

Abstract
The advent of Bcr-Abl tyrosine kinase inhibitors (TKIs) has revolutionized the treatment of CML. However, resistance evolves due to BCR-ABL mutations and other mechanisms. Furthermore, patients with blast crisis (BC) CML are less responsive and quiescent CML stem cells are insensitive to these inhibitors. We found that triptolide, a diterpenoid, at nM concentrations, promoted equally significant death of KBM5 cells, a cell line derived from a Bcr-Abl-bearing BC CML patient and KBM5STI571 cells, an imatinib-resistant KBM5 subline bearing the T315I mutation. Similarly, Ba/F3 cells harboring mutated BCR-ABL were as sensitive as Ba/F3Bcr-Ablp210wt cells to triptolide. Importantly, triptolide induced apoptosis in primary samples from BC CML patients, who showed resistance to Bcr-Abl TKIs in vivo, with less toxicity to normal cells. Triptolide decreased XIAP, Mcl-1, and Bcr-Abl protein levels in K562, KBM5, KBM5STI571 cells and in cells from BC CML patients. It sensitized KBM5, but not KBM5STI571 cells to imatinib. More importantly, triptolide also induced death of quiescent CD34+ CML progenitor cells, a major problem in the therapy of CML with TKIs. Collectively, these results suggest that triptolide potently induces BC CML cell death independent of the cellular responses to Bcr-Abl TKIs, suggesting that triptolide could eradicate residual quiescent CML progenitor cells in TKI-treated patients and benefit TKI-resistant BC CML patients.
Keywords: triptolide, XIAP, Mcl-1, Bcr-Abl, quiescent CD34+ CML cells
Introduction
CML is a lethal myeloproliferative disorder derived from the clonal expansion of transformed hematopoietic stem cells(1) and is characterized by the formation of the BCR-ABL fusion gene coding for a constitutively active tyrosine kinase which is necessary and sufficient for malignant transformation.(2-4) Imatinib, a Bcr-Abl tyrosine kinase inhibitor (TKI), has drastically improved the outcome of patients with CML. However, imatinib is less effective in accelerated phase and blast crisis (BC) CML patients, 45-90% of whom fail to respond or develop resistance within 3-12 months.(5;6) Resistance to TKIs develops in both chronic and advanced CML patients, which leads to the reactivation of Bcr-Abl kinase activity within leukemic cells, through either gene amplification(7-9) or mutations.(7;10-13) New Bcr-Abl TKIs such as nilotinib and dasatinib have been reported to overcome resistance of most mutants but not of the T315I mutation. Regardless, since these agents inhibit tyrosine kinase activity but do not affect protein expression, mutations will likely develop again. Clearly, alternative therapeutic strategies are needed for patients with advanced and TKI-resistant CML.
Further, Bcr-Abl was shown to induce DNA damage and contribute to genomic instability(14) which is believed to be responsible for promoting the transition from the benign chronic to blast phase and providing additional growth and survival advantages for BC CML cells. Therefore, eliminating the Bcr-Abl protein could be an alternative approach to treating CML patients and overcoming resistance to Bcr-Abl TKIs. Several groups have worked toward the development of such strategy.(15-17)
Although the majority of CML progenitors have a higher proliferative capacity than normal progenitors,(18) a subpopulation of CD34+ CML progenitors is quiescent. This cell population constitutes approximately 0.5% of the total CD34+ compartment and is able to engraft NOD/SCID mice and to initiate leukemia, which makes it a candidate CML stem cell.(19;20) An additional problem with current therapies for CML, including TKIs, other signal transduction inhibitors, and conventional chemotherapeutic agents is that they act by inhibiting cell proliferation and inducing apoptosis. Thus, complete cures are rare because TKIs are not effective against quiescent progenitor/stem cells(21;22) and these cells persist even when complete hematological and cytogenetic remissions are achieved.(20) Discontinuation of therapy frequently leads to relapse of the disease.(21) In vitro studies, corroborating these findings, have demonstrated that quiescent CML stem cells are insensitive to imatinib and dasatinib.(21;22) Clearly, the cure of CML depends on the eradication of quiescent stem cells.
Triptolide, a diterpenoid isolated from a Chinese herb, Tripterygium wilfordii Hook.f, has been shown to have anti-tumor properties by suppressing cell growth and inducing apoptosis.(23-26) We recently reported that triptolide induces cell death in AML by inhibiting the RNA and protein levels of XIAP and decreasing the protein level of Mcl-1,(27) two potent cellular antiapoptotic proteins. Mcl-1 was recently reported to be a Bcr-Abl targeted gene in CML cells.(27;28) We also showed that triptolide effectively kills KBM5 and K562 cells, two BC CML cell lines.(27) We here examined the effect of triptolide on imatinib-resistant CML cells and CD34+ quiescent CML progenitor cells. The results show that triptolide potently kills BC CML cells independent of their responses to TKIs, in agreement with a recent report by Shi et al..(29) The results also suggest that triptolide has the potential to target quiescent CD34+ CML progenitor cells.
