


Abstract
We developed an integrated method to identify aptamers with only 10 fixed nucleotides through ligation and removal of primer binding sites within the systematic evolution of ligands by exponential enrichment (SELEX) process. This Tailored-SELEX approach was validated by identifying a Spiegelmer (‘mirror-image aptamer’) that inhibits the action of the migraine-associated target calcitonin gene-related peptide 1 (α-CGRP) with an IC50 of 3 nM at 37°C in cell culture. Aptamers are oligonucleotide ligands that can be generated to bind to targets with high affinity and specificity. Stabilized aptamers and Spiegelmers have shown activity in vivo and may be used as therapeutics. Aptamers are isolated by in vitro selection from combinatorial nucleic acid libraries that are composed of a central randomized region and additional fixed primer binding sites with ∼30–40 nt. The identified sequences are usually not short enough for efficient chemical Spiegelmer synthesis, post-SELEX stabilization of aptamers and economical production. If the terminal primer binding sites are part of the target recognizing domain, truncation of aptamers has proven difficult and laborious. Tailored-SELEX results in short sequences that can be tested more rapidly in biological systems. Currently, our identified CGRP binding Spiegelmer serves as a lead compound for in vivo studies.
INTRODUCTION
Since the invention of in vitro selection of oligonucleotides from combinatorial nucleic acid libraries, also known as systematic evolution of ligands by exponential enrichment (SELEX), the use of these molecules (termed aptamers) as therapeutics, e.g. for the specific interruption of disease-related protein–protein interactions, has been predicted and aspired (1–3). Aptamers usually show binding constants to their respective protein or peptide targets in the same range as most receptor–ligand interactions and exhibit a high substrate specificity (4–6).
In order to move aptamers from the bench to the clinic, several hurdles have to be taken. The most prominent ones are the issues of aptamer stability in biological fluids and production costs. Stabilization of aptamers against abundant nucleases has been improved by the introduction of chemically modified nucleic acid libraries (pre-SELEX modifications) in combination with post-SELEX modifications, that both rely on the substitution of the RNA’s 2′-OH group (7–10). In a second strategy, chiral principles were introduced into the SELEX process in order to generate nuclease resistant aptamers on the basis of l-RNA or l-DNA, so-called Spiegelmers (from the German ‘Spiegel’, meaning mirror) (11). Spiegelmers are identified through in vitro selection of an unmodified d-RNA or d-DNA library against the mirror-image configuration (enantiomer) of a drug target. The selected aptamer sequences are then synthesized in their unnatural enantiomeric configuration as l-RNAs or l-DNAs. Following the rules of symmetry, these Spiegelmers bind to the natural target of interest just like the aptamers bind to the mirror-image selection target (11–13). Aptamers with pre- and post-SELEX modifications as well as Spiegelmers have been reported to be stable for many hours in biological fluids (11,14).
For both strategies, the ability to chemically synthesize the lead candidates is crucial, since neither post-SELEX-modified aptamers nor Spiegelmers can be synthesized enzymatically due to the lack of appropriate enzymes. However, aptamers and Spiegelmers that are identified through the standard SELEX process usually comprise 60–90 nt, since they are typically selected from nucleic acid libraries with 30–40 nt long randomized regions plus fixed primer sites of ∼15–25 nt on each side. Standard chemical oligoribonucleotide synthesis, however, is only efficiently applicable up to 60 nt, with decreasing yields and escalating production costs for every incorporated base.
Therefore, the identified lead oligonucleotides need rational and experimental truncation before they can be further tested in biological systems (15). Whether or not the truncation of a given lead aptamer or Spiegelmer will eventually succeed, may not be foreseen. The flanking fixed regions sometimes participate in forming the scaffold that surrounds the binding interface and may thus not simply be omitted (16–18).
Here we describe a methodology that allows the direct and rapid isolation of target binding RNA sequences that only require 10 fixed nucleotides in addition to the random region. The novel procedure, which we named Tailored-SELEX, relies on customized primers/adapters that are added by ligation before and removed within the amplification processes.
In order to prove the efficiency of Tailored-SELEX, we carried out an in vitro selection approach against the optical antipode of the neuropeptide calcitonin gene-related peptide 1 (α-CGRP) of rat. α-CGRP has been recognized as a potent vasodilator and has recently attracted attention as a novel target in acute migraine treatment (19–22).
MATERIALS AND METHODS
Oligonucleotides and peptides
All oligonucleotides were synthesized at NOXXON Pharma AG using standard phosphoramidite chemistry. C-terminal biotinylated rat d-α-CGRP [all-d-(Ser–Cys–Asn–Thr–Ala–Thr–Cys–Val–Thr–His–Arg–Leu–Ala–Gly–Leu–Leu–Ser–Arg–Ser–Gly–Gly–Val–Val–Lys–Asp–Asn–Phe–Val–Pro–Thr–Asn–Val–Gly–Ser–Glu–Ala–Phe)-ε-biotinyl-Lys-NH2] and rat l-α-CGRP were purchased from Bachem (Bubendorf, Switzerland).
