

Abstract
Peptidylglycine α-hydroxylating monooxygenase (PHM, EC 1.14.17.3) catalyzes the stereospecific hydroxylation of a glycyl α-carbon in a reaction that requires O2 and ascorbate. Subsequent dealkylation of the α-hydroxyglycine by another enzyme, peptidylamidoglycolate lyase (PAL. EC 4.3.2.5), yields a bioactive amide and glyoxylate. PHM is a non-coupled, type II dicopper monooxygenase which activates O2 at only a single copper atom, CuM. In this study, the PHM mechanism was probed using a non-natural substrate, benzaldehyde imino-oxy acetic acid (BIAA). PHM catalyzes the O-oxidative dealkylation of BIAA to benzaldoxime and glyoxylate with no involvement of PAL. The minimal kinetic mechanism for BIAA was shown to be steady-state ordered using primary deuterium kinetic isotope effects. The D(V/K)APPARENT, BIAA decreased from 14.7 ± 1.0 as [O2] → 0 to 1.0 ± 0.2 as [O2] → ∞ suggesting the dissociation rate constant from the PHM·BIAA complex decreases as [O2] increases; thereby, reducing the steady-state concentration of [PHM]free. BIAA was further used to differentiate between potential oxidative Cu/O species using a QM/MM reaction coordinate simulation to determine which species could yield product O-dealkylation that matched our experimental data. The results of this study provided compelling evidence for the presence of a covalently linked CuII-alkoxide intermediate with a quartet spin state responsible BIAA oxidation.
1. Introduction
Peptidylglycine α-amidating monooxygenase (PAM, E.C. 1.14.17.3) catalyzes the final reaction in the biosynthesis of α-amidated peptide hormones and may also have a role in the production of oleamide and other bioactive primary fatty acid amides.1,2 This chemistry is a two-step process with the formation of an intermediate that has been stereospecifically hydroxylated at the α-carbon of a glycine. PAM is bifunctional consisting of two catalytic units: peptidylglycine α-hydroxylating monooxygenase (PHM) and peptidylamidoglycolate lyase (PAL). PHM catalyzes the glycine hydroxylation while PAL catalyzes the carbinolamide dealkylation (Scheme 1a). The relationship between PHM and PAL is multifaceted and species specific. Mammalian PAM is coded for by a single gene; alternative splicing of the PAM mRNA and proteolytic processing of PAM yields a mixture of the bifunctional enzyme and the monofunctional enzymes, PHM and PAL. In insects and cnidarians, bifunctional PAM is not found; only monofunctional PHM and PAL are known, each coded by a distinct gene.3-5 In the mollusk, Lymnae stagnalis, a pentafunctional enzyme is produced with four PHM domains per PAL domain.6 The complexities in the various forms of the PAM/PHM/PAL system are not well understood, but hint at a novel regulatory mechanism for the in vivo production of the amide and carbinolamide products of this chemistry.
Scheme 1a and 1b.


Peptidylglycine α-amidating monooxygenase (PAM) and dopamine β-monooxygenase (DβM) reactions.
