

Abstract
A hallmark of cell-surface processes involving glycans is their multivalent interaction with glycan binding proteins (GBPs). Such multivalent interaction depends critically on the mobility and density of signaling molecules on the membrane surface. While glycan microarrays have been used in exploring multivalent interactions, the lack of mobility and the difficulty in controlling surface density both limit their quantitative applications. Here we apply a fluidic glycan microarray, with glycan density varying for orders of magnitude, to profile cell surface interaction using a model system, the adhesion of Escherichia coli (E. coli) to mannose. We show the quantitative determination of monovalent and multivalent adhesion channels; the latter can be inhibited by nanopartices presenting a high density of mannosyl groups. These results reveal a new E. coli adhesion mechanism: the switching in the FimH adhesion protein avidity from monovalent to multivalent as the density of mobile mannosyl groups increases; such avidity switching enhances binding affinity and triggers multiple fimbriae anchoring. Affinity enhancement towards FimH has only been observed before for oligo-mannose due to the turn on of secondary interactions outside the mannose binding pocket. We suggest that the new mechanism revealed by the fluidic microarray is of general significance to cell surface interactions: the dynamic clustering of simple sugar groups (homogeneous or heterogeneous) on the fluidic membrane surface may simulate the functions of complex glycan molecules.
1. Introduction
A wide range of cell surface processes, such as pathogen recognition and attachment onto host cells, cell-cell communication, and the innate immune response, often occur via the simultaneous interaction between multiple copies of receptor proteins and glycan molecules.1-3 Given the tremendous complexity and variation in glycan moieties, understanding such multivalent cell surface interactions and the development of drugs targeting these interactions can be greatly assisted by large-scale profiling and analysis techniques. One of the most attractive tools in glycomics research is the glycan microarray in which synthetic or natural libraries are immobilized on solid surfaces for the high throughput, large-scale analysis. 4-9 It has been argued that the display of a high density of glycan molecules on the surface of a microarray can facilitate multivalent interaction. Despite these advances, there are two significant limitations associated with current glycan microarray technology.
The first limitation is the lack of mobility. On most glycan microarrays demonstrated to date, glycan moieties are immobilized on solid surfaces. The lack of mobility does not mimic cell surface processes in vivo where glycan groups associated with glycolipids and glycoproteins are in a fluidic lipid bilayer environment. Mobility is believed to be a significant factor in mediating multivalent interactions, e.g., in the dynamic clustering of glycan ligands on the host cell surface1 and in promoting statistical pattern matching.10 The advantage of using the fluidic supported lipid bilayer (SLB) platform in probing multivalent cell surface interactions is well recognized.11,12 An added advantage of the supported lipid bilayer is the preferential orientation of surface glycan moieties imposed by their conjugation to lipid molecules embedded in the SLB. However, the fragile nature of the SLB remains a major obstacle in its application in high throughput analysis, including glycan microarrays. In principle, whole cell based microarrays13 may be used to present glycan moieties in a fluidic environment but controlling glycan density on cell surfaces is a formidable challenge.