In vitro selection procedure
Three complexities of a combinatorial RNA library (5′-GGAC-N40-GACAGG) of 1015 different molecules, that was obtained by in vitro transcription of a synthetic ssDNA library (5′-COMeCOMeTGTC-N40-GTCCTATAGTGAGTCGTATTAGTAGTCGC) in the presence of a 1.5-fold excess of the forward primer (5′-GCGACTACTAATACGACTCACTATAGGAC), were used as the starting library. The last two nucleotides of the 5′ terminus of the DNA library were modified with 2′-methoxy-cytidines in order to reduce the non-templated nucleotide addition at the 3′ terminus of the in vitro transcripts.
Before each selection round, the RNA library was dissolved in 10 mM HEPES/KOH pH 7.4, 150 mM NaCl, 4 mM KCl and denatured (95°C, 5 min). It was then allowed to cool to 23 or 37°C for 15 min and CaCl2 (final 1 mM), MgCl2 (final 1 mM) and Tween (final 0.05%) were added to give the final selection buffer. In the first five rounds the biotinylated rat d-α-CGRP concentration was 10 µM. In later rounds the peptide concentration was consecutively reduced to reach 1 nM in round 14. The selection process was carried out at 23°C for rounds 1–9 and at 37°C for rounds 10–15 (see Fig. 5). Neutravidin Agarose or Streptavidin Ultralink beads (Pierce, Rockford, IL) were utilized as selection matrices for the immobilization of the RNA–peptide complexes. The matrix was equilibrated with selection buffer before use. The selection process included a pre-column to prevent accumulation of non-specific matrix binders (pure selection matrix, 30 min incubation) from round 2 onwards. The binding reaction of RNA and peptide was done in solution for 2 up to 12 h. The complexes of RNA and biotinylated rat d-α-CGRP were immobilized for 30 min on the selection matrix. Non-binding oligonucleotides were removed by washing with 10–25 matrix volumes of selection buffer. Bound RNA was eluted twice (8 M urea, 10 mM EDTA, 65°C, 15 min), extracted with phenol/chloroform/isoamyl alcohol, precipitated with ethanol and dissolved in water.
Figure 5.

Course of the in vitro selection. The histogram shows the course of the in vitro selection. The fraction of the RNA pool eluted from the underivatized streptavidin or neutravidin matrix (yellow bars) and from the identical matrix after capturing RNA:peptide complexes from a solution (green bars) with the indicated peptide concentration (red triangles) is shown. Starting from round 6, selection was usually performed at three different peptide concentrations. Only the data of the minimal successful peptide concentration are shown.
Ligation and amplification
The ligation reaction was done at 25°C for 12 h [71.4 or 357 nM RNA with 36 or 7.2 UT4 DNA ligase/pmolRNA (Fermentas, St Leon-Roth, Germany), respectively, depending on the amount of eluted RNA, 1× ligation buffer (Fermentas), 5% PEG 4000, 0.5 µl of RNaseOUT (Invitrogen, Karlsruhe, Germany), a 20-fold excess of forward adapter and a 20-fold excess of reverse adapters]. The adapters are double-stranded oligonucleotides: forward adapter [forward ligate (RNA, 5′-GCG ACUACUAAUACGACUCACUAUA) plus forward bridge (DNA, 5′-GTCCTATAGTGAGTCG-3′dT) with 2′-3′-dideoxythymidine at the 3′ terminus], reverse adapter 1 [reverse ligate 1 (DNA, pACGCTGAGCTGAACTCG-3′dC, 5′-phosphorylated and 3′-blocked) plus reverse bridge 1 (DNA, 5′-GCGAGT TCAGCUOHCAGCGUOHCCTGTC) with two incorporated ribonucleotides] and reverse adapter 2 [reverse ligate 2 (DNA, 5′-pCGCTGAGCTGAACTCG-3′dC, 5′-phosphorylated and 3′-blocked) plus reverse bridge 2 (DNA, 5′-GCGAGTTCAGCUOHCAGCGIOHCCTGTC) with two incorporated ribonucleotides]. Reverse adapter 2 was added to the ligation reaction (10-fold excess of reverse adapter 1 and 2 each) from round 2 onwards. The reverse transcription reaction was carried out without intermediate purification at a final RNA concentration of 30 or 150 nM for 20 min at 51°C and 10 min at 54°C [1× First strand buffer (Invitrogen), 1× Q solution (Qiagen, Hilden, Germany), 0.5 mM dNTPs (Larova, Teltow, Germany), 4 U/µl Superscript Reverse Transcriptase II (Invitrogen), 10 mM DTT]. The cDNA was directly transferred to the PCR [1–10 nM cDNA, 1× PCR buffer (Roche), 5 µM forward primer, 5 µM reverse primer, 0.2 mM dNTPs (Larova), 0.05 U/µl Taq DNA polymerase (Roche, Mannheim, Germany), annealing temperature at 68°C, 7–15 cycles]. The cleavage of the PCR reverse strand was done by alkaline fission of the ribonucleotides [310 mM (final) NaOH, 10 min at 95°C, neutralization with HCl, buffered with 0.1 mM (final) sodium acetate]. The PCR product was ethanol precipitated before in vitro transcription [80 mM HEPES/KOH pH 7.5, 22 mM MgCl2, 1 mM spermidine, 10 mM DTT, 1.2 µg/µl BSA, [α-32P]GTP (Hartmann, Braunschweig, Germany), 4 mM NTPs (Larova), 32 mM 5′-GMP (Sigma, Taufkirchen, Germany), 1 µl RNaseOUT (Invitrogen), 0.1 U/µl T7 RNA polymerase (Stratagene, La Jolla, CA) at 37°C for 6–12 h]. The transcribed RNA was gel-purified (23). The enriched library from round 15 was ligated and PCR amplified with all DNA primers before cloning and sequencing (GATC, Konstanz, Germany).