Of the two monofunctional enzymes in the PAM/PHM/PAL system, PHM has been the most extensively studied, due to its structural and mechanistic similarity to dopamine β-monooxygenase (DβM).7 Both PHM and DβM catalyze an O2-, copper-, and ascorbate-dependent hydrogen atom transfer and substrate hydroxylation resulting in the stereospecific hydroxylation of their respective substrates (Scheme 1a and 1b). Both enzymes contain a solvent filled active site separating two essential, non-coupled copper atoms.1,8-12 In PHM, the two protein bound copper atoms have different roles in catalysis. One copper atom, CuM, has two Nε-histidines ligands (His240 and His242) and a methionine sulfur ligand (Met314) and is directly involved in dioxygen activation and substrate oxidation. The other copper atom, CuH, has three Nδ-histidine ligands (His107, His108, and His172) and is involved in electron transfer. The PAL domain, unique to the PAM system, is a zinc and calcium-dependent and catalyzes the dealkylation of the carbinolamide to the corresponding amide and glyoxylate (Scheme 1a).13,14
Intriguingly, DβM, without a PAL-like partner, will catalyze oxidative dealkylation similar to the sequential reactions catalyzed by PHM and PAL in bifunctional PAM.15 We report herein the PAL-independent oxidative dealkylation of imino-oxyacetic acids to the corresponding oximes and glyoxylate by PHM. For PHM and DβM, the dealkylation reactions result solely from the oxidation chemistry of their monooxygenase domains. This similarity in dealkylation chemistry provides a novel framework to study the mechanism of PAM catalysis.15-17
The role of oxygen activation in PHM and DβM has been the subject of much study.18-22 The presumed nucleophile responsible for hydrogen abstraction is predominantly considered to be an end-on/η1 copper-superoxo radical species.11,23 Both η1 and η2 copper-superoxo species had been prepared in models, although none exhibited the propensity for both H-abstraction and oxidation chemistry.24-29 Recently, the Karlin group prepared end-on/η1 CuII-superoxo model complexes able to orchestrate both H-abstraction and oxidation chemistry.30-33 Characterization of CuII-superoxo species was a critical advance; however, ambiguities remain as to the identity of the actual oxidant species.30
The two PHM mechanisms which involve a CuII-superoxo nucleophile for substrate activation differ in the initial coordination geometry of the dioxygen species to copper (Scheme 2). The ‘side-on/η2’ mechanism is supported by spectroscopic evidence from side-on/η2 Cu/O species model studies and is predicted to have an antiferromagnetically coupled singlet ground state.25,34 When compared with the end-on/η1 species, the side-on/η2 copperII-superoxo was calculated to be thermodynamically preferred for H-transfer.19,35 Evidence for the end-on/η1 species comes from the PHM crystal structure11 and the work of Blackburn et al.,36 suggesting that this species is the nucleophile responsible for Cα-H abstraction. Both mechanisms include an end-on/η1 CuII-hydroperoxo species following Cα-H bond cleavage. The ‘side-on/η2’ mechanism involves a direct hydroxylation of the Cα-substrate radical, through a ‘water-assisted’ radical recombination reaction resulting in the simultaneous reduction of the CuII-hydroperoxo and Cα-OH product release. Conversely, the CuII-hydroperoxo species from the ‘end-on/η1’ mechanism is reduced via an intra-molecular electron transfer from the other Cu atom in the active site, CuH, yielding a CuII–O· radical. Radical recombination of the substrate and Cu/O radical species produces an inner-sphere alcohol intermediate. Subsequent hydrolysis of the inner-sphere alcohol generates the hydroxylated product.
Scheme 2.

Collection of mechanisms postulated for the hydrogen abstracting and oxidation reactions of peptidylglycine α-amidating monooxygenase (and dopamine β-monooxygenase), respectively.
A highly reduced copper-oxo species is postulated for substrate Cα–H bond cleavage in both ‘copper-oxo’ mechanisms, which uncouple dioxygen reduction from substrate activation. Hybrid QM/MM simulations suggest that a CuIII–oxide/CuII–oxyl species is thermodynamically preferred relative to CuII-superoxo or CuII-hydroperoxo nucleophiles.37-39 The CuIII–oxide/CuII–oxyl species is analogous to the ferryl oxidant (CpI) responsible for C-H activation in cytochrome P450.37,40 Both ‘copper-oxo’ mechanisms proceed from an initial end-on/η1 CuII–superoxo species to a highly reduced copper-oxo species by coupling intra-molecular electron transfer with the acquisition of two protons leading to water release. In ‘copper-oxo pathway A’, the reduced copper-oxo species is CuII-oxyl with two unpaired electrons ferromagnetically coupled to an unpaired electron delocalized within the CuM domain yielding a quartet spin state.37 Here, H-abstraction and hydroxylation are in concert with spin inversion to a doublet ground state allowing substrate oxidation and hydroxylated product release to be directly coupled. In the ‘copper-oxo pathway B’, the CuII –oxyl species is in a triplet ground state as the most thermodynamically favorable species for H-abstraction. Spin inversion yields an antiferromagnetically coupled singlet state to drive concerted H-abstraction with substrate oxidation/product release. Simulations by Crespo et al. favor the singlet state oxidant relative to either quartet or triplet state oxidants.37
Analysis of the four mechanisms outlined in Scheme 2 shows commonality between the ‘side-on/η2’ 35,41 and both ‘copper-oxo’ 38,39 mechanisms. Product release and substrate oxidation are simultaneous, lacking covalent interaction between H-donor and Cu/O acceptor (‘free-product’). The direct hydroxyl transfer reactions of the ‘side-on/η2’ and ‘copper-oxo pathway B’ mechanisms are electronically similar as hydroxyl transfer begins with an antiferromagnetically coupled singlet state. Therefore, as the reaction coordinate proceeds toward substrate oxidation/product release, the transition states of both ‘copper-oxo pathway B’ and ‘side-on/η2’ species become super imposable.