Materials and Methods
Cells and cell culture
KBM5,(30) an imatinib-sensitive BC CML cell line and KBM5STI571, an imatinib-resistant KBM5 subline harboring a T315I mutation(31) in the BCR-ABL gene were cultured in Iscove's modified Dulbecco's medium (Gibco-BRL, Gaithersburg, MD). K562 cells and Ba/F3vec, Ba/F3Bcr-Ablp210wt, Ba/F3Bcr-AblE255K, and Ba/F3Bcr-AblT315I cells (kindly provided by Dr. C. Sawyers, UCLA, Los Angeles, CA) were cultured in RPMI-1640 medium. Both media were supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Medium for Ba/F3vec cells also contained 2 ng/mL mouse recombinant IL-3 (Peprotech Inc., Rocky Hill, NY). Bone marrow (BM) or peripheral blood (PB) samples from CML patients in BC were acquired after informed consent had been obtained according to institutional guidelines and in concordance with the declaration of Helsinki. Mononuclear cells from these samples were purified by Ficoll-Hypaque (Sigma Chemical Co., St. Louis, MO) density-gradient centrifugation and cultured in the same medium as K562 cells.
Cell viability assay
CML cells from either cell lines (0.2×106/mL) or patients with CML (0.5×106/mL) and Ba/F3 cells (0.05×106/mL) were treated with triptolide (Alexis Biochemicals, San Diego, CA) for 24 or 48 hours. KBM5 and KBM5STI571 cells were treated with imatinib (LC Laboratories, Woburn, MA) for 24 to 72 hours. For triptolide and imatinib combination, triptolide was administered 24 hours before imatinib (1 μM) was added and cell death was assessed 24 hours after imatinib treatment (total 48 hours). Apoptotic cell death was analyzed by measuring externalized phosphatidyl serine with the Annexin-V-FLUOS Staining Kit (Roche Diagnostics Corp., Indianapolis, IN) in combination with a vital dye: propidium iodide (PI) or 7-amino-actinomycin D (7-AAD), expressed as percentage of Annexin V+/PI+ or 7-ADD+ for cell lines and as percentage of survival cells (Annexin V-/PI-) for CML patient samples of triptolide treated cells compared to the untreated cells.
Cell viability of CD34+ quiescent and proliferating CML cells
Mononuclear cells (5×106/mL) from BM or PB of patients with BC CML were labeled with 1 μM 5-(and 6-) carboxy-fluorescein diacetate succinimidyl ester (CFSE) as described elsewhere(32) and then cultured in serum-free RPMI-1640 medium supplemented with growth factors (GM-CSF, 200 pg/mL; G-CSF, 1 ng/mL; stem cell factor, 200 pg/mL; LIF, 50 pg/mL; MIP-1α, 200 pg/mL; and IL-6, 1 ng/mL) or co-cultured in RPMI-1640/10%FCS medium with MS-5 cells, a mouse mesenchymal stromal cell (MSC) line known to support primitive human progenitor and to mimic the BM microenvironment.(33-35) Cell proliferation was monitored by flow cytometric measurement of CFSE fluorescence intensity, which halves with each cell division. Quiescent cells were defined as those within a region of CFSE fluorescence of paraformaldehyde-fixed cells on day 0 (CFSEbright). Proliferating cells were defined as those with a fluorescent intensity less than that of the initiating cell population (CFSEdim). After culturing for 4-7 days, cells were treated with triptolide for 24 or 48 hours and then stained with CD34-PE and annexin V-Cy5. Apoptosis of quiescent primitive CD34+ CML cells was defined as annexin V positivity in the CD34+CFSEbright population, while apoptosis of proliferating primitive CD34+ CML cells was defined as annexin V positivity in the CD34+CFSEdim population.
Western blot analysis
XIAP, Mcl-1, and Bcr-Abl protein levels were determined by western blot analysis, as described previously.(36) XIAP antibody was purchased from BD-Transduction lab (San Diego, CA), Mcl-1 antibody from BD-Pharmingen, and c-Abl antibody from Cell Signaling Technology (Danvers, MA). Signals were detected using a PhosphorImager (Storm 860 Version 4.0; Molecular Dynamics, Sunnyvale, CA) and quantitated by ImageJ (NIH, Bethesda, MD). β-actin was included as a loading control.
Real-time RT-PCR
RNA was extracted using Trizol solution (Invitrogen, Carlsbad, CA) and cDNA was generated with random hexamers and AMV reverse transcriptase (Roche Applied Science, Mannheim, Germany) at 42°C for one hour. Real-time PCR was performed in Applied Biosystems 7900HT Fast RT-PCR system using SYBR Green detection method. Reaction mixture contained 0.5 μL cDNA, 0.4 μM each of the forward and reverse primers, 2× SYBR Green master mix (Applied Biosystems, Forster City, CA) in a total of 25 μL. The reaction was initiated at 95°C for 10 minutes, followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C. The specificity of the PCR products was confirmed by dissociation curve analysis with ABI SDS 2.3 software. The forward/reverse primers for Mcl-1 were 5′-GAGGCTGGGATGGGTTTGT-3′/5′-AAAGCCAGCAGCACATTCCT-3′ and XIAP 5′-CCCAAATTGCAGATTTATCAACG-3′/5′-TGCATGTGTCTCAGATGGCC-3′. 18S RNA was used as an internal control. The abundance of each transcript relative to that of 18S was calculated using the 2−ΔCt method, where ΔCt is the mean Ct of the transcript of interest minus the mean Ct of the transcript for 18S.
