

Abstract
ESI-FTICR MS was utilized to characterize a 4Fe-4S containing protein Mycobacterium tuberculosis APS reductase. This enzyme catalyzes the reduction of APS to sulfite and AMP with reducing equivalents from the protein cofactor, thioredoxin. Under nondenaturing conditions, a distribution of the apoprotein, a 2Fe-2S intermediate, and the 4Fe-4S holoprotein were observed. Accurate mass measurements indicated an oxidation state of +2 for the 4Fe-4S cluster, with no disulfide bond in the holoenzyme. Gas-phase stability of the 4Fe-4S cluster was investigated using both in-source and collision induced dissociation, which provided information regarding the relative gas-phase binding strength of iron towards protein ligands and inorganic sulfides. Noncovalent complexes of the holoprotein with several ligands, including APS, thioredoxin and AMP, were also investigated. Calculated values of dissociation constants for the complexes indicate that AMP binds with a higher affinity to the enzyme intermediate than to the free enzyme. The implications of the binary and ternary complexes observed by gas-phase noncovalent interactions in the mechanism of APS reduction are discussed.
Keywords: APS reductase, Iron-sulfur cluster, ESI-FTICR mass spectrometry, Noncovalent complexes, Reaction mechanism
Introduction
Iron-sulfur clusters are important cofactors, often involved in biological oxidoreductive functions [1–4]. These clusters are composed of iron and inorganic sulfide in forms of two to four iron-clusters and typically interact with the apoprotein through ligation between iron and cysteinate (Cys) sulfur atoms [5, 6]. In addition to the most commonly known function of electron transfer [6, 7], iron-sulfur clusters are implicated in other important biological functions, such as binding and activation of substrates [8–10], regulation of oligomerization and stabilization of protein tertiary structure [11–13], storage of irons and sulfides [11, 14, 15], as well as sensing of iron, dioxygens, and superoxides [16, 17]. Traditional techniques to study iron-sulfur cluster containing proteins include metal analysis by inductively coupled plasma spectroscopy [18, 19], cluster diamagnetic nature by electron paramagnetic resonance [20–22], Fe-S bond features by resonance Raman spectroscopy [23, 24] and Mössbauer spectroscopy [25, 26], and structural analysis by magnetic circular dichroism spectroscopy [27] and X-ray absorption [28–30].
More recently, electrospray mass spectrometry (ESI-MS) has been successfully used in the study of iron-sulfur cluster containing proteins, such as rubredoxins [31–33], ferredoxin [34–36], high potential iron-sulfur protein [37], and biotin synthase [38]. The gentle feature of electrospray allows the intact protein-metal cluster complexes to be transferred into the gas-phase without dissociation, and thus mass measurements can be obtained. Amster and coworkers [39] have reported the observation of intact iron-sulfur clusters in small iron-sulfur containing proteins both in positive and negative ion modes. Combined with the high resolution capability of FTICR mass spectrometry, they obtained stoichiometry and the oxidation state of the metal clusters [40–42]. Using either chemical or on-line electrochemical reduction, they also observed changes in the oxidation state of a manganese-substituted iron-sulfur cluster [43]. Robinson et al. [38] reported on the 2Fe-2S cluster in E. coli biotin synthase using nanoflow electrospry Q-TOF mass spectrometry in both positive and negative ion modes. They also investigated the effect of solution conditions on the stability of the protein-cluster complex and the effect of the cluster on dimerization of the protein. Compared to other traditional techniques, ESI-MS has the advantage of generating direct information on the stoichiometry, oxidation state, and stability of the protein bound metal-cluster, as well as offering better assay sensitivity and speed.
APS reductase catalyzes the reduction of adenosine-5′-phosphosulfate (APS) to adenosine-5′-phosphate (AMP) and sulfite with reducing equivalents from the protein cofactor, thioredoxin (Trx) (Figure 1) [44]. It is the first committed step in the process of sulfur assimilation for plants, fungi and many bacteria [45, 46]. The sulfite generated is reduced in a subsequent enzymatic step catalyzed by sulfite reductase to sulfide, the reduced form of sulfur that is required for the biosynthesis of cysteine, methionine and other primary metabolites that incorporate thiols and sulfides. As humans do not have homologous enzymes, bacterial APS reductases represent attractive therapeutic targets. Recently, APS reductase has been identified in a screen for essential genes in Mycobacterium bovis [47], and found to be critical for virulence in a murine model of tuberculosis infection (R. Senaratne, personal communication). APS reductase from Mycobacterium tuberculosis is a monomer with a molecular mass of 28.7 kDa and contains an essential 4Fe-4S cluster that is ligated by at least three cysteines within the sequence motif, -CC-X~80-CXXC-[48]. Interestingly, not all organisms that assimilate sulfate reduce APS as a source of sulfite. Some organisms such as Escherichia coli and Saccharomyces cerevisiae [49, 50] reduce the related metabolite 3′-phosphoadenosine-5′-phosphosulfate (PAPS), but do not possess a metal cluster.
Figure 1.

Reaction scheme of sulfonucleotide reduction
Previously, based on the detection of a covalent intermediate by electrospray mass spectrometry in combination with other biochemical approaches, a two-step mechanism for sulfonucleotide reductases as a class of enzyme was proposed [48]. In that model, the sulfonucleotide undergoes a nucleophilic attack to form an enzyme-thiosulfonate intermediate E-SO3H. Sulfite is then released in a thioredoxin dependent manner. More recently, however, new results suggest that the iron-sulfur cluster is a key constituent of the active site of M. tuberculosis APS reductase and is essential for the reduction of APS [51]. Herein, a more detailed investigation of various non-covalent complexes containing APS reductase from M. tuberculosis, by ESI-FTICR mass spectrometry is presented, which provides specific information about the mechanism of this enzyme. Accurate mass measurement was obtained for the 28.7 kDa 4Fe-4S containing holoenzyme. These data demonstrate that the native holoenzyme does not possess an intramolecular disulfide bond. Binary and ternary protein-ligand and protein-protein noncovalent interactions of the holoenzyme with substrate APS, cofactor Trx, and product AMP were investigated, with corresponding dissociation constants determined. These measurements provide important information on the binding strength of AMP with the native enzyme and with the thiolsulfonate intermediate. Finally, the implications of the observed complexes with respect to the mechanism of APS reduction are discussed.
Experimental Procedures
Materials
APS was purchased from Biolog Life Sciences Institute, ≥ 95% (Bremen, Germany). AMP was purchased from Sigma Chemical Company (St. Louis, MO). Molecular biology grade DTT was purchased from Invitrogen (Carlsbad, CA). E. coli thioredoxin protein was purchased from EMD Biosciences (San Diego, CA). Mycobacterium tuberculosis APS reductase protein was expressed in BL21(DE3) (Novagen) cells and purified using metal affinity chromatography as previously described [48]. All other chemicals were purchased from Sigma and were of the highest purity available (≥ 95%).
Sample Preparation for Mass Spectrometry
Aliquots (100 μL) of purified APS reductase or thioredoxin were buffer exchanged into 50 mM ammonium acetate (NH4OAc), pH 7.5, using Amicon 10,000 Da molecular weight cut off centrifugal filters with the temperature of the centrifuge set at 4°C. Protein concentrations, after buffer exchange, were determined by Bradford assay, with a correction factor derived from quantitative amino acid analysis used for APS reductase as described previously [48]. All ligands were prepared as 1 mM stock solutions in 50 mM NH4OAc and appropriate volumes of ligand, buffer and enzyme were mixed and incubated for 15 min to generate noncovalent complexes. The mixture was chilled on ice and infused into the ESI-FTICR mass spectrometer for analysis. The syringe pump and spray chamber were wrapped with ice bags to prevent protein precipitation.