Conversely, the ‘end-on/η1’ mechanism is defined by an inner-sphere alcohol intermediate produced through a radical recombination reaction.7,20 The CuII–oxyl radical species in the ‘end-on/η1’ mechanism is unique in that the reductive cleavage of O-O bond in the the CuII–hydroperoxo moiety results from an intra-molecular reduction followed by homolysis to yield the active species. Unlike the ferryl (FeIV=O; CpI) complex of P450, the CuII–oxyl lacks the strong electron delocalization into the d–orbitals of the metal, allowing full radical stabilization in the oxygen 2π* orbital.41 Formation of the CuII–oxyl species by reduction of the CuII–hydroperoxo followed by homolytic O-O bond cleavage suggests that this cupryl species must exist as a quartet, with well-defined radical character on the oxygen.
The hypothesis of the present study was that the PHM catalyzed reaction proceeds through an ‘inner sphere alcohol’ intermediate. To test this hypothesis we designed a novel PHM substrate, benzaldehyde imino-oxy acetic acid (BIAA), which would undergo PAL-independent dealkylation. Compared to other known PHM substrates that release a hydroxylated product1, the unique ability of BIAA to decompose during oxygen insertion allows isolation of the substrate oxidation step. Characterization of BIAA dealkylation demonstrated that this chemistry is exclusively dependent on PHM-catalyzed oxidation (and independent of product release), allowing hybrid QM/MM simulations of substrate radical oxidation to be accurately modeled. Our data provides compelling evidence that the PHM reaction does, in fact, proceed via an inner sphere alcohol intermediate.
Materials and Methods
Materials
Benzamide, α-hydroxyhippurate, benzaldoxime (BOX), bromoacetic acid, [α-2H2]-bromoacetic acid, hydroxylamine hydrochloride, benzaldehyde, and rabbit muscle lactate dehydrogenase were purchased from Sigma-Aldrich. Carboxymethylhydroxylamine was purchased from TCI. Bovine liver catalase was purchased from Worthington. N-dansyl-Tyr-Val-Gly was purchased from Fluka Biochimika. Recombinant type A rat medullary thyroid carcinoma bifunctional PAM was produced and purified as described42 and was a gift from Unigene Laboratories, Inc. (Fairfield, NJ, see www.unigene.com). All other reagents were of the highest quality available from commercial suppliers.
Synthesis of Benzaldehyde Imino-Oxy Acetic Acid (BIAA)
Benzaldehyde was converted to the corresponding benzaldoxime using a published procedure.43 Oxime (4.1 mmole) was dissolved in 20 mL of H2O containing 5 eq. of NaOH, the reaction stirred at room temperature for 45 minutes, after which 1.5 equivalents of [α-H2 or α-2H2]-bromoacetic acid was slowly added in small increments with continued stirring. Upon completion (as determined by TLC using a 1:3 hexanes:ethyl acetate as the mobile phase) the reaction was acidified with dilute HCl, the BIAA was collected in a Büchner funnel, and solvent removed by washing the solid with petroleum ether. BIAA was recrystallized twice with benzene-petroleum ether yielding a white crystalline solid. Compound purity was analyzed by RP-HPLC and 1H and 13C NMR. [α-H2]-BIAA: 1H NMR analysis (250MHz, MeOD-d4) δ4.96 (singlet, 2H, CH2, α-methylene), δ7.66-7.87 (m, 5H, ArH), δ8.48 (singlet, 1H, H-C=N). 13C NMR analysis (62.5MHz, MeOD-d4) δ171.841 (C=O, carboxylic acid), δ149.486 (C=N, imine), δ130.911 (Ar, C-1), δ129.188 (Ar, C-4), δ127.673 (Ar, C-3, C-5), δ126.136 (Ar, C-2, C-6), δ69.325 (CH2, α-methylene), m.p. 93-94°C.