Statistical analysis
Experiments were performed three times and the results expressed as mean ± standard deviation. For experiments with patient samples, the results were expressed as mean ± standard error. Statistical significance was denoted at p<0.05, where applicable, using Student's t-test. Triptolide concentrations that induced annexin V positivity in 50% of cells (IC50) was calculated using Calcusyn software (Biosoft, Ferguson, MO).
Results
Triptolide induces cell death independent of BCR-ABL mutations and cellular response to imatinib
We showed previously that triptolide potently induces apoptosis in various acute leukemic cells including KBM5 and K562 cells,(27) two BC CML cell lines. To evaluate whether triptolide is effective in imatinib-resistant CML cells, we treated KBM5 and KBM5STI571 cells with triptolide for 48 hours. As shown in Fig 1A, KBM5STI571 cells and KBM5 cells had similar sensitivities to triptolide. We then treated Ba/F3vec, Ba/F3Bcr-Ablp210wt, Ba/F3Bcr-AblE255K, and Ba/F3Bcr-AblT315I cells with triptolide. We found that it invariably killed Ba/F3 cells harboring either the wild type (wt) or mutant BCR-ABL genes at 48 hours (Fig 1B). We concluded that triptolide induces cell death independent of BCR-ABL mutation status and cellular response to imatinib. Of note, CML cells with a T315I mutation in the BCR-ABL gene are resistant not only to imatinib but also to most other more potent Bcr-Abl TKIs.
Figure 1.

Triptolide potently induces cell death in BC CML cells independent of cellular responses to imatinib. (A) KBM5 and KBM5STI571 cells and (B) Ba/F3vec, Ba/F3Bcr-Ablp210wt, Ba/F3Bcr-AblE255K, and Ba/F3Bcr-AblT315I cells were treated with various concentrations of triptolide for 48 hours. (C) Blasts from patients with CML were treated for 24 hours. Cell death was determined by annexin V staining.
Triptolide induces death of blasts from patients with BC CML in vitro independent of patients' responses to imatinib and other Bcr-Abl TKIs in vivo
Seven samples of mononuclear cells were obtained from six patients with BC CML and treated with triptolide in vitro. The characteristics of these patients and their in vivo responses to treatments are illustrated in Table 1. Interestingly, apart from patient 4, who had not been treated with imatinib or other TKIs, all patients were resistant to imatinib. Three of six patients (1, 3, and 5) were also resistant to nilotinib, a more potent Bcr-Abl TKI. In addition, patient 5 was also treated with dasatinib, a dual Bcr-Abl and Src inhibitor, but had failed to achieve a cytogenetic remission. As shown in Table 1, other than M351T mutation detected in BCR-ABL gene in patient 1, no mutations were found in other patients. Nevertheless, most of them were insensitive to TKIs in agreement with literature reports. Regardless of their response to TKIs, all samples were sensitive to triptolide in vitro. As shown in Fig 1C and Table 1, triptolide induced significant cell death in all samples studied at 24 hours, indicating that triptolide induces death of CML blast cells in vitro independent of patients' clinical responses to imatinib and other Bcr-Abl TKIs. It is important to point out that with 50 or 100 nM of triptolide, approximately 40% or 50% of cells, respectively, lost viability in those samples, while our previous study showed that at the same concentrations, >90% or about 75% of CD34+ cells, respectively, from normal BM samples were viable.(27)
Table 1. Characteristics of CML Patients Treated with Triptolide in vitro.
| in vitro triptolide treatment | |||||||
|---|---|---|---|---|---|---|---|
| patient | blast % | sample source | in vivo treatments and responses at the time of sampling | Bcr-Abl mutation status | BC lineage | IC50 (nM) | western blot |
| 1 | 49 | PB | Resistant to imatinib and nilotinib | M351T | myeloid | 32.5 | XIAP |
| 2 | 62 | PB | Resistant to imatinib | wt | myeloid | 178.9 | XIAP, Mcl-1, Bcr-Abl |
| 3 | 80 | PB | Resistant to imatinib, nilotinib and other therapies | wt | myeloid | 84.1 | - |
| 3a | 83 | BM | wt | 87.9 | - | ||
| 4 | 91 | PB | Hydrea, later died | wt | myeloid | 43.0 | - |
| 5 | 87 | PB | no cytogenetic remission with imatinib, dasatinib, or nilotinib | wt | myeloid | 111.7 | XIAP, Mcl-1, Bcr-Abl |
| 6 | 64 | PB | failed imatinib and other chemotherapies | wt | bilineage | 206.2 | XIAP, Mcl-1, Bcr-Abl |
Triptolide decreases antiapoptotic XIAP and Mcl-1 as well as CML-causative Bcr-Abl levels
Next, we investigated the involvement of apoptosis regulators in triptolide-induced cell death in CML cells. Triptolide at 100 nM decreased XIAP and Mcl-1 mRNA levels in a time-dependent manner in K562 cells (Fig 2A). Lou Y et al.(37) had reported that triptolide decreased BCR-ABL RNA levels in K562 cells. We therefore analyzed XIAP, Mcl-1, and Bcr-Abl protein levels in triptolide-treated CML cells. Results showed that triptolide indeed induced dose- and time-dependent decreases in XIAP, Mcl-1, and Bcr-Abl protein levels in K562 cells (Fig 2B). To demonstrate that triptolide has a similar effect on imatinib resistant cells, we measured XIAP, Mcl-1, and Bcr-Abl protein levels in triptolide treated KBM5 and KBM5STI571 cells. As shown in Fig 2C, triptolide treatment resulted in decreases of XIAP, Mcl-1, and Bcr-Abl protein levels in both cell lines. The responses of KBM5 and KBM5STI571 cells to imatinib were also verified and shown in Fig 2C. We next determined the levels of these proteins in triptolide-treated primary blast cells from CML patients and Fig 2D shows that XIAP, Mcl-1, and Bcr-Abl protein levels decreased in a dose-dependent manner. These were the same samples that were used in the in vitro triptolide experiments shown in Fig 1C. Because of the limited number of cells available in some samples, XIAP protein levels were determined in four and Bcr-Abl and Mcl-1 protein levels in three of the seven samples (Table 1).