Bioassay
The bioassay was performed with SK-N-MC human neuroblastoma cells (DSMZ, Braunschweig, Germany) which endogenously express the CGRP1 receptor. Cells were grown at 37°C and 5% CO2 in DMEM (1 mg/l glucose), further containing 10% heat-inactivated fetal calf serum, 4 mM l-alanyl-l-glutamine, 50 U/ml penicillin, 50 µg/ml streptomycin, and, for experiments, seeded at 4 × 104 in 96-well microtiter plates. The Spiegelmers were pre-incubated in different concentrations with 1 nM rat α-CGRP in Hank’s balanced salt solution plus 1 mg/ml BSA (60 min, 37°C). Twenty minutes before the stimulation, the cells were pre-treated with 1 mM isobutyl-1-methylxanthin (IBMX). IBMX (1 mM) was also added to the CGRP/Spiegelmer solutions. For stimulation, the medium was removed and the CGRP/Spiegelmer mixtures were added to the cells. After 30 min stimulation at 37°C, the cells were lysed and the cAMP content of the cell extract was quantified using cAMP-Screen™ System kits (Applied Biosystems, Foster City, CA). The luminescence signal was detected by a POLARstar Galaxy multi-detection microplate reader (BMG, Offenburg, Germany).
RESULTS
The conventional SELEX process, which comprises alternating steps of selection and amplification, is carried out with oligonucleotide libraries consisting of 60–90 nt. The method we have devised is performed with a 50 nt long library that consists of a 40 nt long randomized region plus a total of 10 fixed flanking nucleotides. The fixed regions are necessary for an efficient ligation of the primer binding sites that are needed for the amplification steps. These oligonucleotides (referred to as ligates) are enzymatically ligated to the core library with the assistance of bridging oligonucleotides (bridges) that are complementary to one of the ligates and the respective fixed sequence of the RNA library (Fig. 1A).
Figure 1.

(A) Cartoon of the RNA library and the double-stranded adapters each containing a ligate and an oligonucleotide bridge, before the ligation of the primer binding sites. The library consists of a randomized region that is flanked by 4 and 6 nt long stretches of fixed sequence (green). They serve as hybridization sites for the bridging oligonucleotides of the pre-annealed double-stranded adapters. The forward ligate contains a T7 RNA polymerase promoter at its 3′ end. Reverse bridge 1 is also used as a PCR reverse primer. Two nucleotides in the reverse bridge 1 are uridines (U) which allow for primer removal under alkaline conditions. Forward bridge and reverse ligates contain a 3′ terminal 2′-3′-dideoxynucleotide (3′H) to prevent them from mispriming in the PCR. (B) Up to 50% of all run-off transcripts contain a non-templated nucleotide (N) at their 3′ ends (red). In order to ligate these species, an alternative adapter 2 was designed. It consists of a reverse ligate 2 that lacks the 5′ terminal adenosine (A) and a reverse bridge 2 that offers the universal base inosine for hybridization opposite to the additional nucleotide. Thus, the overall length of the library does not increase.
All reactions that contribute to the amplification, including ligation and removal of the primers before the next selection round, may be pursued in a single reaction vessel without the need for additional purification steps (Fig. 2).
Figure 2.

Flowchart of the tailored selection scheme. The amplification steps of the Tailored-SELEX process may be performed within a single tube. The process begins with the ligation of primer binding sites to those species within the nucleic acid pool that bind to a target of interest. The ligation is assisted by deoxyoligonucleotide bridges that span the ligation site. The reverse bridges also serve as cDNA and PCR primer in the subsequent reactions. The reverse strand of the PCR product is then cleaved under alkaline conditions at a predetermined cleavage site that was introduced via the reverse primer. After neutralization and an optional ethanol precipitation, the truncated reverse strand serves as a template for the in vitro transcription which is followed by RNA purification by PAGE or by DNase treatment and spin column filtration.
Ligation
Since the ligation of the primer binding sites takes place with a finite number of molecules that survived the selection step, high ligation yields are desirable in order to propagate the binding sequences. This is particularly true in the first rounds, where only a few copies of each molecule are present. Ligation yields of 80% were attained with highly concentrated T4 DNA ligase and a DNA bridge assisted ligation strategy (24). The bridging is achieved by the overhanging reverse strands of double-stranded adapter molecules that are complementary to the fixed nucleotides of the library. Four and six fixed nucleotides at the 5′ and 3′ ends, respectively, proved to be the minimum required length of the fixed sequence parts. Nevertheless, three fixed nucleotides are necessary on the 5′ end to accommodate the transcription initiation sequence GG-purine. Furthermore, the ligation strategy proved functional not only for RNA, but also for 2′-F-pyrimidine-RNA.