The second limitation of glycan microarray technology is the difficulty in controlling surface glycan density in an immobilized state. The nature of multivalent interaction dictates that the adhesion probability is a strong function of surface glycan density.1-3 Changes in glycan density may even lead to the switching in binding selectivity due to the turn-on of secondary interactions.14 In order to establish a mechanistic understanding, e.g., quantifying the valency (number of binding pairs) and binding constants involved, one must quantitatively control and vary surface glycan density over a broad range. While synthetic methodologies have been devised to control surface glycan density,14 doing this over many orders of magnitude in glycan density is no easy task. For example, Barth et al. used a poly(ethylene glycol) brush to immobilize mono-, tri-, and high mannose and studied the adhesion of E. coli.15 These authors were only able to vary the surface mannoside density by a factor of ∼4, ranging from 3.8×104 to 1.6×105 μm-2, and the density of adhered E. coli already reached saturation at the lowest glycan density for one mono- and one tri-mannoside. Even if one varies the density over a much broad range, quantifying surface glycan density over many orders of magnitude is beyond most analytical techniques. As an example, Disney and Seeberger explored the effects of glycan density on cell and pathogen adhesion using glycan microarrays, but failed to correlate the amount of cell/pathogen adhesion directly with the density of glycans on the surface.16 Instead, these authors had to settle with an indirect and qualitative correlation with the solution concentration of functional glycan molecules used in the covalent immobilization reaction. Similar difficulties in determining immobilized glycan density are encountered in other quantitative applications of carbohydrate microarrays, e.g., in the works of Wong and coworkers,17,18 Gildersleeve and coworkers,19 and others.20,21
Here we demonstrate a fluidic glycan microarray for quantitative analysis in glycomics. This is based on a robust supported lipid bilayer (SLB) technology,22 which differs from previous demonstrations of fluidic glycan microarrays where the lack of robustness required surface patterning from microfabrication and stringent sample handling.23 Achieving air-stability allows us to easily fabricate glycan microarrays from spotting of lipid solutions on a planar substrate. e.g., glass slide or coverslip. Controlling surface glycan density can be quantitatively achieved by simply adjusting the concentrations of glycol lipids in the lipid mixture for the preparation of small unilamellar vesicle (SUV) solutions. For a single glycan type, we can prepare microarrays in a broad density range, as illustrated in Fig. 1, to obtain a complete binding curve. We can also extend this approach to multiple glycan molecules to explore the complex glycan universe. In the present study, we illustrate the approach using the model system of E. coli with type 1 pilus mutants and a mannose microarray with density varying over two orders of magnitude (0.002 – 0.3 nm-2). Note that we can easily extend this to a much broader density range by successive dilution and mixing. We use an E. coli strain, ORN178, with strong affinity for mannose.24 We use as negative control another E. coli strain, ORN208, with a point mutation is the lectin binding domain resulting in little affinity for mannose.24 We show the successful determination of valency and binding constants for E-coli adhesion. This quantitative finding reveals a new multivalent adhesion mechanism for FimH on E-coli to multiple mannose on the fluidic cell membrane mimicking surface.
Fig. 1.
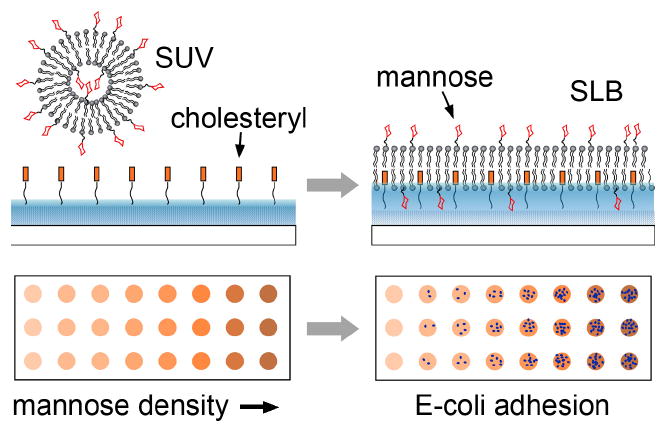
Upper: the formation of a glycan presenting supported lipid bilayer (SLB) surface from a small unilamellar vesicle (SUV) solution. Lower: schematic illustration of a glycan density gradient microarray for pathogen adhesion.
2. Results
The microarray is based on a fluidic and air-stable supported lipid bilayer (SLB) technology developed at MicroSurfaces, Inc.22 This approach utilizes tethered cholesteryl groups incorporated into the bottom leaflet of the supported bilayer lipid membrane, as shown in Fig. 1. The incorporation of cholesterols increases the stability and rigidity of lipid bilayer membranes. This cholesterol function is combined with covalent tether to impart the desired robustness. Achieving air-stability allows us to easily fabricate cell membrane mimicking microarrays from robotic spotting of SUV solutions. The SUV solution is from lipid mixtures of Egg phophatidylcholine (EggPC), mannose-conjugated 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (Ma-PTE), and 0.5% of Texas Red-dihexadecanoyl-phosphatidylethanolamine (TR-DHPE). We verify the fluidity of the SLB spots by fluorescence recovery after photobleaching (FRAP).22 We incorporate various concentrations of mannose-linked lipids in the lipid mixture to fabricate a density gradient microarray, with precisely known mannose density in each spot. The microarray is then incubated with a suspension of E. coli ORN178 bacteria for up to 2.5 hours. After washing, the number density of E-coli adhered to each surface is counted. We use E-coli ORN208 as negative control. Details on experimental methods are available as Online Supporting Material.