Figure 2.
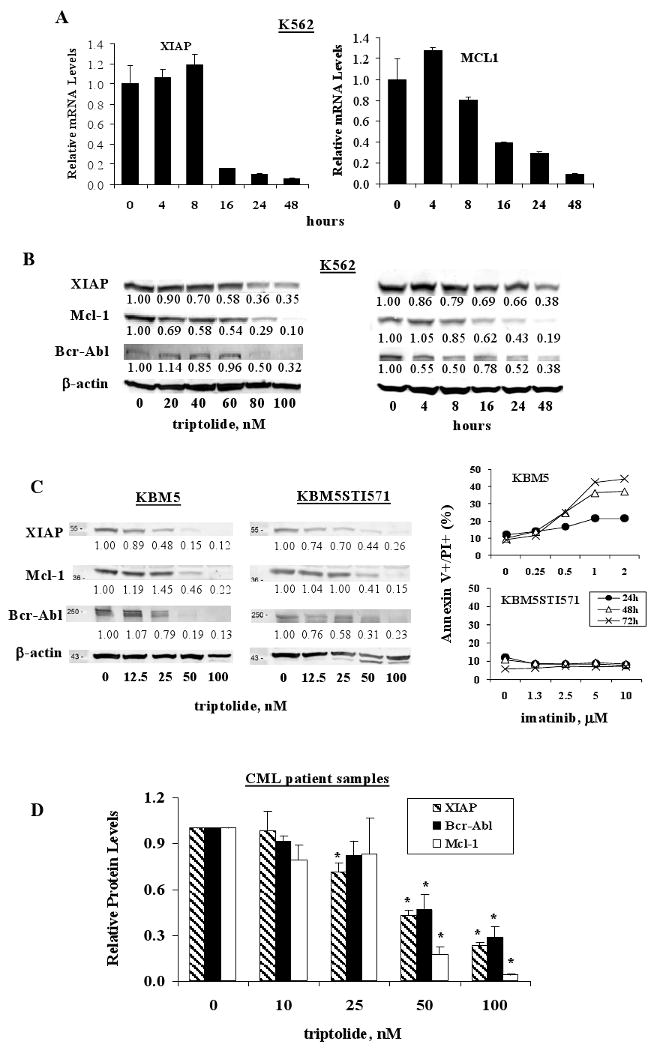
Triptolide decreases XIAP, Mcl-1, and Bcr-Abl levels in CML cells. (A) K562 cells were treated with 100 nM of triptolide. XIAP and Mcl-1 mRNA levels were determined at various time points by Real-time RT-PCR. (B) K562 cells were treated with various concentrations of triptolide for 48 hours or with 100 nM of triptolide for various times. (C) KBM5 and KBM5STI571 cells were treated with triptolide for 24 hours and with imatinib for 24 to 72 hours and (D) cells from patients with CML were treated with triptolide for 24 hours. XIAP, Mcl-1, and Bcr-Abl protein levels were determined by western blot analysis and cell death was determined by annexin V staining.
Triptolide sensitizes CML cells to imatinib-induced cell death
Based on the observation that triptolide decreases the levels not only of antiapoptotic proteins XIAP and Mcl-1 but also of the CML-causative Bcr-Abl protein, we hypothesized that the combination of triptolide and imatinib would be more effective at inducing death in CML cells than each of the agents alone. To test this hypothesis, we treated KBM5 cells with both triptolide and imatinib and did not observe any sensitization. This was probably due to the fact that imatinib primarily inhibits cell growth at low concentrations and with short treatment which diminishes the killing effect of triptolide. We then treated KBM5 cells with triptolide for 24 hours and then added 1 μM imatinib for additional 24 hours. This combination treatment was more effective in inducing cell death in KBM5 cells (Figure 3, IC50=15.4 ± 0.6 nM, compared with IC50=24.3 ± 2.8 nM for triptolide alone). Triptolide did not sensitize KBM5STI571 cells to imatinib (results not shown).
Figure 3.

Combination of triptolide and imatinib enhances death in KBM5 cells. KBM5 cells were treated with triptolide for 24 hours and then plus 1 μM imatinib for an additional 24 hours. Cell death was determined by annexin V staining.
