:3953–3959. doi: 10.1523/JNEUROSCI.23-09-03953.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenberg D, Litonius E, Halldner L, Johansson B, Fredholm BB, Porkka-Heiskanen T. Sleep and its homeostatic regulation in mice lacking the adenosine A1 receptor. J Sleep Res. 2003;12:283–290. doi: 10.1046/j.0962-1105.2003.00367.x. [DOI] [PubMed] [Google Scholar]
- Stickgold R, Walker MP. Memory consolidation and reconsolidation: what is the role of sleep? Trends Neurosci. 2005;28:408–415. doi: 10.1016/j.tins.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM, Boison D. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience. 2006;142:125–137. doi: 10.1016/j.neuroscience.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Welker E, Fritschy JM, Tobler I. Regional pattern of metabolic activation is reflected in the sleep EEG after sleep deprivation combined with unilateral whisker stimulation in mice. Eur J Neurosci. 2004;20:1363–1370. doi: 10.1111/j.1460-9568.2004.03583.x. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–208. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- Walker MP, Stickgold R. Sleep-dependent learning and memory consolidation. Neuron. 2004;44:121–133. doi: 10.1016/j.neuron.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Tegner J, Constantinidis C, Goldman-Rakic PS. Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. Proc Natl Acad Sci U S A. 2004;101:1368–1373. doi: 10.1073/pnas.0305337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Yanagisawa M. To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci. 2001;24:429–458. doi: 10.1146/annurev.neuro.24.1.429. [DOI] [PubMed] [Google Scholar]
- Winocur G. Radial-arm-maze behavior by rats with dorsal hippocampal lesions: effect of cuing. J Comp Physiol Psychol. 1982;96:155–169. doi: 10.1037/h0077882. [DOI] [PubMed] [Google Scholar]
- Wisor JP, O'Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, Edgar DM, Franken P. A role for cryptochromes in sleep regulation. BMC Neurosci. 2002;3:20. doi: 10.1186/1471-2202-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]