Fig. 2a shows an example of a 3×4 array of SLB spots (fluorescence microscopy) containing 0.1, 1, 5, and 10% mannose. For each mannose density, there are three duplicates. The mannose density array is incubated with E. coli suspension (4×106 cells/ml) at room temperature. After extensive washing, the number of adsorbed E-coli cells is counted under an optical microscope. Figs. 2b & 2c show two representative images of E. coli ORN178 cells adsorbed on 1% and 10% mannose SLB surfaces. Each E. coli cell appears as an elongated and isolated spot of μm dimension. More sample images are available in the Online Supporting Material.
Fig. 2.
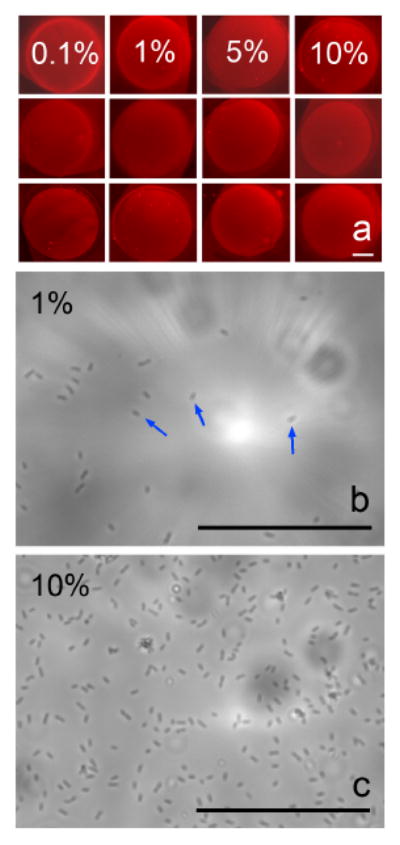
(a) Fluorescence microscope images of a 3×4 lipid bilayer microarray containing the indicated percentage of mannose-linked lipids (0.1-10%) in each column. The red color comes from TR-DHPE; (b) & (c) Zoomed-in optical microscope image (inside the boundary of each SLB spot) of E-coli 178 adsorbed on the lipid bilayer spots containing 1% and 10% mannose. The adsorbed E-coli appears as elongated spots, as illustrated by blue arrows. Scale bars = 50 μm.
Fig. 3 summarizes quantitatively the density of adsorbed E. coli ORN178 cells as a function of mannose density on the SLB microarray. We use a logarithmic scale to clearly show data in the low cell density region. The error bar on each data point is the standard deviation obtained from 10-20 repeated measurements. The binding curve is characterized by two distinct regions. At mannose density < 5%, we see a low cell density region which rises with mannose density and saturates at ∼0.005 cells/μm2. At high mannose density (≥ 5%), the cell density rises rapidly and saturates at ∼0.03 cells/μm2. For negative control, we see no E. coli ORN208 adsorption at low mannose density, but a measurable adsorption (0.001 cell/μm2) at mannose density > 5%. The saturation density for ORN208 is only 3% of that for ORN178.
Fig. 3.
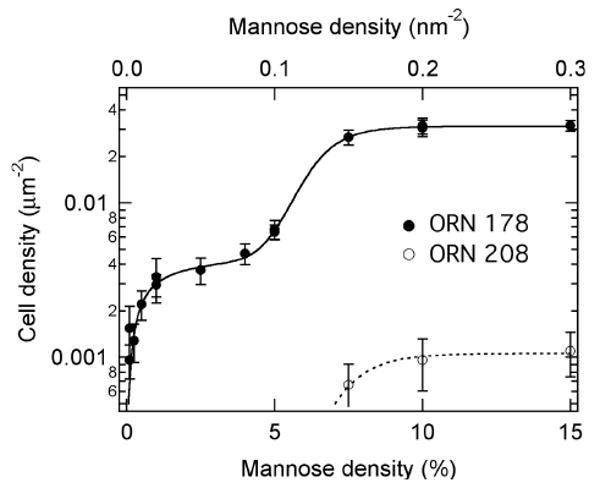
Number density of adsorbed E-coli cells (solid and open circles for ORN178 and ORN208 strains, respectively) as a function of mannose density on the lipid membrane surface. The solid and dotted curves are fits to equation (3).