Triptolide induces death of quiescent CD34+ primitive CML cells
To test the effect of triptolide on the viability of quiescent CD34+ CML cells, which are known to be resistant to TKIs,(21;22) we first cultured CFSE-labeled cells from five CML patients (Table 2) with medium supplemented with a cocktail of growth factors (patients 1 and 2) or with MS-5 stromal cells (patients 3-5) for 4-7 days. Cell proliferation was tracked by flow cytometric analysis of CFSE fluorescence intensity and shown on the left panel of Fig 4 (gated on live cells by scattering of bulk population for patient 1 who had <2% CD34+ cells and of CD34+ cells for other patients). As illustrated in Fig 4, proliferating cells which represent the majority of blasts from all five patients were sensitive to triptolide, consistent with the results shown in Fig 1C. Quiescent CD34+ cells were in general as sensitive as proliferating CD34+ cells to triptolide: in four of the five patient samples (patients 1, 3-5), proliferating and quiescent cells responded similarly, undergoing the same degree of apoptosis. Only in cells from patient 2, quiescent CD34+ cells were resistant, while proliferating CD34+ cells were sensitive to triptolide. The sensitivity of CD34+ cells to triptolide was not affected by pre-culturing these cells with growth factors or with MS-5 cells (Fig 4). Note as shown in Table 2, 4 out of 5 patients had mutation in BCR-ABL gene and patient 4 had T315I mutation.
Table 2. Characteristics of Patients with CML whose CD34+ Cells were Used for Testing Triptolide Sensitivity.
| patient | source | blast % | treatment and responses at the time of sampling | Bcr-Abl mutation status | BC lineage |
|---|---|---|---|---|---|
| 1 | PB | 59 | resistant to or relapsed from various treatment including Interferon, imatinib, dasatinib, fludarabine+Ara-C bid, and intravenous homoharringtonine + imatinib | F317L | myeloid |
| 2 | PB | 17 | resistant to various therapies including Interferon + low dose Ara-C, imatinib, nilotinib, and dasatinib | wt | myeloid |
| 3 | PB | 58 | resistant to imatinib and nilotinib | F317L | lymphoid |
| 4 | PB | 58 | achieved short hematological remission with Idarubicin+Ara-C / imatinib but soon after had evidence of Ph+ chromosome, resistant to dasatinib and MK-0457 | T315I | myeloid |
| 5 | BM | 60 | achieved hematological response with imatinib and dasatinib, achieved and then lost cytogenetic response to dasatinib, was on bosutinib | V299L | myeloid |
Figure 4.
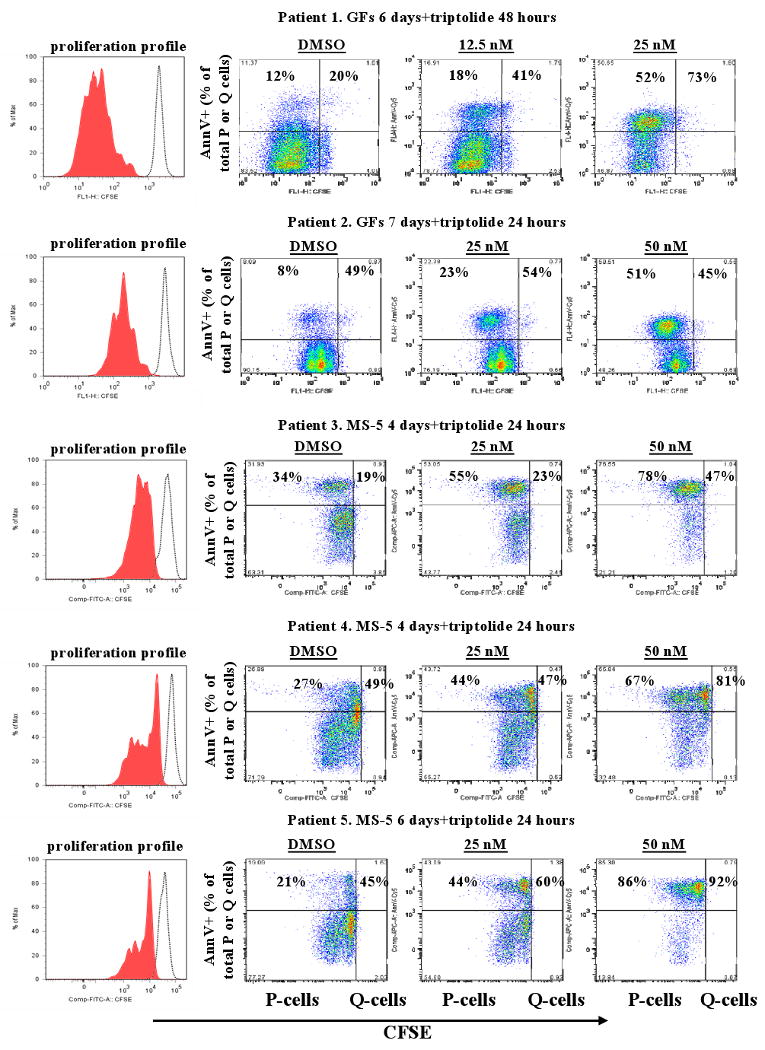
Triptolide induces the death of both quiescent and proliferating CD34+ cells from patients with CML. Profiles of CFSE-labeled cells from patients with CML were shown on the left panel. Shaded area represents proliferation profile at the time of annexin V assay while open area shows the fluorescence profiles of cells before starting the culture. Cells were cultured with growth factors or co-cultured with MS-5 cells for 4-7 days and then treated with triptolide for 24 or 48 hours. Apoptosis in quiescent and proliferating CD34+ cells was determined using annexin V staining and shown as % positivity of the respective populations. GF, growth factor; P, proliferating; Q, quiescent.