Mass spectrometry
All mass spectrometry data were acquired on a Bruker APEX II FT-ICR mass spectrometer equipped with an actively shielded 7 tesla superconducting magnet and an Apollo electrospray source (Billerica, MA). Instrumental details can be found in previous publications [48, 51]. For accurate mass measurement, the FTICR mass spectra were internally calibrated against carbonic anhydrase. For tandem mass spectrometry, the ion of interest was isolated by correlated sweeps using correlated harmonic excitation fields (CHEF). Argon was then pulsed into the cell to a pressure of ~1×10−7 mbar and the ions were collisionally activated for 2 sec using sustained off-resonance irradiation (SORI) at 1000 Hz above the ion’s cyclotron frequency. The activation energy was tuned for best precursor-product distribution and kept constant for all experiments unless mentioned otherwise.
Accurate mass measurement
The molecular masses of APS reductase and its associated complexes were measured by using the average mass method as previously reported by Zubarev et al.[52, 53]. Briefly, portions of the isotopic distribution above 50% relative intensity of the highest isotopic peak were used to calculate the average m/z of each charge state, and the average mass of the neutral molecule was determined based on all resolvable charge states. A correction factor (Δm50%) of 0.5 Da was then added to derive the true average mass (Mexp). To determine the number of disulfides and the oxidation state of the iron-sulfur cluster, a modified method reported by Johnson et al. [39, 41] was used. The calculated average mass (Mcal) is determined based on the elemental composition of the apoprotein plus the components in the metal center, assuming that all cysteins are in reduced form and the metal cluster has an oxidation state of zero. Mcal and Mexp are then compared, and the difference been the two values are attributed to the number of disulfide bonds and the oxidation state of the cluster. For each disulfide bond, two hydrogen atoms are lost, therefore a 2 Da difference is expected. If the metal cluster carries an oxidation state of 1+, one proton must be removed in order to retain the neutral molecule. Therefore, for an iron-sulfur protein with a disulfide number of m and a cluster oxidation state of n+, the difference between Mcal and Mexp is expected to be mx2+n. After the mass difference is determined, the final assignments of m and n are made considering other available information. See results and discussion for more details.
Results
MS characterization of APS reductase
The ESI mass spectrum of APS reductase sprayed from 50 mM NH4OAc, pH 7.5 is shown in Figure 2A. With the nondenaturing solution condition used, three major charge states ranging from 9+ to 11+ were observed. Figure 2B represents an expansion of the 10+ charge state, in which three m/z values were measured, representing three distinctive forms of the protein: the apoprotein, a 2Fe-2S containing protein and the holoprotein with 4Fe-4S bound. Isotopically resolved spectra of the 28.7 kDa protein in nondenaturing conditions were obtained, which allowed for the assignment of disulfide number and oxidation state of the metal center in each protein form. To minimize the error in mass measurement, the average mass method as reported by Zubarev et al. [52, 53] was used.
Figure 2.
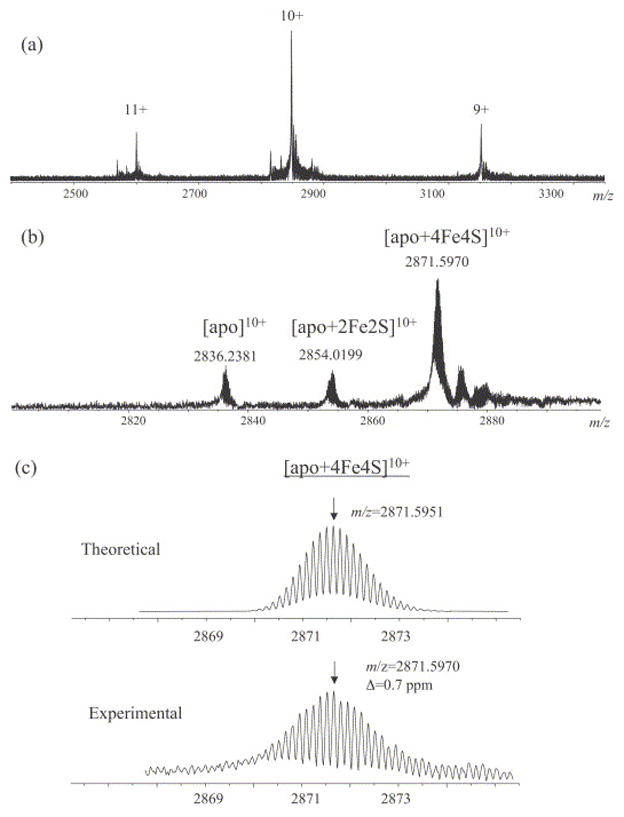
ESI mass spectra of 10 μM APS reductase in 50 mM NH4OAc. (A) Overall charge state distribution. (B) Expansion of the 10+ charge state showing the ions corresponding to the apo, apo+2Fe2S, and apo+4Fe4S forms. (C) Comparison of the measured isotopic distribution for the [apo+4Fe4S]10+ ion and the theoretical simulation.
All high resolution spectra were internally calibrated against the most abundant isotopic peaks of carbonic anhydrase. After calibration, the measured average mass for the 4Fe-4S containing holoenzyme was found to be 28,706.3 Da, which is 2 Da lower than the theoretical value of 28,708.2 Da calculated based on its chemical formula (C1245H1951N363O381S12Fe4). As described in the experimental section, the −2 Da mass difference could be attributed to either a disulfide bond or a 2+ oxidation state of the metal center. However, if a disulfide bond is present in the holoenzyme, the 4Fe-4S cluster would be assigned an oxidation state of zero, which is highly unlikely. Therefore, the more reasonable interpretation for this mass difference is that the holoenzyme is in the reduced form and the cluster has an oxidation state of 2+.
The average mass of the apoprotein was measured to be 28,352.7 Da, and the calculated average mass for the protein based on its amino-acid sequence (C1245H1951N363O381S8) is 28,356.6 Da. The −4 Da difference is consistent with the formation of two disulfides between the four free Cys residues that normally coordinate the cluster. The formation of disulfide bonds between Cys ligands after cluster removal has been reported in several other iron-sulfur cluster containing proteins [38, 41]. Similarly, the measured average mass of the 2Fe-2S containing intermediate was found to be 2 Da lower than the calculated value based on its chemical formula (C1245H1951N363O381S10Fe2). This mass difference is most likely due to the expected 2+ oxidation state for the 2Fe-2S cluster, which results from partial dissociation of the intact 4Fe-4S complex. This type of cluster decomposition has been observed in other iron-sulfur cluster containing proteins [54]. In a complete 4Fe-4S cluster, two irons are expected to have a formal charge of +2 while the other two are +3; each iron would be coordinated to one Cys ligand and three inorganic sulfides. When two irons and two sulfides dissociate from the cluster, each of the remaining two irons would be ligated by two adjacent Cys ligands and two inorganic sulfides.
To verify the accuracy of mass measurement, the experimental isotopic distribution of each ion was compared to the theoretical simulation. As shown in Figure 2C, the deviation between the measured and theoretical mass to charge ratios for the most abundant isotopic peak of the [apo+4Fe4S]10+ ion is 0.7 ppm, indicating that the mass assignment of the holoprotein is correct. The same verification was carried out for all species at all observed charge states. The calculated masses for the neutral molecule of all protein forms were compared to the theoretical values, as summarized in Table 1.
Table 1.