We now analyze the bimodal binding curve using a simple Langmuir adsorption model17, 25 assuming two types of cell adsorption with different valency:
| (1) |
| (2) |
where [C] = 4.0×106 cells/ml = 6.6 fM (1fM = 10-15 M), is the solution phase bacteria concentration; θM is the surface mannose density; θC1 and θC2 are the surface densities of adsorbed bacteria with valency of n1 and n2, respectively. θC1o and θC2o are the maximum densities of the two types of adsorbed bacteria. Each term in parenthesis represents the available surface sites for bacteria adsorption. K1 and K2 are the equilibrium constants. From these two equilibrium equations, we have:
| (3) |
where θC = θC1 + θC2 is the total density of adsorbed cells. A least-squares non-linear fit to the experimental data in Fig. 3 yields n1 = 1.0 ± 0.3 and n2 = 10.6 ± 0.5, with θC1o =0.0048 ± 0.003 μm-2 and θ C2o =0.031 ± 0.002 μm-2. The error bars are obtained from uncertainties in the fitting. We conclude from this quantitative analysis that there are two types of adsorbed E. coli cells: one involves monovalent binding to one surface mannose group and other with multivalent binding to 10-11 surface mannose groups.
These fits also give equilibrium constants (K1 and K2) that contain units of θm-n We can convert these to the familiar dissociation constants (Kd) for a particular surface mannose density.
| (4) |
As an example, at θM =0.2 nm-2 (the high density region), Kd1 = 1.0±0.2 (10-22 M) and Kd2 = 1.3±0.3 (10-27 M) for ORN178 and Kd2′=4.9±0.8 (10-26 M) for ORN208. Thus, at this surface mannose density, multivalent cell adhesion for ORN178 is 40 times more effective than that of ORN208 and 105 times more effective than that of monovalent ORN178 adhesion. When the mannose density is at the low end, e.g., θM =0.02 nm-2, we have Kd1 = 1.0×10-21 M, Kd2 = 5.2×10-17 M for ORN178. Here, the monovalent channel dominates. There is no measurable ORN208 adhesion in the monovalent channel.
We can effectively inhibit the multivalent adhesion channel to the SLB surface using nanoparticles presenting multiple copies of mannose groups (∼300 per particle) as competitive inhibitors. These mannose presenting nanoparticles are added to the E-coli solution as competitive inhibitors for binding to the mannose-presenting SLB surface. Fig. 4 shows the number density of adsorbed E. coli ORN178 cells on the fluidic SLB surface (7.5% mannose) as a function of nanoparticle concentration in the solution. The surface cell density decreases from 0.023 μm-2 to 0.005 μm-2 with increasing nanoparticle concentration to ∼8 pM (pM = 10-12 M). Increasing nanoparticle concentration higher than 8 pM does not lead to further inhibition and the surface cell density remains at ∼0.005 μm-2, which is equal to the θC1o value within experimental uncertainty. Thus we conclude that inhibition by mannose presenting nanoparticles occurs for the multivalent adsorption channel, not the nearly monovalent channel. The IC50 value for nanoparticle inhibition is ∼4 pM. Note that efficient inhibition of E-coli adhesion requires the multivalent presentation of mannose groups on each nanoparticle surface. We have tested free mannose in the solution phase and do not find inhibition effect for mannose concentration as high as 1 mM.
Fig. 4.
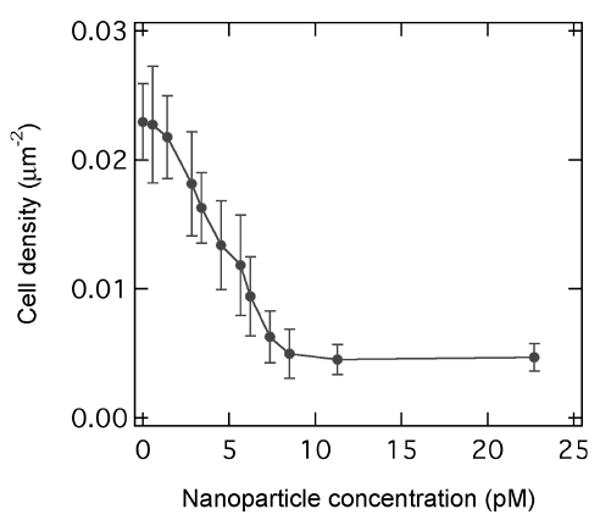
Number density of adsorbed ORN178 E-coli cells on the lipid membrane surface containing 7.5% mannose as a function of solution phase nanoparticle (inhibitor) concentration.