Discussion
Our study demonstrated that triptolide potently induces apoptosis of BC CML cells from both cell lines and primary patient samples, independent of the BCR-ABL mutation status and response to imatinib and other TKIs, at least in part, by decreasing XIAP, Mcl-1, and Bcr-Abl proteins. The triptolide-induced decrease in these protein levels is likely part of the mechanism by which triptolide induces death of Bcr-Abl-expressing cells and overcomes TKI resistance. More importantly, triptolide promotes the death not only of proliferating but also of quiescent CD34+ primitive CML cells.
Generation of the Bcr-Abl fusion gene is the causative genetic defect in early stage CML. With disease progression, additional chromosomal and molecular changes occur and blasts from patients with BC CML show enhanced proliferation and survival. Hence, it is not surprising that imatinib and other TKIs are not very effective in patients with advanced CML. Triptolide decreases not only Bcr-Abl protein level, but also the levels of XIAP and Mcl-1, two potent antiapoptotic proteins. These attributes of triptolide give it a distinct advantage over imatinib and other TKIs which affect only proliferating cells; in particular in decreasing Bcr-Abl protein levels, and by lowering the apoptotic threshold and activating the apoptotic cascade. This ability of triptolide to decrease Bcr-Abl, XIAP, and Mcl-1 levels also explains why triptolide-induced cell death is independent of cellular response to imatinib or other second-generation Bcr-Abl TKIs and occurs in quiescent cells. Mcl-1 expression was reported to be dependent on BCR-ABL in CML,(27;28) therefore the decrease in Bcr-Abl levels by triptolide may result in further decrease in Mcl-1 levels in triptolide-treated CML cells. In our experiments, Bcr-Abl protein decreased prior to Mcl-1 (Fig 2B) supporting the notion that Mcl-1 is a downstream target of Bcr-Abl.
Ba/F3vec cells, the vector control without Bcr-Abl, were also killed by triptolide. These cells were cultured in a medium supplemented with IL-3 which activates various survival pathways of the cells. Since triptolide also induces cell death by decreasing survival proteins XIAP and Mcl-1 independent of Bcr-Abl, it is not surprising that they are sensitive to triptolide.
We have previously reported that although triptolide inhibits colony formation of normal BM cells, normal CD34+ cells are less sensitive than AML blasts to triptolide.(27) This is probably due to the fact that apoptotic pathways are often deregulated in leukemia cells and these cells are more than normal cells dependent on XIAP, Mcl-1, Bcr-Abl in the case of CML, and other antiapoptotic proteins for survival, as supported by the oncogene addiction theory.(38) Given the resistance of quiescent CML stem cells to most anticancer agents, the ability of triptolide to promote the death of quiescent CD34+ CML cells warrants the clinical development of this agent. It was reported that PG490-88, a water-soluble derivative of triptolide, at 0.25 mg/kg markedly decreased tumor growth and at 0.5 and 0.75 mg/kg doses caused profound tumor regression without apparent toxicities in nude mouse human tumor xenograft models.(39) A decrease in white blood cells was reported in clinical trials with triptolide or extracts of Tripterygium wilfordii.(40;41) A phase I clinical trial with a water-soluble derivative of triptolide in solid tumors is presently ongoing and importantly, a clinical Phase I trial in France has determined the MTD for triptolide and reported 3 CRs out of 26 patients with AML.(42) The importance of the hematopoietic microenvironment in the maintenance and differentiation of hematopoietic progenitor cells has been recognized only in the past few years. Both in vivo and in vitro the growth, survival, and differentiation of hematopoietic cells have been found to require direct contact with MSCs, which produce various cytokines and chemokines.(43-47) MSC interacts similarly with leukemic cells in vivo and provide a protective microenvironment that enables leukemic cells to proliferate and survive. For these reasons, a cocktail of growth factors is commonly used for the maintenance of CD34+ CML progenitor cells in vitro, as was done in the current study. We found that the sensitivity of CD34+ cells to triptolide is not affected by pre-culturing these cells with either the cocktail (Fig 4, patients 1 and 2) or with MS-5 cells (Fig 4, patients 3-5).
Taken together, results show that triptolide has the potential of complementing the activity of imatinib and other TKIs by effective killing bulk CML cells in blast crisis and depleting quiescent as well as proliferating CD34+ primitive CML progenitor cells, independent of their sensitivity to TKIs. Thus, triptolide could be a novel agent in patients with CML in accelerated and blast crisis, in patients not responsive to TKIs including those with the T315I mutation, and in eradicating quiescent CD34+ CML progenitor cells.
Our findings of effects of triptolide on CML are in agreement with Shi et al's results(29) that triptolide decreases Bcr-Abl, XIAP, and Mcl-1 protein levels in CML cell lines and our studies extend these results to primary CML samples. Importantly, we here demonstrate that triptolide sensitizes to imatinib-induced cell death and has major activity in quiescent primitive CD34+ CML progenitor cells.
Acknowledgments
We would like to thank Elena S. Vess, Bradley S. Tadlock, Betty L. Notzon, and Angelique L. Geehan for helping with the manuscript preparation.