Design of the forward adapter
The forward adapter consists of the forward ligate (the oligonucleotide that is ligated to the RNA) and the forward bridge. The adapter has a recessive 3′ end that is complementary to the four fixed nucleotides at the 5′ end of the library. While the forward bridge was made of DNA, the forward ligate was made from RNA, since RNA is a better substrate in the subsequent reverse transcription step. The forward adapter was chosen to contain a T7 RNA polymerase promoter directly upstream of the ligation site. The forward bridge was made up of only 17 nt to prevent hindrance of cDNA synthesis during reverse transcription at elevated temperatures.
Unused adapter molecules are carried over into the PCR, because no purification step between ligation and amplification is foreseen. PCR mispriming, however, is a common event during the amplification of nucleic acid libraries, due to the heterogeneous nature of the randomized region and the high primer concentrations commonly used. In order to avoid elongation of the forward bridge and potential ligation side products, we introduced a 2′-3′-dideoxy nucleotide at its 3′ end.
Design of the reverse adapters
Two different kinds of reverse adapters—both made predominantly from DNA—were used. The reverse adapter 1 was designed to accommodate RNA molecules with the 6 nt long 3′ fixed region at its 5′ recessive end. The reverse ligate 1 carries a 5′ phosphate since it is the phosphate donor in the ligation reaction. The 3′ end is blocked by a 2′-3′-dideoxynucleotide in analogy to the forward bridge. The reverse bridge 1 was not blocked since it serves as cDNA and PCR primer. In order to dispose of the primer region before the next selection step, two ribonucleotides were incorporated in the reverse bridge 1 so as to create predetermined breaking points at alkaline pH.
However, up to 50% of the products of run-off in vitro transcriptions with T7 RNA polymerase contain an additional nucleotide at their 3′ ends (25). This phenomenon, which is known as 3′ microheterogeneity, strongly confines the 3′ ligation efficiency of the bridge-assisted ligation because no undistorted duplex can be formed at the ligation site (26).
Hence, for an efficient ligation of the inevitable N + 1 transcripts that are formed within the repetitive SELEX process, we designed a reverse adapter 2. It consists of the 5′ phosphorylated 3′ blocked reverse ligate 2 that lacks the first 5′ nucleotide in order to accommodate the additional base of the N + 1 transcripts (Fig. 1B). Hybridized to the reverse ligate 2 is the reverse bridge 2, which is identical to reverse bridge 1 in all but one position: we substituted the uridine at the 3′ cleavage site with the universal nucleobase analog inosine that can base pair with any base at the transcript’s N + 1 position, so that T4 DNA ligase will ligate the paired ends. Analogous to the uridine in the reverse bridge 1, the inosine nucleotide also served as a predetermined fission point at alkaline pH. By using an equimolar mixture of the reverse adapters 1 and 2, overall ligation yields were improved from 40 up to 80% (Fig. 3).
Figure 3.
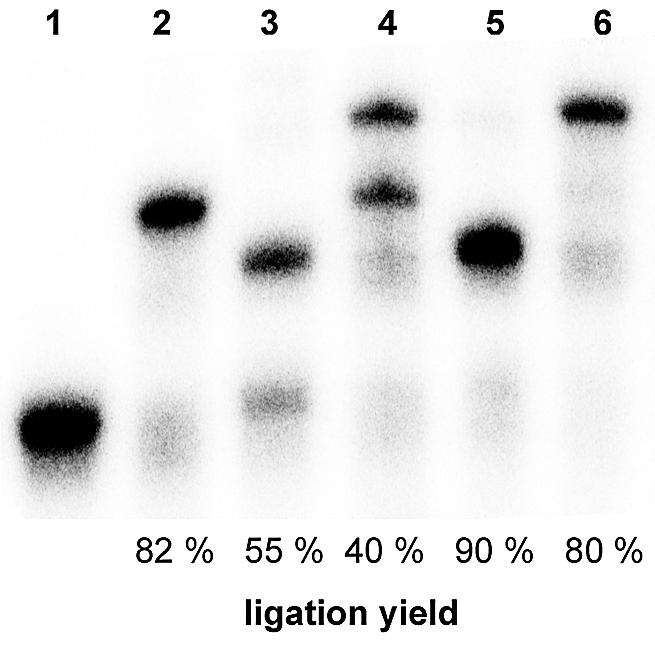
Improved ligation yield with special adapters. The autoradiogram shows the bridge-assisted ligation of primer binding sites to a 32P-labeled in vitro transcribed RNA library (mixture of 50 and 51mer due to the partial non-templated addition of a single nucleotide) (1). While ligation to the RNA’s 5′ end with the forward adapter (plus 25 nt) was acceptable (2), ligation to the 3′ end with reverse adapter 1 only (plus 18 nt) (3) and the overall ligation yield (plus 43 nt) (4) were insufficient, because N + 1 transcripts were not ligated. By using an equimolar mixture of reverse adapters 1 and 2, the 3′ ligation efficiency (5) and the overall ligation yield (6) were markedly improved.