3. Discussions
Previous studies indicate that E. coli binds to a mannose-presenting surface via FimH proteins at the tips of type 1 fimbriae.26 The FimH protein possesses a monomannose (1M) binding pocket, but adhesion is assisted by interactions in regions immediately outside this pocket. It is well established that FimH exhibits 20× enhanced specific adhesion to oligosaccharides, particularly trimannose (3M).27 In fact, the static incubation condition used here (no shear flow) is known to favor multivalent binding to 3M than monovalent binding to 1M.28 With this background in mind, we can now interpret the quantitative finding in the present study.
On the fluidic SLB surface at low mannose density (< 0.1/nm2), there is weak, monovalent binding: only one fimbriae on a bacteria binds one mannose on the fluidic membrane surface. This is the n1 = 1 (first order) adsorption channel in Fig. 3. Note that this finding does not mean that only one fimbriae can bind to the surface. E. coli adhesion is a dynamic process and this first order channel suggests that, on the average, one fimbriae binds to one surface mannose at every instant of time.
As the mannose density reaches above the critical value of 0.1/nm2, we see a rapid rise in the multivalent (n2 = 10-11) adhesion channel. The turn-on of this multivalent channel is not due to a switch from one pilus to 10-11 pili tethering each E. coli cell. There is a mismatch in dimensions: at this critical density, the inter-mannose distance is ∼ 4nm, two orders of magnitude shorter than the inter-pili distance on the E. Coli cell surface. Thus, the turn-on of multi-pili tethering should have occurred at much lower surface mannose density. Instead, this multivalent channel must be attributed to a switch in the FimH avidity from monovalent to trivalent. As the mannose density increases, the close distance of mannose groups on the lipid membrane surface, combined with the mobility of the lipid bilayer, allows multivalent binding to each fimbriae. At the critical mannose density (average distance of ∼4 nm), there is need for only minor rearrangement to enable the multivalent adhesion of each FimH protein to multiple mannose groups. Given the preference and affinity of FimH protein to the covalently linked trimannose,27,28 we believe each fimbriae likely binds to a cluster of three mannose groups on the fluidic lipid membrane surface. The enhanced interaction of the mannose cluster with each fimbriae triggers the anchoring of multiple fimbriae. The observed number of mannose involved in the adhesion of each E-Coli cell, n2 = 10.6 ± 0.5, corresponds to the anchoring of 3-4 fimbriae, each with trivalent bonding to 3M.
While there is no measurable adsorption of ORN208 in the monovalent region at mannose density < 0.1/nm2 as expected from the much weaker binding of the mutated FimH protein to mannose,24 there is a measurable amount of adsorption in the multivalent region, with the ORN208 E. coli cell density 30× lower than that of ORN178. At the high mannose density limit, the turn on of secondary interactions with additional mannose outside of the FimH binding pocket results in appreciable adhesion of the ORN208 E. coli cell. This finding underscores the importance of such quantitative analysis in a broad density range to understand the complex process of cell adhesion.
The multivalent mechanism proposed here for FimH adhesion differs from past proposals of multivalent interaction to trimannose or other oligomannose moieties. The new mechanism proceeds via multivalent interaction between FimH and non-covalent mannose clusters, instead of covalently linked oligo-mannose. This mechanism may be of general significance to the cell surface where mobility and the presence of various glycan moieties allow synergistic interaction with GBPs. Note that in the present example the clustering of mannose groups on the surface of the fluidic supported lipid bilayer is expected to be mechanistically different than the random clustering of mannose on an immobilized surface. The former allows dynamic arrangement during the binding event, as expected for many multivalent cell surface processes, while the latter only provides limited local movement allowed by the chemical tethers. This dynamic advantage of the supported lipid bilayer platform for multivalent interactions has been succinctly argued before11,12 and we hypothesize that it will be of general significance to the study of cell surface processes. Unfortunately, the difficulty with preparing immobilized mannose in such a broad density range prevented us from establishing an unambiguous proof of this hypothesis.