Supported in part by grants from Elsa Pardee Foundation to BZC and the National Institutes of Health (P01 CA49639, P01 CA55164, and CA16672) to MA
Footnotes
Authors have no conflict of interest to disclose
Reference List
- 1.Jandl JH. Blood: Pathophysiology. Boston: Blackwell Scientific Publication, Inc.; 1991. [Google Scholar]
- 2.Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci U S A. 1990;87(17):6649–6653. doi: 10.1073/pnas.87.17.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 4.Elefanty AG, Hariharan IK, Cory S. bcr-abl, the hallmark of chronic myeloid leukaemia in man, induces multiple haemopoietic neoplasms in mice. EMBO J. 1990;9:1069–1078. doi: 10.1002/j.1460-2075.1990.tb08212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantarjian HM, Cortes J, O'Brien S, Giles FJ, Albitar M, Rios MB, et al. Imatinib mesylate (STI571) therapy for Philadelphia chromosome-positive chronic myelogenous leukemia in blast phase. Blood. 2002;99:3547–3553. doi: 10.1182/blood.v99.10.3547. [DOI] [PubMed] [Google Scholar]
- 6.Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–3539. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 7.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 8.le Coutre P, Tassi E, Varella-Garcia M, Barni R, Mologni L, Cabrita G, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 2000;95:1758–1766. [PubMed] [Google Scholar]
- 9.Weisberg E, Griffin JD. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell lines. Blood. 2000;95:3498–3505. [PubMed] [Google Scholar]
- 10.Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–3475. doi: 10.1182/blood.v99.9.3472. [DOI] [PubMed] [Google Scholar]
- 11.von Bubnoff N, Schneller F, Peschel C, Duyster J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002;359:487–491. doi: 10.1016/S0140-6736(02)07679-1. [DOI] [PubMed] [Google Scholar]
- 12.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Lai JL, Philipp N, Facon T, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–1018. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 13.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 14.Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T, Gloc E, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104(12):3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 15.Bartholomeusz GA, Talpaz M, Kapuria V, Kong LY, Wang S, Estrov Z, et al. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood. 2007;109(8):3470–3478. doi: 10.1182/blood-2006-02-005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiotsu Y, Soga S, Akinaga S. Heat shock protein 90-antagonist destabilizes Bcr-Abl/HSP90 chaperone complex. Leuk Lymphoma. 2002;43(5):961–968. doi: 10.1080/10428190290021371. [DOI] [PubMed] [Google Scholar]
- 17.Cobaleda C, Sanchez-Garcia I. In vivo inhibition by a site-specific catalytic RNA subunit of RNase P designed against the BCR-ABL oncogenic products: a novel approach for cancer treatment. Blood. 2000;95(3):731–737. [PubMed] [Google Scholar]
- 18.Eaves C, Cashman J, Eaves A. Defective regulation of leukemic hematopoiesis in chronic myeloid leukemia. Leuk Res. 1998;22(12):1085–1096. doi: 10.1016/s0145-2126(98)00113-1. [DOI] [PubMed] [Google Scholar]
- 19.Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood. 1999;94(6):2056–2064. [PubMed] [Google Scholar]
- 20.Elrick LJ, Jorgensen HG, Mountford JC, Holyoake TL. Punish the parent not the progeny. Blood. 2005;105(5):1862–1866. doi: 10.1182/blood-2004-08-3373. [DOI] [PubMed] [Google Scholar]
- 21.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 22.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML, but does not eliminate the quiescent fraction. Blood. 2006;107:4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 23.Shamon LA, Pezzuto JM, Graves JM, Mehta RR, Wangcharoentrakul S, Sangsuwan R, et al. Evaluation of the mutagenic, cytotoxic, and antitumor potential of triptolide, a highly oxygenated diterpene isolated from Tripterygium wilfordii. Cancer Lett. 1997;112(1):113–117. doi: 10.1016/S0304-3835(96)04554-5. [DOI] [PubMed] [Google Scholar]
- 24.Yang S, Chen J, Guo Z, Xu XM, Wang L, Pei XF, et al. Triptolide inhibits the growth and metastasis of solid tumors. Mol Cancer Ther. 2003;2(1):65–72. [PubMed] [Google Scholar]
- 25.Kiviharju TM, Lecane PS, Sellers RG, Peehl DM. Antiproliferative and proapoptotic activities of triptolide (PG490), a natural product entering clinical trials, on primary cultures of human prostatic epithelial cells. Clin Cancer Res. 2002;8(8):2666–2674. [PubMed] [Google Scholar]
- 26.Yang Y, Liu Z, Tolosa E, Yang J, Li L. Triptolide induces apoptotic death of T lymphocyte. Immunopharmacology. 1998;40(2):139–149. doi: 10.1016/s0162-3109(98)00036-8. [DOI] [PubMed] [Google Scholar]