Reverse transcription, PCR and alkaline fission
Reverse transcription was performed at the highest possible temperature in order to resolve RNA secondary structures and to ease the removal of the forward bridge, which might interfere with cDNA synthesis.
Twenty to 100 pmol of PCR product accumulated from 10 fmol to 5 pmol of template per 100 µl of PCR within 7–15 cycles. The forward primer resembles the forward ligate but additionally extends into the GC-rich 5′ fixed region of the nucleic acid pool to improve priming efficiency. The PCR reverse primer is identical to the reverse bridge. It contains two ribonucleotides to provide for alkaline fission. The second ribonucleotide is incorporated in order to prevent the cleaved reverse primer from re-hybridizing to the forward strand that might lead to a possible read-through of the RNA polymerase.
Cleavage of the PCR product under alkaline conditions leads to strands of unequal length, as the reverse strand lacks the unwanted reverse primer site (Fig. 4).
Figure 4.
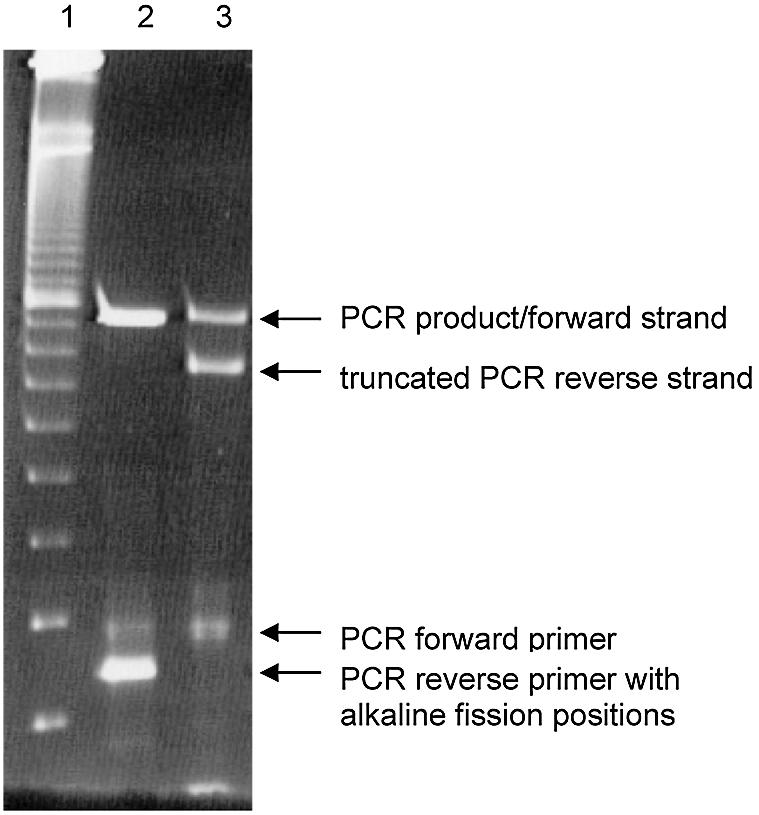
Alkaline fission of the PCR product. The reverse strand of the PCR product is truncated by alkaline fission at the ribonucleotide positions within the reverse primer. The procedure removes both the incorporated and unincorporated reverse primer that is now dispensable. (1) Ten base pair size marker, (2) PCR product before alkaline treatment, (3) PCR product after alkaline fission of the reverse strand and the unincorporated reverse primer.
In vitro transcription
The cleaved PCR product then serves as a template for the in vitro transcription. Since the T7 RNA polymerase promoter is located in the forward primer and the forward ligate upstream of the ligation site, the transcripts commence with the identical initiation sequence that is equal to the 5′ fixed sequence of the nucleic acid library. The sequence was designed to provide for a high transcription yield (27) and a good ligation efficiency. Additionally, we included an excess of guanosine monophosphate as an initiation nucleotide into the transcription reaction, so that the majority of the transcripts contain a 5′ monophosphate that is required for the next ligation step.
Identification of α-CGRP binding Spiegelmers
As a proof of concept, we carried out 15 rounds of in vitro selection against the unnatural enantiomer of rat α-CGRP. After the fifth selection round, enrichment of binding sequences became visible. In subsequent selection rounds, the peptide concentration was successively decreased from the initial 10 µM to 30 nM in round 9, without loss of pool binding. We also increased the stringency by switching from room temperature to 37°C after round 9. In round 15, no more progress could be achieved (Fig. 5). With regard to pool binding or variations in sequence length, the system proved to be stable within the conducted 15 rounds of selection/amplification. The enriched pool of round 15 was cloned and 38 sequences were determined, 14 of which were different (Fig. 6). Their length varied between 48 and 52 nt, which is probably due to errors of the polymerases involved. By sequence alignment we identified four motifs that occurred in every sequence, either alone or in combination with each other. One of these motifs was found to occur in three families both as a full-length motif (24 nt) or as a truncated version (11 nt).
Figure 6.