Accurate mass measurements of APS reductase and its associated complexes
| Enzyme forms | Mexp (Da)a | Mtheor (Da)b | ΔM (ppm) | disulfide number | cluster oxidation state |
|---|---|---|---|---|---|
| Apo | 28352.376±0.073 | 28352.235 | +5.0 | 2 | N/A |
| Apo+2Fe-2S | 28530.192±0.107 | 28530.078 | +3.9 | 0 | +2 |
| Apo+4Fe-4S (E) | 28705.995±0.058 | 28705.879 | +4.0 | 0 | +2 |
| E-SO3H | 28785.971±0.064 | 28785.836 | +4.7 | 0 | +2 |
| E·AMP | 29053.051±0.075 | 29052.902 | +5.1 | 0 | +2 |
| E-SO3H·AMP | 29133.029±0.077 | 29132.898 | +4.5 | 0 | +2 |
The average experimental value of the most abundant isotopic mass over the charge state distribution. The most abundant isotopic mass was assigned after the average mass was first determined as described in the experimental section. Four measurements were made for each mass assignment. All spectra were internally calibrated against carbonic anhydrase.
The most abundant isotopic mass determined from theoretical simulation.
Noncovalent interaction of APS reductase and associated ligands
Figure 3A represents the ESI mass spectrum acquired for a mixture of 10 μM APS reductase and 5 μM APS. Four major ion clusters were observed, with the m/z values for the 10+ charge state ions measured at 2871.5970, 2879.5913, 2906.3042, and 2914.2998. The ion with the lowest m/z value corresponds to the free holoenzyme (E). A second ion results in a mass shift of +80 Da versus the holoenzyme, and represents a thiosulfonate-modified enzyme (E-SO3H). The third ion is observed at 347 Da higher than the holoenzyme, and corresponds to a noncovalent complex between the holoenzyme and AMP (E·AMP). The product AMP is formed once APS is reduced with concomitant transfer of the sulfate group to the enzyme. The last ion, with a calculated mass shift of +427 Da compared to the holoenzyme, is identified as the noncovalent complex of E-SO3H with AMP. Based on the results of the accurate mass measurements listed in Table 1, the oxidation state of the cluster in each of the four enzyme forms was determined to be +2, which indicates that there is no electron transfer during substrate binding and intermediate formation. This data is consistent with previous electron paramagnetic resonance spectra of APS reductase observed in the presence or absence of APS or AMP [51]. It is apparent that AMP could bind both to the intact holoenzyme and to the covalent thiosulfonate enzyme-intermediate, but possibly with different affinities. The dissociation constant of E·AMP (Kd1) and that of ESO3H·AMP (Kd2) can be expressed as:
| (1) |
| (2) |
Figure 3.
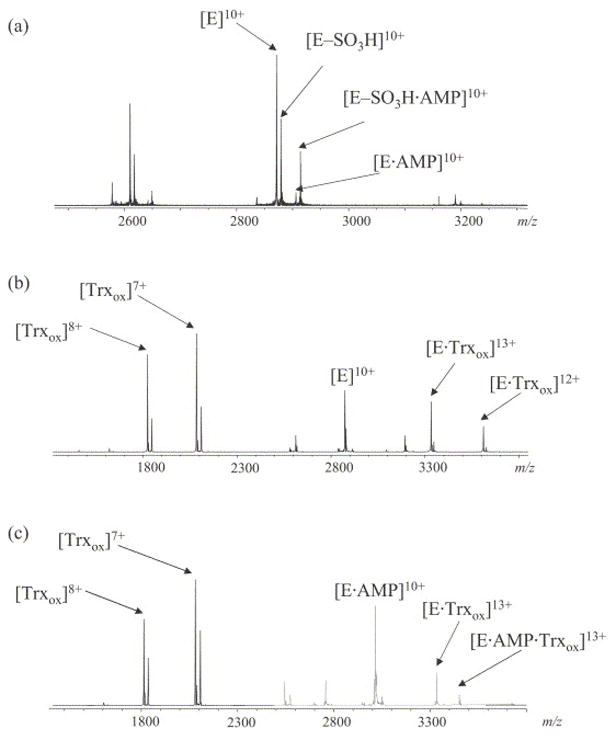
ESI mass spectra of 10 μM APS reductase with different ligands (A) APS reductase with 5 μM APS. Ions corresponding to various charge states of intact enzyme (E), thiosulfate intermediate (E–SO3H), noncovalent complex of enzyme with AMP (E·AMP) and noncovalent complex of intermediate with AMP (E–SO3H·AMP) are labeled. (B) APS reductase with 2.5 μM Trx showing the formation of a binary complex. (C) APS reductase with 2.5 μM Trx and 20 μM AMP showing the formation of a ternary complex.
Therefore, the ratio between Kd1 and Kd2 is:
| (3) |
The concentrations of the four different enzyme associated forms can be represented by their ion abundances measured in the same spectrum, assuming that they have approximately the same ionization efficiencies. As has been noticed previously for other protein-ligand noncovalent interaction systems [55, 56], the relative ratio between bound and unbound protein ions are slightly different at different charge states. Therefore, the normalized sum of ion abundances over all resolvable charge states was used to for each form. The corresponding data used in the measurement of relative dissociation constant of E·AMP versus ESO3H·AMP are listed in Table 2. The calculated value of Kd1/Kd2, using equation 3, is 2.67±0.25 (n=4), which indicates that AMP binds with a higher affinity to the enzyme-intermediate than to the free enzyme. This affinity difference is consistent with biochemical data that suggest a conformational change in the active site of APS reductase upon covalent intermediate formation [51]. However, Kd measurements were not reported previously and thus this is the first time that this effect has been quantitatively reported.
Table 2.
Ion abundances used in the calculation of relative dissociation constant1
| measurement | Normalized Ion abundances | E | E-SO3H | E·AMP | E-SO3H·AMP | Kd1/Kd2 |
|---|---|---|---|---|---|---|
| 1 | I10+/10+I11+/11 | 599964.3 | 384986.8 | 146615.9 | 246015.0 | 2.61 |
| 2 | I10+/10+I11+/11 | 784145.4 | 484102.8 | 177272.2 | 274522.8 | 2.51 |
| 3 | I10+/10+I11+/11 | 691255.5 | 448656.4 | 155019.4 | 254743.6 | 2.53 |
| 4 | I10+/10+I11+/11 | 755920.9 | 417363.6 | 157638.5 | 264646.4 | 3.04 |
| average | 2.67 | |||||
| SD | 0.25 |
Ion abundances in each measurement were the averages of 32 scans.
Protein-protein interactions between APS reductase and Trx were also investigated. Trx is a protein cofactor with a molecular weight of 12.5 kDa and a redox active disulfide bond. Under aerobic conditions, the disulfide bond is primarily oxidized. Pre-incubating Trx with DTT reduced the disulfide bond as evidenced by a +2 Da shift in the corresponding mass spectrum (data not shown). Figure 3B shows the spectrum acquired for a mixture of APS reductase and oxided Trx (Trxox) under aerobic conditions. In addition to ions corresponding to the holoenzyme and Trxox, a new series of ions were observed with m/z values observed at 3330.19 and 3607.64, which correspond to the noncovalent complex between the enzyme and Trxox (E·Trxox) at charge states 13+ and 12+, respectively (theoretical values are 3330.26 and 3607.70, respectively). Unlike the phenomenon observed in noncovalent complexation with small molecule ligands, binding of a protein (MW> 10 kDa) alters the charge states of the reductase to higher values. The most abundant ion for free Trxox detected in the same spectrum is represented by the 7+ charge state (measured m/z value of 2083.06 with theoretical calculated value of 2083.08, oxidized form), and the most abundant ion for free APS reductase has a charge state of 10+. The total apparent charge for the protein-protein complex is less than the sum of the apparent charge for each free protein. This effect is mostly due to a higher ordered structure of the binary complex, with some chargeable residues in each component shielded in the binding interface, as has been observed in the investigations of other protein-protein interactions [57]. No significant change was observed in the resulting spectrum when Trx was reduced by the addition of DTT to the binary complex. Figure 3C shows the spectrum acquired for a ternary mixture of APS reductase, Trx and AMP. Binary complexes of APS reductase with AMP and Trx were observed, as expected. In the same spectrum, an additional ion with an m/z value of 3356.89 was observed, which corresponds to the ternary complex E·AMP·Trxox at the 13+ charge state (theoretical value is 3356.95).