We believe that affinity enhancement of GPBs to oligo- and high-glycan structures can in general be simulated by the dynamic clustering of simple carbohydrate groups on the fluidic membrane surface. As another example, C-type lectin receptor DC-SIGN (dendritic cell – specific intercellular adhesion molecule-grabbing nonintegrain) binds high mannose moieties on HIV-1 virus, a process essential to HIV-1's ability to escape the immune surveillance.29 The actual mechanism in multivalent binding of DC-SIGN to mannose is complex: trimannoside binds DC-SIGN more strongly than monomannose does and binding is further enhanced when addition mannose moieties are present.30 The ability to precisely vary the density of surface glycan groups allows us not only to obtain the quantitative binding curves (Fig. 3) and to screen for multivalent inhibitors (Fig. 4), but also to identify changes in affinity and selectivity. The easy fabrication of cell membrane mimicking microarrays with the potential of quantitative incorporation of not just one, but multiple glycan molecules with surface densities over many orders of magnitudes should make this technological platform a workhorse in the burgeon field of glycomics.
4. Conclusions
We apply a fluidic glycan microarray to quantitatively profile and characterize cell surface interactions using a model system, the adhesion of E. coli to mannose. We show the determination of monovalent and multivalent adhesion channels; the latter can be inhibited by nanopartices presenting a high density of mannosyl groups. We found that the FimH adhesion protein at the tip of each fimbriae on the E. coli cell surface switches avidity from monovalent to multivalent as the density of mobile mannosyl groups increases; the avidity switching enhances binding affinity and triggers multiple fimbriae anchoring. In view of previous findings that affinity enhancement towards FimH occurs for oligo-mannose due to the turn on of secondary interactions, we conclude that the dynamic clustering of mannosyl groups on the fluidic membrane surface can simulate the functions of complex oligo- and high-mannose molecules. We propose that this mechanism is of general significance to cell surface interactions where dynamic clustering of simple glycans (homogeneous or heterogeneous) may lead to affinity enhancement, a function traditionally believed to be reserved for oligo- or high-glycans.
Supplementary Material
Acknowledgments
This work was supported by SBIR phase-I grants from the National Institute of Health (1R43AI077161) and the National Science Foundation (IIP_0810474) awarded to MicroSurfaces, Inc. We thank Ocean Nanotech for supplying magnetic nanoparticles.
Footnotes
Supporting Information Available: Details on Experimental Methods and Complete Ref. 8 are available online. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Bertozzi R, Kiessling LL. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 3.Raman R, Raguram S, Venkataraman G, Paulson JC, Sasisekharan R. Nature Methods. 2005;2:817–824. doi: 10.1038/nmeth807. [DOI] [PubMed] [Google Scholar]
- 4.Kiessling LL, Cairo CW. Nature Biotech. 2002;20:234–235. doi: 10.1038/nbt0302-234. [DOI] [PubMed] [Google Scholar]
- 5.Houseman BT, Mrksich M. Chem Biol. 2002;9:443–454. doi: 10.1016/s1074-5521(02)00124-2. [DOI] [PubMed] [Google Scholar]
- 6.Fuhui S, Feizi T, Galustain C, Lawson AM, Chai W. Nature Biotech. 2002;20:1011–1017. doi: 10.1038/nbt735. [DOI] [PubMed] [Google Scholar]
- 7.Wang D, Liu S, Trummer BJ, Deng C, Wang A. Nature Biotech. 2002;20:275–281. doi: 10.1038/nbt0302-275. [DOI] [PubMed] [Google Scholar]