[α-2H2]-BIAA: 1H NMR analysis (400MHz, Me2SO-d6): δ7.397-7.589 (m, 5H, ArH), δ8.302 (singlet, 1H, H-C=N). 13C NMR analysis (100MHz, Me2SO-d6) δ171.660 (C=O, carboxylic acid), δ150.455 (C=N, imine), δ130.865 (Ar, C-1), δ129.502 (Ar, C-4), δ127.688 (Ar, C-3, C-5), δ125.644 (Ar, C-2, C-6), m.p. 96-97°C.
Standard PAM Reaction Conditions
All reactions were performed at 37.0 ± 0.1 °C in the following solution: 100 mM MES/NaOH (pH 6.0), 30 mM NaCl, 1% (v/v) EtOH, 0.001% (v/v) Triton X-100, 1.0 μM Cu(NO3)2, 5.0 mM sodium ascorbate, and 5.75 μg/mL bovine liver catalase, 217 μM O2 and the desired oxidizable substrate. Morrison and Billett44 showed that the ambient O2 concentration at 1 atm and 37 °C is 217 μM
Reverse-Phase HPLC (RP-HPLC) Analysis of PAM Catalyzed Reactions
A RP-HPLC separation for N-benzoylglycine, α-hydroxy-N-benzoylglycine, benzamide, BIAA, and BOX was performed at 50 °C using a 65% (v/v) 50 mM potassium phosphate pH 6.0, 30% (v/v) methanol, and 5% (v/v) acetonitrile with a flow rate of 1.0 mL/min. Analytes were detected at 248 nm (Figure A, supporting information,). A Hewlett Packard HP-1100 HPLC equipped with a Phenomenex C18-Hypersil column (100 × 4.6 mm) attached to a HP-variable wavelength detector was employed for all HPLC separations. Standard curves for N-benzoylglycine, α-hydroxy-N-benzoylglycine, benzamide, BIAA, and BOX were prepared from the corresponding peak area values.
Characterization of Oxidation Product from BIAA
PAM (360 nM) was added to initiate the oxidation of 4.0 mM BIAA under standard reaction conditions, reaction volume = 0.5 mL. After a 20 hr. incubation, the reaction was diluted by addition of 8 mL of H2O, extracted with ethyl acetate (20 mL × 3), rinsed with brine, and dried over MgSO4. The product was isolated by flash column chromatography and concentrated under reduced pressure. A small fraction of the BIAA oxidation product was first evaluated by RP-HPLC by retention time comparison to commercially available benzaldoxime. The remainder of the BIAA oxidation product was analyzed by 13C NMR using a Bruker 250 MHz NMR.
Determination of Glyoxylate Stoichiometry
The stoichiometry of BIAA oxidation to glyoxylate formation was determined under standard reaction conditions using 5.0 mM BIAA as the oxidizable substrate. BIAA oxidation was initiated by the addition of 80 nM PAM. At the desired time, a 40 μL reaction aliquot was removed and the reaction terminated by the addition of trifluoroacetic acid (TFA) yielding a final concentration of 1% (v/v) TFA. Conversion of BIAA to benzaldoxime was monitored by RP-HPLC. All the RP-HPLC samples were diluted to a constant BOX concentration of 48 μM (mid-point of the BOX standard curve) for glyoxylate analysis. The concentration of glyoxylate in the acid quenched samples was determined by the lactate dehydrogenase-mediated reduction of glyoxylate to glycolate coupled to the oxidation of NADH to NAD+ (Δε340 = 6.22 × 103 M-1 cm-1). Glyoxylate containing samples were combined with 7.6 units/mL lactate dehydrogenase in 100 mM potassium phosphate pH 7.0 and 220 μM NADH and incubated for 1 hr. at 37 ± 0.1 ° C. The increase in absorbance at 340 nm was measured using a JASCO V-530 UV-VIS spectrophotometer.