APS reductase was further incubated with both substrates APS and Trx to investigate the possible existence of a ternary complex associated with the intermediate. Figure 4A shows the spectrum acquired for the ternary mixture with APS at a limiting concentration. Under these conditions, free enzyme is the major species whereas the intermediate is in relatively lower abundance, and the following complexes were observed: E·AMP, E-SO3H·AMP, E·Trxox and E·AMP·Trx ox. When the concentration of APS in the mixture was increased, the majority of the enzyme was converted to the intermediate form as shown in Figure 4B. Under these conditions, the oxidized Trx was found to bind to the intermediate as well, with [E-SO3H·Trxox]13+ observed at m/z 3336.32 (theoretical value is 3336.35). In addition to the ternary complex of E·AMP·Trxox, another ternary complex E-SO3H·AMP·Trx ox was also observed in this spectrum, with the m/z of the 13+ charge state ion measured at 3363.00 (theoretical value is 3363.11). When DTT was added to the reaction mixture to reduce Trx, all complexes associated with the intermediate were reduced to those associated with the free enzyme.
Figure 4.
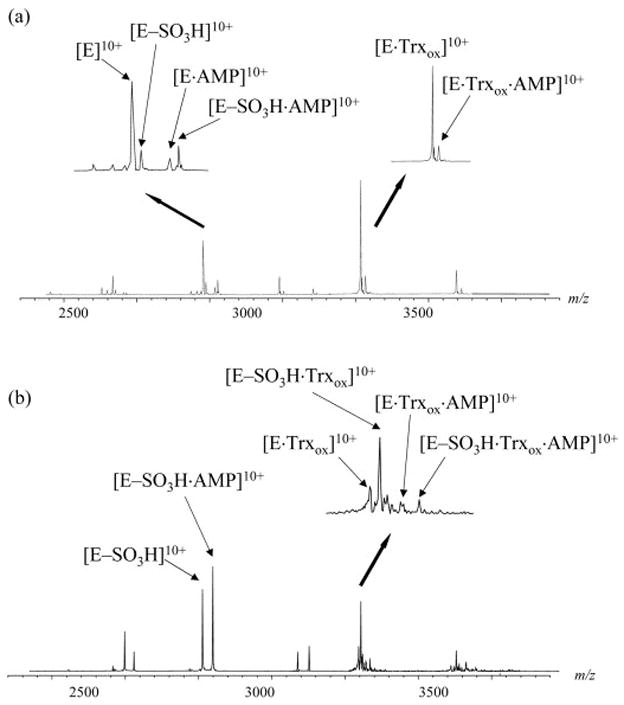
ESI mass spectra of 10 μM APS reductase incubated with 2.5 μM Trx and different concentration of APS. (A) [APS]=2.5 μM. (B) [APS]=7.5 μM. Insets are expansion of the 10+ charge state region showing the formation of binary and ternary complexes.
Gas-phase stability of the iron-sulfur cluster
The gas-phase stability of the iron-sulfur cluster was evaluated using in-source dissociation by changing the capillary exit voltages. At capillary exit voltages below 180 V, good electrospray mass spectra could not be obtained for this 28.7 kDa iron-sulfur cluster containing protein, probably due to inadequate desolvation. When the capillary exit voltage was varied between 180 V and 240 V, no significant difference was observed in the corresponding spectra and the relative ratio among the apo, 2Fe-2S and 4Fe-4S forms remained relatively constant (data not shown). This result suggests that the intermediate and apo forms observed in the mass spectra are not due to the ESI process, but represent different solution populations. In addition, as pointed out by Hernandez et al. [38], disulfide bonds will be formed between adjacent cluster ligands if the cluster is dissociated in solution, whereas free thiols would be formed due to in-source dissociation of the cluster. The accurate mass measurements described above indicate that the apoprotein contains two disulfide bonds. When the capillary exit voltages were increased above 240 V, dissociation products were observed as shown in Figure 5. In these experiments, intact APS reductase and its thiosulfonate intermediate, which was prepared by incubation with at least 5 molar excess of APS, were analyzed at capillary exit voltages of 240 V, 280 V, and 320 V with all other instrumental parameters kept constant. For the intact enzyme, four well-resolved dissociation products were detected at higher capillary exit voltages with measured mass differences of 34, 68, 102, and 136 Da from the precursor. The most likely explanation for these dissociation products is loss of one to four S2− from the intact cluster. In order for the product ions to be observed at the same charge state as the precursor ions, two additional protons need to be lost, therefore the total mass of the neutral loss (n × SH2) is n × 34, n=1–4, which agrees well with the measured values. For the mixture of APS reductase with excess APS, the two major ions shown in Figure 5 (trace A, right panel) represent the thiosulfonate intermediate and AMP bound intermediate. At capillary exit voltage of 280 V, sequential losses of SH2 were observed for both precursor ions with losses of four SH2 being the most abundant product ions. At a capillary exit voltage of 320 V, AMP was found to dissociate from the noncovalent complex and only the thiosulfonate intermediate with loss of 4×SH2 was observed in the spectrum.
Figure 5.
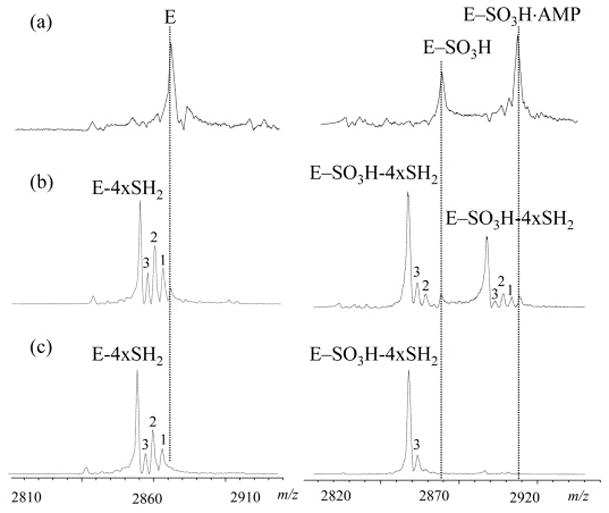
Expansion (10+ charge state) of ESI mass spectra acquired for APS reductase (left) and APS reductase pre-incubated with 5 molar excess APS (right) at capillary exit voltages of 240 V (A), 280 V (B) and 320 V (C). Peaks labeled with 1, 2 and 3 represent dissociation products with loss of 1 to 3-SH2 from the corresponding precursor ions, which are aligned with the dotted lines.
In order to obtain complementary information, sustained off-resonance irradiation collision induced dissociation (SORI-CID) spectra were also acquired for intact APS reductase (E), the enzyme-thiosulfonate intermediate (E-SO3H), and noncovalent complexes E-SO3H·AMP and E·AMP, as shown in Figure 6. In each experiment, the 10+ charge state ion of the corresponding protein/complex was isolated and then dissociated at the same collision energy. For E, the major product ion observed corresponds to loss of 4×SH2 from the precursor ion. In addition to the loss of the inorganic sulfide ion, further loss of the covalently bound sulfite was also observed for E-SO3H. When the same experiment was repeated for E-SO3H·AMP or E·AMP, only one product ion was observed, representing the loss of 4×SH2 from the corresponding precursor ion. Dissociation of AMP from the complexes was not detected until the collision energy was increased to higher values, which is consistent with the results obtained from in-source dissociation.