- 27.Carter BZ, Mak DH, Schober WD, McQueen T, Harris D, Estrov Z, et al. Triptolide induces caspase-dependent cell death mediated via the mitochondrial pathway in leukemic cells. Blood. 2006;108(2):630–637. doi: 10.1182/blood-2005-09-3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105(8):3303–3311. doi: 10.1182/blood-2004-02-0749. [DOI] [PubMed] [Google Scholar]
- 29.Shi X, Jin Y, Cheng C, Zhang H, Zou W, Zheng Q, et al. Triptolide Inhibits Bcr-Abl Transcription and Induces Apoptosis in STI571-resistant Chronic Myelogenous Leukemia Cells Harboring T315I Mutation. Clin Cancer Res. 2009;15(5):1686–1697. doi: 10.1158/1078-0432.CCR-08-2141. [DOI] [PubMed] [Google Scholar]
- 30.Beran M, Pisa P, O'Brien S, Kurzrock R, Siciliano M, Cork A, et al. Biological properties and growth in SCID mice of a new myelogenous leukemia cell line (KBM-5) derived from chronic myelogenous leukemia cells in the blastic phase. Cancer Res. 1993;53:3603–3610. [PubMed] [Google Scholar]
- 31.Ricci C, Scappini B, Divoky V, Gatto S, Onida F, Verstovsek S, et al. Mutation in the ATP-binding pocket of the ABL kinase domain in an STI571-resistant BCR/ABL-positive cell line. Cancer Res. 2002;62:5995–5998. [PubMed] [Google Scholar]
- 32.Holtz MS, Slovak ML, Zhang F, Sawyers CL, Forman SJ, Bhatia R. Imatinib mesylate (STI571) inhibits growth of primitive malignant progenitors in chronic myelogenous leukemia through reversal of abnormally increased proliferation. Blood. 2002;99(10):3792–3800. doi: 10.1182/blood.v99.10.3792. [DOI] [PubMed] [Google Scholar]
- 33.Itoh K, Tezuka H, Sakoda H, Konno M, Nagata K, Uchiyama T, et al. Reproducible establishment of hemopoietic supportive stromal cell lines from murine bone marrow. Exp Hematol. 1989;17:145–153. [PubMed] [Google Scholar]
- 34.Issaad C, Croisille L, Katz A, Vainchenker W, Coulombel L. A murine stromal cell line allows the proliferation of very primitive human CD34++/CD38- progenitor cells in long-term cultures and semisolid assays. Blood. 1993;81:2916–2924. [PubMed] [Google Scholar]
- 35.Konopleva M, Konoplev S, Hu W, Zaritskey AY, Afanasiev BV, Andreeff M. Stroma cells prevent apoptosis of AML cells by upregulation of anti-apoptotic proteins. Leukemia. 2002;16:1713–1724. doi: 10.1038/sj.leu.2402608. [DOI] [PubMed] [Google Scholar]
- 36.Carter BZ, Milella M, Altieri DC, Andreeff M. Cytokine-regulated expression of survivin in myeloid leukemia. Blood. 2001;97:2784–2790. doi: 10.1182/blood.v97.9.2784. [DOI] [PubMed] [Google Scholar]
- 37.Lou YJ, Jin J. Triptolide down-regulates bcr-abl expression and induces apoptosis in chronic myelogenous leukemia cells. Leuk Lymphoma. 2004;45(2):373–376. doi: 10.1080/1042819031000139710. [DOI] [PubMed] [Google Scholar]
- 38.Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3(8):448–457. doi: 10.1038/ncponc0558. [DOI] [PubMed] [Google Scholar]
- 39.Fidler JM, Li K, Chung C, Wei K, Ross JA, Gao M, et al. PG490-88, a derivative of triptolide, causes tumor regression and sensitizes tumors to chemotherapy. Mol Cancer Ther. 2003;2(9):855–862. [PubMed] [Google Scholar]
- 40.Lipsky PE, Tao XL. A potential new treatment for rheumatoid arthritis: thunder god vine. Semin Arthritis Rheum. 1997;26(5):713–723. doi: 10.1016/s0049-0172(97)80040-6. [DOI] [PubMed] [Google Scholar]
- 41.Wu SX, Guo NR. Clinical observation on effect of triptolide tablet in treating patients with psoriasis vulgaris. Chin J Integr Med. 2005;11(2):147–148. doi: 10.1007/BF02836473. [DOI] [PubMed] [Google Scholar]
- 42.Harousseau JLH, Dombret HD, Pigneux AP, Michallet MM, Brandely MB. Phase I study of F60008, a triptolide derivative, in patients with refractory or relapsing acute leukemias. 13th Congress of the European Hematology Association (EHA); 2008. [Google Scholar]
- 43.Dexter TM. Regulation of hemopoietic cell growth and development: experimental and clinical studies. Leukemia. 1989;7:469–474. [PubMed] [Google Scholar]
- 44.Dorshkind K. Regulation of hemopoiesis by bone marrow stromal cells and their products. Annu Rev Immunol. 1990;8:111–137. doi: 10.1146/annurev.iy.08.040190.000551. [DOI] [PubMed] [Google Scholar]
- 45.Asosingh K, Gunthert U, Bakkus MH, De Raeve H, Goes E, Van Riet I, et al. In vivo induction of insulin-like growth factor-I receptor and CD44v6 confers homing and adhesion to murine multiple myeloma cells. Cancer Res. 2000;60:3096–3104. [PubMed] [Google Scholar]
- 46.Shimakura Y, Kawada H, Ando K, Sato T, Nakamura Y, Tsuji T, et al. Murine stromal cell line HESS-5 maintains reconstituting ability of ex vivo-generated hematopoietic stem cells from human bone marrow and cytokine-mobilized peripheral blood. Stem Cells. 2000;18:183–189. doi: 10.1634/stemcells.18-3-183. [DOI] [PubMed] [Google Scholar]
- 47.Koller MR, Oxender M, Jensen TC, Goltry KL, Smith AK. Direct contact between CD34+lin- cells and stroma induces a soluble activity that specifically increases primitive hematopoietic cell production. Exp Hematol. 1999;27:734–741. doi: 10.1016/s0301-472x(98)00080-0. [DOI] [PubMed] [Google Scholar]