Aligned sequences from the CGRP binding RNA pool after selection round 15. The numbers indicate the individual sequence’s frequency of occurrence. The fixed sequence parts were shaded in gray. Four different conserved motifs were identified as indicated by the background colors. The blue motif was found to occur as a 24mer or in part as an 11mer if it is flanked by the magenta-colored 16 nt long split-motif. The blue motif appeared close to the 5′ end, in the middle or close to the 3′ end of the randomized region. While the green and magenta motifs were unique to the in vitro selection at 37°C, the blue and yellow motifs were also frequent in room temperature selections that were carried out in parallel (sequences not shown). The sequences within each group seem to have partly arisen from an identical ancestor sequence with Taq polymerase-induced mutations.
Biological activity of the Spiegelmers
Nine sequences were synthesized as Spiegelmers, without the need for prior truncation and further modification for their testing in cell culture. All Spiegelmers tested blocked rat α-CGRP-induced cAMP formation with an IC50 of 500 nM or better, but with no clear preference for any of the four primary sequence motifs. The best inhibition was observed with Spiegelmer STAR-F12 with an IC50 of 3 nM (Fig. 7). The inverse Sequence STAR-F12inv served as a control and showed no inhibitory effect. While the 5′ fixed sequence of the Spiegelmer STAR-F12 was needed for target binding, the six 3′ terminal fixed nucleotides could be removed. The resulting 42mer STAR-F12-Δ43–48 had an IC50 of 3 nM, the same inhibition potency as the full-length Spiegelmer.
Figure 7.

Dose–response curve for the Spiegelmer STAR-F12 and its 3′ truncated version STAR-F12-Δ43–48. IC50 values are indicated by the arrows. The Spiegelmer at different concentrations was pre-incubated with 1 nM rat α-CGRP at 37°C for 1 h. Intracellular cAMP formation in SK-N-MC cells was then stimulated for 30 min at 37°C. After cell lysis, cAMP was quantified in a chemiluminescent immunoassay.
Since the relationship between intracellular cAMP production and the concentration of free extracellular CGRP was linear under our experimental conditions, the dissociation constant KD almost equals the IC50. However, due to the almost equimolar concentrations of Spiegelmer and CGRP at the IC50, the apparent IC50 is slightly higher than the dissociation constant KD. The calculation of KD following the equation KD = IC50 – 0.5 × [CGRP] leads to a dissociation constant of 2.5 nM.
DISCUSSION
Since the invention of the SELEX process, a large number of aptamers and several Spiegelmers active against a variety of biologically relevant targets have been generated (6). However, the transition from the biochemical and biophysical laboratory into living systems like cell culture, animals or even man, has been achieved only in a small number of them so far (28). One of the challenges is still the biological stability of oligonucleotides. Even for 2′-F- or 2′-NH2-pyrimidine-RNA aptamers, the introduction of additional 2′ modifications at as many purine positions as possible is desirable. Such post-SELEX-modified aptamers can only be produced by chemical methods. Spiegelmers are biostable due to their unnatural configuration and require chemical synthesis as well. This is not accomplished easily since the partially stabilized aptamer candidates that are identified by the SELEX process are between 60 and 90 nt in length and to date only shorter RNAs with <60 nt are efficiently produced chemically at reasonable costs. As yields decrease with every coupling step, shorter sequences will be preferable for economical reasons, as soon as larger batches for animal tests are to be produced. Therefore, time-consuming experimental truncation processes that do not always lead to the desired results need to be carried out before Spiegelmers or biostable aptamers are available.
We have developed the Tailored-SELEX strategy to afford the identification of short aptamers by ligation and removal of primer binding sites within the amplification part of the selection-amplification scheme. The candidates are only 10 nt longer than the randomized region of the initial library.
Alternative strategies, e.g. blocking of primer binding sites by hybridization of complementary oligonucleotides, fishing of truncated sequences with full-length complementary strands and further ligation strategies, have been proposed by Toole et al. and Pagratis et al. in the patent literature (29,30).
Tailored-SELEX is simple and requires only a little more hands-on time than a conventional SELEX process. The two extra steps, namely ligation of primers and alkaline fission of the PCR product, compare favorably with the time needed for post-SELEX truncation protocols. This holds especially true for the generation of aptamers against flexible peptides that require a structurally rigid binding partner. In these instances, the aptamer provides a scaffold and an epitope for high affinity binding, which is often governed by an induced fit mechanism by the peptide (31). Since the length of the aptamer’s target binding sequence could generally not be influenced by the experimental set-up, truncated peptide-directed aptamers were found to involve at least 50 nt, which is substantially longer than truncated protein-directed aptamers. An exception is provided by the truncated aptamers binding the arginine-rich core motif of the HIV-1 Rev-protein, which are only 27 and 35 nt in length, probably because binding of arginines to nucleic acids is very advantageous (32).