Figure 6.
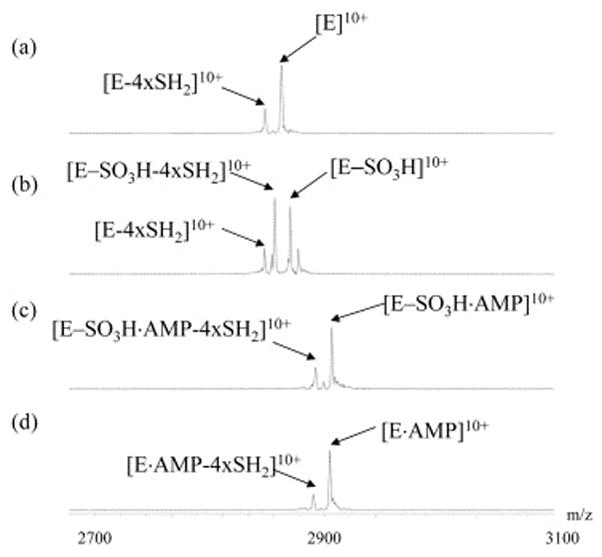
SORI-CID spectra of (A) intact enzyme, (B) thiosulfate intermediate, (C) AMP bound thiosulfate intermediate, and (D) AMP bound enzyme. In each spectrum, the 10+ charge state precursor ion was isolated and subjected to CID as described in the experimental section.
Discussion
Characterization of the 4Fe-4S cluster containing APS reductase by ESI-FTICR MS
ESI-FTICR mass spectrometry was utilized to investigate a 4Fe-4S cluster containing protein, M. tuberculosis APS reductase. Under nondenaturing conditions, the intact holoprotein was observed. The apoprotein without the cofactor and a 2Fe-2S intermediate were also detected, which result from complete or partial dissociations of the metal center due to solution oxidation. The relative ratio among different protein forms could be approximated based on their relative ion abundances in the spectra. With isotopically resolved spectra acquired using ESI-FTICR, the oxidation state of the cluster and the disulfide status for each protein form were determined. The assignments of oxidation states for the 4Fe-4S and 2Fe-2S clusters are consistent with previous electron paramagnetic resonance (EPR) spectroscopy results [51]. Furthermore, cysteine labeling studies do not support the existence of disulfide bonds in the holoenzyme [48]. Compared to EPR and cystein labeling experiments, accurate mass measurement consumes less protein sample and assay time. Notably, M. tuberculosis APS reductase is thus far the largest iron-sulfur cluster containing protein for which high-resolution mass spectra were obtained. Careful tuning of the instrumental parameters is very crucial to observing good quality spectra for the intact holoprotein and its associated complexes on the FTICR mass spectrometer,. Since APS reductase is relatively unstable at room temperature, it is also important to keep the syringe and spray chamber at low temperature during mass analysis.
Tandem mass spectra acquired using both in-source dissociation and SORI-CID provide further evidence for the composition of the iron-sulfur cluster in APS reductase and its associated covalent/noncovalent complexes. The loss of inorganic sulfide ions, together with two additional protons, has been reported for another iron-sulfur cluster containing protein E. coli Biotin synthase [38]. In that story, the authors attribute the observation to the lability of inorganic sulfides, which can be inserted into dethiobiotin to synthesize biotin. For M. tuberculosis APS reductase, loss of iron or the intact cluster was not observed at the highest dissociation energy tested, using both in-source dissociation and SORI-CID. These data indicate that in the gas-phase, the interaction between Fe2+/Fe3+ and the protein ligands is stronger than that between the cluster ions (Fe2+/Fe3+ and inorganic S2−). It is interesting to note that it requires higher energy to dissociate the noncovalently bound AMP compared to inorganic sulfides in the metal cluster. This is due to either a stronger interaction between the holoenzyme and AMP, or to a gas-phase effect through which the interaction between positively charged residues on APS reductase and negatively charged AMP is strengthened. The latter effect has been observed previously in noncovalent spermine-peptide complexes. In that study, the gas-phase interaction between oppositely charged groups was found to be unusually strong [58].
The Mechanism of Sulfonucleotide Reduction
The mechanism of sulfonucleotide reductases has been the subject of much debate over the last decade. Two different models have been proposed, with the main conflict concerning the requirement of pre-reduction of the reductase by the protein cofactor Trx. In the early model, based on initial steady-state kinetics of S. cerevisiae and E. coli PAPS reductases [49], thioredoxin was proposed to reduce an intermolecular disulfide bond between the two C-terminal cysteine residues on the homodimer. One of the liberated thiolates would subsequently execute a nucleophilic attack on the sulfur atom in PAPS, generating a thiosulfonate intermediate. In the final step, the second thiolate would facilitate sulfite release via reformation of the intermolecular disulfide bond. Based on the detection of a stable covalent intermediate between the enzyme and APS, we recently reported a common mechanism that is shared by at least three different reductases (M. tuberculosis APS reductase, E. coli PAPS reductase and P. aruginosa APS reductase [48]), which is also consistent with data previously reported by Weber et al. for A. thalianina APS reductase [59]. In the first step of this model, an absolutely conserved cysteine reside carries out nucleophilic attack on the sulfonucleotide sulfate group, yielding a thiosulfonate intermediate. In the second step, thioredoxin is required for the reduction of the thiosulfonate bond to release sulfite. For monomeric M. tuberculosis APS reductase, the presence or absence of an intramolecular disulfide bond is one way to distinguish between the two possible mechanisms. Accurate mass measurements described in this current study indicate that the 4Fe-4S cluster containing holoenzyme does not have a disulfide bond, thus supporting our proposed mechanism. In our preliminary model, the reaction catalyzed by APS reductase was delineated into two major steps: intermediate formation and intermediate breakdown. To shed light onto other steps during the reaction and thus gain a more comprehensive understanding of the mechanism of APS reduction, noncovalent complexes formed during the reaction were investigated using ESI-FTICR mass spectrometry.
The detection of a stable substituted enzyme (E-SO3H) indicated that APS reduction follows a ping pong mechanism. However, as described above, gas-phase noncovalent complexes of E-SO3H·AMP (converted from E·APS, Figure 3A) and E·Trx (Figure 3B and 3C) were both detected, indicating that the two substrates can associate with APS reductase in the absence of each other. Minimally, these results suggest that the enzyme does not function through a classical ping-pong mechanism [60], in which the second substrate only binds to the enzyme-intermediate, but not to the intact enzyme. Results in the current study also show that the product AMP binds both to the holoenzyme and the thiosulfonate enzyme-intermediate as supported by the observation of noncovalent complexes E·AMP (Figure 3A, Figure 3C and Figure 4A) and E-SO3H·AMP (Figure 3A and Figure 4). These types of complexes have been observed in other non-classical ping-pong systems as well [61, 62]. Finally, several ternary complexes that are predicted for a non-classical ping-pong system according to Segel at al. [60] were also observed in our assay. These complexes include E·AMP·Trxox (Figure 3C and Figure 4) and E-SO3H·AMP·Trxox (Figure 4B).