- 8.Blixt A, et al. Proc Natl Acad Sci USA. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paulsen JC, Blixt O, Collins BE. Nature Chem Biol. 2006;2:238–248. doi: 10.1038/nchembio785. [DOI] [PubMed] [Google Scholar]
- 10.Rai P, Padala C, Poon V, Saraph A, Basha S, Kate S, Tao K, Mogridge J, Kane RS. Nature Biotech. 2006;24:582–586. doi: 10.1038/nbt1204. [DOI] [PubMed] [Google Scholar]
- 11.Groves JT, Dustin ML. J Immuno Methods. 2003;278:19–32. doi: 10.1016/s0022-1759(03)00193-5. [DOI] [PubMed] [Google Scholar]
- 12.Shi J, Yang T, Kataoka S, Zhang Y, Diaz AJ, Cremer PS. J Am Chem Soc. 2007;129:5954–5961. doi: 10.1021/ja069375w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castela D, Pitavala A, Debilya MA, Gidrol X. Drug Discovery Today. 2006;11:616–622. doi: 10.1016/j.drudis.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 14.Horan N, Yan L, Isobe H, Whitesides GM, Kahne D. Proc Natl Acad Sci USA. 1999;96:11782–11786. doi: 10.1073/pnas.96.21.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barth KA, Coullerez G, Nilsson LM, Castelli R, Seeberger PH, Vogel V, Textor M. Adv Funct Mater. 2008;18:1459–1469. [Google Scholar]
- 16.Disney MD, Seeberger PH. Chem Biol. 2004;11:1701–1707. doi: 10.1016/j.chembiol.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 17.Wang CC, Huang YL, Ren CT, Lin CW, Hung JT, Yu JC, Yu AL, Wu CY, Wong CH. Proc Natl Acad Sci USA. 2008;105:11661–11666. doi: 10.1073/pnas.0804923105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang PH, Wang SK, Wong CH. J Am Chem Soc. 2007;129:11177–11184. doi: 10.1021/ja072931h. [DOI] [PubMed] [Google Scholar]
- 19.Manimala JC, Roach TA, Li Z, Gildersleeve JC. Angew Chem Int Ed. 2006;118:3689–3692. doi: 10.1002/anie.200600591. [DOI] [PubMed] [Google Scholar]
- 20.Nimrichter L, Gargir A, Gortler M, Altstock RT, Shtevi A, Weisshaus O, Fire E, Dotan N, Schnaar RL. Glycobiology. 2004;14:197–203. doi: 10.1093/glycob/cwh022. [DOI] [PubMed] [Google Scholar]
- 21.Shin I, Park S, Lee MR. Chem Eur J. 2005;11:2894–2901. doi: 10.1002/chem.200401030. [DOI] [PubMed] [Google Scholar]
- 22.Deng Y, Wang Y, Holtz B, Li JY, Traaseth N, Veglia G, Stottrup B, Elde R, Pei D, Guo A, Zhu XY. J Am Chem Soc. 2008;130:6267–6271. doi: 10.1021/ja800049f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamazaki V, Sirenko O, Schafer RJ, Nguyen L, Gutsmann T, Brade L, Groves JT. BMC Biotech. 2005;5(18):1–11. doi: 10.1186/1472-6750-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris SL, Spears PA, Havell EA, Hamrick TS, Horton JR, Orndorff PE. J Bacter. 2001;183:4099–4102. doi: 10.1128/JB.183.13.4099-4102.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adamson AW, Gast AP. Physical chemistry of surfaces. 6th. Wiley; New York: 1997. [Google Scholar]
- 26.Soto GE, Hultgren SJ. J Bacter. 1999;181:1059–1071. doi: 10.1128/jb.181.4.1059-1071.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sokurenko EV, Chesnokova V, Doyle RJ, Hasty DL. J Biol Chem. 1997;272:17880–17886. doi: 10.1074/jbc.272.28.17880. [DOI] [PubMed] [Google Scholar]
- 28.Nilsson LM, Thomas WE, Trintchina E, Vogel V, Sokurenko EV. J Biol Chem. 2006;281:16656–16663. doi: 10.1074/jbc.M511496200. [DOI] [PubMed] [Google Scholar]
- 29.Van Kooyk Y, Geijtenbeek TBH. Nature Rev Immun. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- 30.Feinberg H, Castelli R, Drickamer K, Seeberger PH, Weis WI. J Biol Chem. 2007;282:4202–4209. doi: 10.1074/jbc.M609689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.