The calculated affinity (KD = 2.5 nM at 37°C) of the best rat α-CGRP binding Spiegelmer (STAR-F12) almost reaches the binding constant of the neuropeptide to its receptor (EC50 = 1 nM). The dissociation constants can be compared well with those of aptamers that were directed against larger proteins such as IgE (10 nM, 37°C) (17), tyrosine phosphatase (18 nM, room temperature) (18) or complement C5 (20 and 2 nM after reselection, 37°C) (33). Peptide-directed aptamers usually display higher dissociation constants for their respective targets, as reported for aptamers against substance P (190 nM, room temperature) (16), the arginine-rich peptide fragment of HIV Rev (19 nM, 4–7°C) (34), K-Ras derived farnesylated peptide (139 nM, 23°C) (35) and neuropeptide Y (370 nM at 37°C) (36) or the Spiegelmers against vasopressin (900 nM, room temperature) (12), GnRH (20–100 nM, room temperature) (13,37) or a 25 amino acid domain of Staphylococcal Enterotoxin B (200 nM, room temperature) (38). A different measure has to be applied to aptamers directed to nucleic acid and heparin binding proteins. Due to the favorable biophysical nature of these targets, resulting aptamers frequently show picomolar dissociation constants (6).
In conclusion, the above data confirm that Tailored-SELEX is suitable to identify not only short, but also high-affinity, aptamers/Spiegelmers. Additionally, the reported methods are ready to be implemented into an automated selection protocol. Further studies will address target and species specificity of our nuclease-resistant rat α-CGRP binding Spiegelmer in cell culture. Its potential to act as a CGRP antagonist in vivo and its feasibility in the treatment of migraine will be tested in animal models in the near future.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Nura Sayed Suleiman and Angelika Dahlke for their technical assistance, Axel Wettich for his ideas, Petra Burgstaller, the oligonucleotide synthesis team at NOXXON Pharma AG, Steffen Helmling and Lara Zilberkweit for their critical reviews of the manuscript and Christian Mihm for the artwork in Figure 1. We are also grateful for the help of Clemens Gillen and the molecular pain research group at Grünenthal GmbH, Aachen, Germany.
REFERENCES
- 1.Ellington A.D. and Szostak,J.W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 2.Tuerk C. and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 3.Ellington A.D. and Szostak,J.W. (1992) Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature, 355, 850–852. [DOI] [PubMed] [Google Scholar]
- 4.Gold L., Polisky,B., Uhlenbeck,O. and Yarus,M. (1995) Diversity of oligonucleotide functions. Annu. Rev. Biochem., 64, 763–797. [DOI] [PubMed] [Google Scholar]
- 5.Conrad R., Keranen,L.M., Ellington,A.D. and Newton,A.C. (1994) Isozyme-specific inhibition of protein kinase C by RNA aptamers. J. Biol. Chem., 269, 32051–32054. [PubMed] [Google Scholar]
- 6.James W. (2000) Aptamers. In Meyers,R.A. (ed.), Encyclopedia of Analytical Chemistry. John Wiley & Sons, Chichester, UK, pp. 4848–4871. [Google Scholar]
- 7.Lin Y., Qiu,Q., Gill,S.C. and Jayasena,S.D. (1994) Modified RNA sequence pools for in vitro selection. Nucleic Acids Res., 22, 5229–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eaton B.E. and Pieken,W.A. (1995) Ribonucleosides and RNA. Annu. Rev. Biochem., 64, 837–863. [DOI] [PubMed] [Google Scholar]
- 9.Ruckman J., Green,L.S., Beeson,J., Waugh,S., Gillette,W.L., Henninger,D.D., Claesson-Welsh,L. and Janjic,N. (1998) 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem., 273, 20556–20567. [DOI] [PubMed] [Google Scholar]
- 10.Green L.S., Jellinek,D., Bell,C., Beebe,L.A., Feistner,B.D., Gill,S.C., Jucker,F.M. and Janjic,N. (1995) Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial factor. Chem. Biol., 2, 683–695. [DOI] [PubMed] [Google Scholar]
- 11.Klussmann S., Nolte,A., Bald,R., Erdmann,V.A. and Furste,J.P. (1996) Mirror-image RNA that binds D-adenosine. Nat. Biotechnol., 14, 1112–1115. [DOI] [PubMed] [Google Scholar]
- 12.Williams K.P., Liu,X.H., Schumacher,T.N., Lin,H.Y., Ausiello,D.A., Kim,P.S. and Bartel,D.P. (1997) Bioactive and nuclease-resistant l-DNA ligand of vasopressin. Proc. Natl Acad. Sci. USA, 94, 11285–11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leva S., Lichte,A., Burmeister,J., Muhn,P., Jahnke,B., Fesser,D., Erfurth,J., Burgstaller,P. and Klussmann,S. (2002) GnRH binding RNA and DNA Spiegelmers: a novel approach toward GnRH antagonism. Chem. Biol., 9, 351–359. [DOI] [PubMed] [Google Scholar]