In the above mass spectra of protein-ligand mixtures, only the holoenzyme with a mature 4Fe-4S cluster forms the corresponding complexes, whereas the apoenzyme and the intermediate containing 2Fe-2S cluster do not. These results indicate that the noncovalent complexes observed in the gas-phase represent specific solution-phase binding effects. However, the equilibrium binding assays provide no information on the binding sequence for formation of a ternary complex, i.e., information on whether two ligands bind randomly, or in a specific order is required. It has been demonstrated that APS could be reduced with concurrent enzyme-intermediate formation in the absence of Trx [48]. However, it is not clear whether binding of Trx to the intact enzyme could also lead to final product formation, or if E·Trx is solely a non-productive complex; i.e. Trx is released when APS binds, then rebinds to E-SO3H·AMP to complete the reaction. It is not possible to distinguish between the two scenarios based on the noncovalent interaction results obtained in the current study. Considering other available biochemical data, the latter scenario seems more reasonable. Previous results indicate that APS reductase experiences a conformational rearrangement upon intermediate formation, and the thiosulfonate intermediate is stable against small molecule reductants [48, 51]. Therefore one can imagine that the intermediate is stabilized by the conformational restraint, which only relaxes upon Trx binding and action. However, without detailed structural information, the other possibility can not be completely excluded.
Based on the above information, a more detailed mechanism is proposed for M. tuberculosis APS reductase as shown in Figure 7. In this mechanism, APS and Trx bind independently to the intact APS reductase, forming binary complexes E-SO3H·AMP and E·Trx. Subsequent binding of the other substrate forms the ternary complex. However, the extent of APS binding towards E·Trx to form the intermediate (indicated with a dotted arrow and question mark) remains to be tested. Once APS is bound, the nucleophilic cysteine (Cys 249) attacks its sulfuryl group, yielding the substituted enzyme-thiosulfonate intermediate. This is followed by the reduction of the intermediate by reduced Trx to release sulfite and recycle APS reductase to its original form. The product AMP could be released in a random fashion either before or after the action of Trx. In the scenario where AMP remains bound until Trx attacks, a conformational change of the intermediate to prevent AMP from dissociation would be expected, which is consistent with our previous results [51]. Detailed structural information is required to test these hypotheses, and will be the subject of future investigations.
Figure 7.
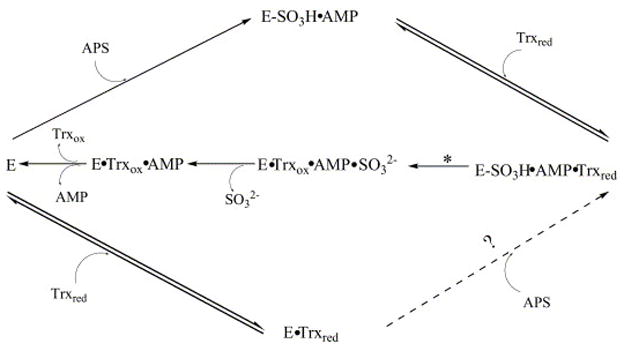
A revised mechanism for APS reductase. The step marked with a star represents the chemical step of sulfite release. All steps shown in solid arrows have been proved to exist. The step shown in a dotted arrow and question mark remains to be tested.
Conclusion
Accurate mass measurement of the 4Fe-4S cluster containing APS reductase holoprotein indicates that there is no disulfide bond in the native enzyme and the cluster has an oxidation state of +2. Accurate mass measurements of the holoenzyme, the enzyme-thiosulfonate intermediate and both with AMP bound indicate that there is no electron-transfer effect in the cluster during substrate binding and reduction. Extensive information was obtained by investigating the noncovalent complexes between the holoenzyme of APS reductase and various ligands. Based on the ion abundance observed in the same spectrum, relative binding affinities of the ligand towards different forms of the enzyme were determined. A more detailed mechanism is proposed for APS reductase given the binary and ternary complexes observed during the reaction. Gas-phase stability of the cluster in different enzyme forms was also investigated, which provides further information about the composition of the cluster in APS reductase and its associated complexes. The current research demonstrates the potential of ESI mass spectrometry in combination with high resolution FTICR in the investigation of complicated biological systems. Better understanding of the reaction mechanism of APS reductase assists in inhibitor design towards the enzyme, and is also crucial to further elucidate the role of APS reductase in M. tuberculosis infection.
Acknowledgments
This work was supported by NIH grant GM 63581 (to JAL) and AI 51622 (to CRB). KSC is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-1783-03). We thank Yonghao Yu for helpful discussion on the accurate mass measurement.
The following abbreviations have been used in this text
- AMP
adenosine-5′-phosphate
- APS
adenosine-5′-phosphosulfate
- NH4OAc
ammonium acetate
- DTT
dithiothreitol
- E
enzyme
- ESI-MS
electrospray mass spectrometry
- 4Fe-4S
four iron-four sulfur cluster
- FT-ICR
Fourier transform ion-cyclotron resonance
- Trx
thioredoxin
- 2Fe-2S
two iron-two sulfur cluster
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beinert H, Holm RH, Munck E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 2.Frazzon J, Dean DR. Formation of iron-sulfur clusters in bacteria: an emerging field in bioinorganic chemistry. Curr Opin Chem Biol. 2003;7:166–173. doi: 10.1016/s1367-5931(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 3.Mortenson LE, Valentine RC, Carnahan JE. An electron transport factor from Clostridium Pasteurianum. Biochem Biophys Res Commun. 1962;7:448–452. doi: 10.1016/0006-291x(62)90333-9. [DOI] [PubMed] [Google Scholar]
- 4.Rees DC, Howard JB. The interface between the biological and inorganic worlds: Iron-sulfur metalloclusters. Science. 2003;300:929–931. doi: 10.1126/science.1083075. [DOI] [PubMed] [Google Scholar]
- 5.Agar JN, Krebs C, Frazzon J, Huynh BH, Dean DR, Johnson MK. IscU as a scaffold for iron-sulfur cluster biosynthesis: Sequential assembly of [2Fe-2S] and [4Fe-4S] clusters in IscU. Biochemistry. 2000;39:7856–7862. doi: 10.1021/bi000931n. [DOI] [PubMed] [Google Scholar]
- 6.Lovenerg W, editor. Iron-Sulfur Proteins. Academic Press; New York: 1973. [Google Scholar]
- 7.Johnson MK. In: Encyclopedia of Inorganic Chemistry. Kind RB, editor. 1994. pp. 1896–1915. [Google Scholar]
- 8.Beinert H, Kennedy MC, Stout CD. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chemical Reviews. 1996;96:2335–2373. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 9.Flint DH, Allen RM. Iron-sulfur proteins with nonredox functions. Chemical Reviews. 1996;96:2315–2334. doi: 10.1021/cr950041r. [DOI] [PubMed] [Google Scholar]
- 10.Sinegina L, Wikstrom M, Verkhovsky MI, Verkhovskaya ML. Activation of isolated NADH: ubiquinone reductase I (complex I) from Escherichia coli by detergent and phospholipids. Recovery of ubiquinone reductase activity and changes in EPR signals of iron-sulfur clusters. Biochemistry. 2005;44:8500–8506. doi: 10.1021/bi050134v. [DOI] [PubMed] [Google Scholar]
- 11.Beinert H, Kennedy MC. Aconitase, A 2-Faced Protein - Enzyme And Iron Regulatory Factor. FASEB J. 1993;7:1442–1449. doi: 10.1096/fasebj.7.15.8262329. [DOI] [PubMed] [Google Scholar]
- 12.Kakuta Y, Horio T, Takahashi Y, Fukuyama K. Crystal structure of Escherichia coli Fdx, an adrenodoxin-type ferredoxin involved in the assembly of iron-sulfur clusters. Biochemistry. 2001;40:11007–11012. doi: 10.1021/bi010544t. [DOI] [PubMed] [Google Scholar]
- 13.Leal SS, Teixeira M, Gomes CM. Studies on the degradation pathway of iron-sulfur centers during unfolding of a hyperstable ferredoxin: cluster dissociation, iron release and protein stability. J Biol Inorg Chem. 2004;9:987–996. doi: 10.1007/s00775-004-0599-z. [DOI] [PubMed] [Google Scholar]
- 14.Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, Hentze W, Rouault TA, Andrews NC, Hediger MA. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001;509:309–316. doi: 10.1016/s0014-5793(01)03189-1. [DOI] [PubMed] [Google Scholar]
- 15.Thauer RK, Schonheit P. In: Iron-Sulfur Proteins. Spiro TG, editor. Wiley-Interscience; New York: 1982. pp. 329–341. [Google Scholar]
- 16.Beinert H, Kiley P. Redox control of gene expression involving iron-sulfur proteins. Change of oxidation-state or assembly disassembly of Fe-S clusters? FEBS Lett. 1996;382:218–219. doi: 10.1016/0014-5793(96)00140-8. [DOI] [PubMed] [Google Scholar]
- 17.Kiley PJ, Beinert H. Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster. Fems Microbiology Reviews. 1998;22:341–352. doi: 10.1111/j.1574-6976.1998.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 18.Cross M, Xiao ZG, Maes EM, Czernuszewicz RS, Drew SC, Pilbrow JR, George GN, Wedd AG. Removal of a cysteine ligand from rubredoxin: assembly of Fe2S2 and Fe(S-Cys)(3)(OH) centres. J Biol Inorg Chem. 2002;7:781–790. doi: 10.1007/s00775-002-0355-1. [DOI] [PubMed] [Google Scholar]
- 19.Rubio LM, Flores E, Herrero A. Purification, cofactor analysis, and site-directed mutagenesis of Synechococcus ferredoxin-nitrate reductase. Photosynthesis Research. 2002;72:13–26. doi: 10.1023/A:1016078700839. [DOI] [PubMed] [Google Scholar]
- 20.Lieder KW, Booker S, Ruzicka FJ, Beinert H, Reed GH, Frey PA. S-adenosylmethionine-dependent reduction of lysine 2,3-aminomutase and observation of the catalytically functional iron-sulfur centers by electron paramagnetic resonance. Biochemistry. 1998;37:2578–2585. doi: 10.1021/bi972417w. [DOI] [PubMed] [Google Scholar]
- 21.Rothery RA, Bertero MG, Cammack R, Palak M, Blasco F, Strynadka NCJ, Weiner JH. The catalytic subunit of Escherichia coli nitrate reductase A contains a novel [4Fe-4S] cluster with a high-spin ground state. Biochemistry. 2004;43:5324–5333. doi: 10.1021/bi049938l. [DOI] [PubMed] [Google Scholar]
- 22.Yano T, Dunham WR, Ohnishi T. Characterization of the Delta mu(H)+-sensitive ubisemiquinone species (SQ(Nf)) and the interaction with cluster N2: New insight into the energy-coupled electron transfer in complex I. Biochemistry. 2005;44:1744–1754. doi: 10.1021/bi048132i. [DOI] [PubMed] [Google Scholar]
- 23.Kim SK, Rahman A, Bick JA, Conover RC, Johnson MK, Mason JT, Hirasawa M, Leustek T, Knaff DB. Properties of the cysteine residues and iron-sulfur cluster of the assimilatory 5′-adenylyl sulfate reductase from Pseudomonas aeruginosa. Biochemistry. 2004;43:13478–13486. doi: 10.1021/bi048811t. [DOI] [PubMed] [Google Scholar]
- 24.Maes EM, Knapp HJ, Czernuszewicz RS, Hendrickson DN. Ligand conformational effects on the resonance Raman signature of [Fe4S4(SAryl)(4)](2−) clusters. J Phys Chem B. 2000;104:10878–10884. [Google Scholar]
- 25.Popescu CV, Bates DM, Beinert H, Munck E, Kiley PJ. Mossbauer spectroscopy as a tool for the study of activation/inactivation of the transcription regulator FNR in whole cells of Escherichia coli. Proc Natl Acad Sci U S A. 1998;95:13431–13435. doi: 10.1073/pnas.95.23.13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walters EM, Garcia-Serres R, Jarneson GNL, Glauser DA, Bourquin F, Manieri W, Schurmann P, Johnson MK, Huynh BH. Spectroscopic characterization of site-specific [Fe4S4] cluster chemistry in ferredoxin: thioredoxin reductase: Implications for the catalytic mechanism. J Am Chem Soc. 2005;127:9612–9624. doi: 10.1021/ja051909q. [DOI] [PubMed] [Google Scholar]
- 27.Beinert H. Spectroscopy of succinate dehydrogenases, a historical perspective. Biochim Biophys Acta-Bioenerg. 2002;1553:7–22. doi: 10.1016/s0005-2728(01)00235-3. [DOI] [PubMed] [Google Scholar]
- 28.Arendsen AF, Hadden J, Card G, McAlpine AS, Bailey S, Zaitsev V, Duke EHM, Lindley PF, Krockel M, Trautwein AX, Feiters MC, Charnock JM, Garner CD, Marritt SJ, Thomson AJ, Kooter IM, Johnson MK, van den Berg WAM, van Dongen W, Hagen WR. The “prismane” protein resolved: X-ray structure at 1.7 angstrom and multiple spectroscopy of two novel 4Fe clusters. J Biol Inorg Chem. 1998;3:81–95. [Google Scholar]
- 29.Sagi I, Bunker G, Hochman Y, Carmeli C, Zeng MT. Determination by X-ray absorption of redox induced structural changes in iron-sulfur cluster F-x in photosystem I. Acta Physica Polonica A. 1997;91:871–875. [Google Scholar]
- 30.Solomon EI, Hedman B, Hodgson KO, Dey A, Szilagyi RK. Ligand K-edge X-ray absorption spectroscopy: covalency of ligand-metal bonds. Coord Chem Rev. 2005;249:97–129. [Google Scholar]
- 31.Petillot Y, Forest E, Mathieu I, Meyer J, Moulis JM. Analysis, By Electrospray-Ionization Mass-Spectrometry, Of Several Forms Of Clostridium-Pasteurianum Rubredoxin. Biochem J. 1993;296:657–661. doi: 10.1042/bj2960657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaquinod M, Leize E, Potier N, Albrecht AM, Shanzer A, Vandorsselaer A. Characterization Of Noncovalent Complexes By Electrospray Mass-Spectrometry. Tetrahedron Lett. 1993;34:2771–2774. [Google Scholar]
- 33.Meyer J, Gagnon J, Sieker LC, Vandorsselaer A, Moulis JM. Rubredoxin From Clostridium-Thermosaccharolyticum - Amino-Acid-Sequence, Mass-Spectrometric And Preliminary Crystallographic Data. Biochem J. 1990;271:839–841. doi: 10.1042/bj2710839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breton JL, Duff JLC, Butt JN, Armstrong FA, George SJ, Petillot Y, Forest E, Schafer G, Thomson AJ. Identification Of The Iron-Sulfur Clusters In A Ferredoxin From The Archaeon Sulfolobus-Acidocaldarius - Evidence For A Reduced [3fe-4s] Cluster With Ph-Dependent Electronic-Properties. Eur J Biochem. 1995;233:937–946. doi: 10.1111/j.1432-1033.1995.937_3.x. [DOI] [PubMed] [Google Scholar]