- 14.Pagratis N.C., Bell,C., Chang,Y.F., Jennings,S., Fitzwater,T., Jellinek,D. and Dang,C. (1997) Potent 2′-amino- and 2′-fluoro-2′-deoxyribonucleotide RNA inhibitors of keratinocyte growth factor. Nat. Biotechnol., 15, 68–73. [DOI] [PubMed] [Google Scholar]
- 15.White R.R., Sullenger,B.A. and Rusconi,C.P. (2000) Developing aptamers into therapeutics. J. Clin. Invest., 106, 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nieuwlandt D., Wecker,M. and Gold,L. (1995) In vitro selection of RNA ligands to substance P. Biochemistry, 34, 5651–5659. [DOI] [PubMed] [Google Scholar]
- 17.Wiegand T.W., Williams,P.B., Dreskin,S.C., Jouvin,M.H., Kinet,J.P. and Tasset,D. (1996) High-affinity oligonucleotide ligands to human IgE inhibit binding to Fc epsilon receptor I. J. Immunol., 157, 221–230. [PubMed] [Google Scholar]
- 18.Bell S.D., Denu,J.M., Dixon,J.E. and Ellington,A.D. (1998) RNA molecules that bind to and inhibit the active site of a tyrosine phosphatase. J. Biol. Chem., 273, 14309–14314. [DOI] [PubMed] [Google Scholar]
- 19.Wimalawansa S.J. (1996) Calcitonin gene-related peptide and its receptors: molecular genetics, physiology, pathophysiology and therapeutic potentials. Endocr. Rev., 17, 533–585. [DOI] [PubMed] [Google Scholar]
- 20.Renfrey S., Downton,C. and Featherstone,J. (2003) The painful reality. Nature Rev. Drug Discov., 2, 175–176. [DOI] [PubMed] [Google Scholar]
- 21.Lassen L.H., Haderslev,P.A., Jacobsen,V.B., Iversen,H.K., Sperling,B. and Olesen,J. (2002) CGRP may play a causative role in migraine. Cephalalgia, 22, 54–61. [DOI] [PubMed] [Google Scholar]
- 22.Edvinsson L. (2003) New therapeutic target in primary headaches—blocking theCGRP receptor. Expert Opin. Ther. Targets, 7, 377–383. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, NY. [Google Scholar]
- 24.Kleppe K., Van de Sande,J.H. and Khorana,H.G. (1970) Polynucleotide ligase-catalyzed joining of deoxyribo-oligonucleotides on ribopolynucleotide templates and of ribo-oligonucleotides on deoxyribopolynucleotide templates. Proc. Natl Acad. Sci. USA, 67, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kao C., Rudisser,S. and Zheng,M. (2001) A simple and efficient method to transcribe RNAs with reduced 3′ heterogeneity. Methods, 23, 201–205. [DOI] [PubMed] [Google Scholar]
- 26.Moore M.J. and Query,C.C. (1998) Uses of site specifically modified RNAs constructed by RNA-ligation. In Smith,C.W.J. (ed.), RNA:Protein Interactions: A Practical Approach. Oxford University Press, Oxford, UK, Vol. 192, pp. 75–108. [Google Scholar]
- 27.Milligan J.F. and Uhlenbeck,O.C. (1989) Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol., 180, 51–62. [DOI] [PubMed] [Google Scholar]
- 28.Vater A. and Klussmann,S. (2003) Toward third-generation aptamers: Spiegelmers and their therapeutic prospects. Curr. Opin. Drug Discov. Devel., 6, 253–261. [PubMed] [Google Scholar]
- 29.Toole J., Griffin,L., Bock,L., Latham,J., Muenchau,D.D. and Krawczyk,S. (1992) Patent WO 92/14843.
- 30.Pagratis N., Gold,L., Shtatland,T. and Javornik,B. (2000) Patent WO 00/56930.
- 31.Hermann T. and Patel,D.J. (2000) Adaptive recognition by nucleic acid aptamers. Science, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- 32.Ye X., Gorin,A., Frederick,R., Hu,W., Majumdar,A., Xu,W., McLendon,G., Ellington,A. and Patel,D.J. (1999) RNA architecture dictates the conformations of a bound peptide. Chem. Biol., 6, 657–669. [DOI] [PubMed] [Google Scholar]
- 33.Biesecker G., Dihel,L., Enney,K. and Bendele,R.A. (1999) Derivation of RNA aptamer inhibitors of human complement C5. Immunopharmacology, 42, 219–230. [DOI] [PubMed] [Google Scholar]
- 34.Xu W. and Ellington,A.D. (1996) Anti-peptide aptamers recognize amino acid sequence and bind a protein epitope. Proc. Natl Acad. Sci. USA, 93, 7475–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilbert B.A., Sha,M., Wathen,S.T. and Rando,R.R. (1997) RNA aptamers that specifically bind to a K Ras-derived farnesylated peptide. Bioorg. Med. Chem., 5, 1115–1122. [DOI] [PubMed] [Google Scholar]
- 36.Proske D., Hofliger,M., Soll,R.M., Beck-Sickinger,A.G. and Famulok,M. (2002) A Y2 receptor mimetic aptamer directed against neuropeptide Y. J. Biol. Chem., 277, 11416–11422. [DOI] [PubMed] [Google Scholar]
- 37.Wlotzka B., Leva,S., Eschgfaller,B., Burmeister,J., Kleinjung,F., Kaduk,C., Muhn,P., Hess-Stumpp,H. and Klussmann,S. (2002) In vivo properties of an anti-GnRH Spiegelmer: an example of an oligonucleotide-based therapeutic substance class. Proc. Natl Acad. Sci. USA, 99, 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purschke W.G., Radtke,F., Kleinjung,F. and Klussmann,S. (2003) A DNA Spiegelmer to staphylococcal enterotoxin B. Nucleic Acids Res., 31, 3027–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]