- 35.Armengaud J, Gaillard J, Forest E, Jouanneau Y. Characterization Of A 2[4fe-4s] Ferredoxin Obtained By Chemical Insertion Of The Fe-S Clusters Into The Apoferredoxin-Ii From Rhodobacter-Capsulatus. Eur J Biochem. 1995;231:396–404. doi: 10.1111/j.1432-1033.1995.tb20712.x. [DOI] [PubMed] [Google Scholar]
- 36.Petillot Y, Forest E, Meyer J, Moulis JM. Observation Of Holoprotein Molecular-Ions Of Several Ferredoxins By Electrospray-Ionization-Mass Spectrometry. Anal Biochem. 1995;228:56–63. doi: 10.1006/abio.1995.1314. [DOI] [PubMed] [Google Scholar]
- 37.Johnson KA, Amster IJ. First observation by mass spectrometry of a 3+oxidation state for a [4Fe-4S] metalloprotein: An ESI-FTICR mass spectrometry study of the high potential iron-sulfur protein from Chromatium vinosum. J Am Soc Mass Spectrom. 2001;12:819–825. doi: 10.1016/S1044-0305(01)00263-X. [DOI] [PubMed] [Google Scholar]
- 38.Hernandez H, Hewitson KS, Roach P, Shaw NM, Baldwin JE, Robinson CV. Observation of the iron-sulfur cluster in Escherichia coli biotin synthase by nanoflow electrospray mass spectrometry. Anal Chem. 2001;73:4154–4161. doi: 10.1021/ac0102664. [DOI] [PubMed] [Google Scholar]
- 39.Johnson KA, Verhagen M, Adams MWW, Amster IJ. Differences between positive and negative ion stabilities of metal-sulfur cluster proteins: an electrospray ionization Fourier transform ion cyclotron resonance study. Int J Mass Spectrom. 2001;204:77–85. [Google Scholar]
- 40.Taylor PK, Kurtz DM, Amster IJ. Electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry of multimeric metalloproteins. Int J Mass Spectrom. 2001;210:651–663. [Google Scholar]
- 41.Johnson KA, Verhagen M, Brereton PS, Adams MWW, Amster IJ. Probing the stoichiometry and oxidation states of metal centers in iron-sulfur proteins using electrospray FTICR mass spectrometry. Anal Chem. 2000;72:1410–1418. doi: 10.1021/ac991183e. [DOI] [PubMed] [Google Scholar]
- 42.Johnson KA, Brereton PS, Verhagen M, Calzolai L, La Mar GN, Adams MWW, Amster IJ. A gallium-substituted cubane-type cluster in Pyrococcus furiosus ferredoxin. J Am Chem Soc. 2001;123:7935–7936. doi: 10.1021/ja0160795. [DOI] [PubMed] [Google Scholar]
- 43.Johnson KA, Shira BA, Anderson JL, Amster IJ. Chemical and on-line electrochemical reduction of metalloproteins with high-resolution electrospray ionization mass spectrometry detection. Anal Chem. 2001;73:803–808. doi: 10.1021/ac001004p. [DOI] [PubMed] [Google Scholar]
- 44.Lampreia J, Pereira AS, Moura JJG. Adenylylsulfate Reductases From Sulfate-Reducing Bacteria. Methods Enzymol. 1994;243:241–260. [Google Scholar]
- 45.Kopriva S, Buchert T, Fritz G, Suter M, Benda RD, Schunemann V, Koprivova A, Schurmann P, Trautwein AX, Kroneck PMH, Brunold C. The presence of an iron-sulfur cluster in adenosine 5′-phosphosulfate reductase separates organisms utilizing adenosine 5′-phosphosulfate and phosphoadenosine 5′-phosphosulfate for sulfate assimilation. J Biol Chem. 2002;277:21786–21791. doi: 10.1074/jbc.M202152200. [DOI] [PubMed] [Google Scholar]
- 46.Bick JA, Dennis JJ, Zylstra GJ, Nowack J, Leustek T. Identification of a new class of 5′-adenylylsulfate (APS) reductases from sulfate-assimilating bacteria. J Bacteriol. 2000;182:135–142. doi: 10.1128/jb.182.1.135-142.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci U S A. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carroll KS, Gao H, Chen H, Stout CD, Leary JA, Bertozzi CR. A conserved Mechanism for Sulfonucleotide Reduction. PLoS Biology. 2005;3:1418–1435. doi: 10.1371/journal.pbio.0030250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berendt U, Haverkamp T, Prior A, Schwenn JD. Reaction-Mechanism Of Thioredoxin - 3′-Phospho-Adenylylsulfate Reductase Investigated By Site-Directed Mutagenesis. Eur J Biochem. 1995;233:347–356. doi: 10.1111/j.1432-1033.1995.347_1.x. [DOI] [PubMed] [Google Scholar]
- 50.Schwenn JD, Krone FA, Husmann K. Yeast Paps Reductase - Properties And Requirements Of The Purified Enzyme. Arch Microbiol. 1988;150:313–319. doi: 10.1007/BF00408300. [DOI] [PubMed] [Google Scholar]
- 51.Carroll KS, Gao H, Chen H, Leary JA, Bertozzi CR. Investigation of the Iron-Sulfur Cluster in Mycobacterium Tuberculosis APS Reductase: Implications for Substrate Binding and Catalysis. Biochem. doi: 10.1021/bi051344a. in press. [DOI] [PubMed] [Google Scholar]
- 52.Yu Y, Sweeney MD, Saad OM, Crown SE, Handel TM, Leary JA. Chemokine-Glycosaminoglycan binding: Specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J Biol Chem. 2005;280:32200–32208. doi: 10.1074/jbc.M505738200. [DOI] [PubMed] [Google Scholar]
- 53.Zubarev RA, Demirev PA, Hakansson P, Sundqvist BUR. Approaches and limits for accurate mass characterization of large biomolecules. Anal Chem. 1995;67:3793–3798. [Google Scholar]
- 54.Berndt C, Lillig CH, Wollenberg M, Bill E, Mansilla MC, de Mendoza D, Seidler A, Schwenn JD. Characterization and reconstitution of a 4Fe-4S adenylyl sulfate/phosphoadenylyl sulfate reductase from Bacillus subtilis. J Biol Chem. 2004;279:7850–7855. doi: 10.1074/jbc.M309332200. [DOI] [PubMed] [Google Scholar]
- 55.Gao H, Yu Y, Leary JA. Mechanism and Kinetics of Metalloenzyme Phosphomannose Isomerase: Measurement of Dissociation Constants and Effect of Zinc Binding Using ESI-FTICR Mass Spectrometry. Anal Chem. 2005;77:5596–5603. doi: 10.1021/ac050549m. [DOI] [PubMed] [Google Scholar]
- 56.Wang W, Kitova EN, Klassen JS. Influence of solution and gas phase processes on protein-carbohydrate binding affinities determined by nanoelectrospray Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2003;75:4945–4955. doi: 10.1021/ac034300l. [DOI] [PubMed] [Google Scholar]
- 57.Loo JA. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 58.Feng R. Proceedings of the 43rd ASMS Conference on Mass Spectrometry and Allied Topics; Atlanta, GA. 1995. p. 1264. [Google Scholar]
- 59.Weber MMS, Brunold C, Kopriva S. Sulfate assimilation in higher plants: characterization of a stable intermediate in the adenosine 5′-phosphosulfate redutase reaction. Eur J Biochem. 2000;267:3647–3653. doi: 10.1046/j.1432-1327.2000.01394.x. [DOI] [PubMed] [Google Scholar]
- 60.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Jon Wiley & Sons; New York: 1975. [Google Scholar]
- 61.Pi N, Yu Y, Mougous JD, Leary JA. Observation of a hybrid random ping-pong mechanism of catalysis for NodST: A mass spectrometry approach. Protein Science. 2004;13:903–912. doi: 10.1110/ps.03581904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu Y, Kirkup CE, Pi N, Leary JA. Characterization of Novcovalent Protein-Ligand Complexes and Associated Enzyme Intermediates of GlcNac-6-O-Sulfotransferase by Electrospray Ionization FT-ICR Mass Spectrometry. J Am Soc Mass Spectrom. 2004;15:1400–1407. doi: 10.1016/j.jasms.2004.06.002. [DOI] [PubMed] [Google Scholar]