Determination of Oxygen Stoichiometry during BIAA Oxidation
Amperometric analysis was used to correlate total [O2] consumed during PAM catalysis under conditions of limiting [BIAA]. The PAM-dependent consumption of O2 was followed using a Yellow Springs Instrument Model 53 oxygen monitor equipped with a polarographic oxygen electrode interfaced with a personal computer using a Dataq Instruments analogue/digital converter (model DI-154RS) as previously described in McIntyre et al.45
Background levels of [O2] consumed obtained in the absence of enzyme were subtracted from the total [O2] after the addition of PAM.46,47 Standard reactions were initiated with 2.0 nM PAM with the BIAA concentrations being 20, 35, 75, or 100 μM. For each initial [BIAA], the concentration of O2 (217 μM) was in excess and, thus, BIAA was the limiting reagent. The total [O2] consumed at each initial [BIAA] was determined from the asymptote of the O2 consumption progress curve using SigmaPlot 8.0.
Measurement of Noncompetitive Kinetic Isotope Effects
Initial rates of [α-H2]- and [α-2H2]-BIAA were compared independently as a function of initial [O2]. Working stocks of BIAA were calibrated by correlating the O2 consumption to 20 μM BIAA for both substrates under standard reaction conditions. Variation in initial [O2] was accomplished by mixing different proportions of an O2 and N2 with the final [O2] was calibrated versus 217 μM [O2] at 37.0 ± 0.1 °C after a 4 min. equilibration.48 Enzyme-dependent rates of O2 consumption were obtained after initiation of the reaction with a ∼110 nM (4-5 μL) of PAM. Background rates of O2 consumption obtained in the absence of PAM were subtracted from the enzyme-dependent rates. Values for both D(VMAX/KM)APPARENT, BIAA and D(VMAX/KM)APPARENT, OXYGEN were attained from the ratio of protio/deuterio kinetic parameters (equation 1) using Kaleidagraph™.
| Equation 1 |
Values for the D(VMAX/KM)APPARENT, BIAA were also determined directly at a single BIAA concentration below the KM,APPARENT values for [O2], as the observed rate/[substrate] at low substrate concentration is expressed in the units of a second order rate constant, mM-1 s-1 (equation 2). The D(VMAX/KM)APPARENT,BIAA was calculated by dividing the (VMAX/KM)APPARENT,BIAA values obtained for protiated substrate by the deuterated substrate. The reported error is the standard error.
| Equation 2 |
The plot of D(VMAX/KM)APPARENT, BIAA versus [O2] was fit to a hyperbolic curve and analyzed by the least squares method, while the D(VMAX/KM)APPARENT, OXYGEN versus [BIAA] was described as the mean isotope effect ± standard deviation. The full initial rate data matrix for both α-diprotiated and α-dideuterated substrates versus [O2] concentration were analyzed separately and fit to both equation 3 (steady-state preferred) and equation 4 (equilibrium preferred) using the programs of Cleland.49 Single point D(VMAX/KM)APPARENT, BIAA determinations were excluded from this analysis
| Equation 3 |
| Equation 4 |
Rates from equations 3 and 4 are comprised of the substrate concentrations; BIAA and O2, the maximal initial velocity; VMAX, Michaelis constants; KM,BIAA and KM,O2, and the dissociation constant for BIAA, KD,BIAA. Further analysis of D(VMAX/KM) APPARENT, BIAA vs. [O2] and D(VMAX/KM)APPARENT, OXYGEN vs. [BIAA] data were performed to extrapolate values for D(VMAX/KM)APPARENT, OXYGEN, as [O2] → 0 μM and the value of D(VMAX/KM)APPARENT, OXYGEN, as [O2] → ∞ μM. The D(VMAX/KM) APPARENT, OXYGEN as [O2] → 0 μM values were determined through non-linear regression analysis fitting re-plot data to equation 5. The values for D(VMAX/KM)APPARENT,OXYGEN as [O2] → ∞ μM were calculated from the reciprocal expression of equation 5, shown with the expression in equation 6. 50
| Equation 5 |
| Equation 6 |
Quantum Mechanical/Molecular Mechanics Reaction Coordinate
The crystal structure of reduced peptidylglycine α-hydroxylating monooxygenase (PHM) was acquired from the protein databank (1sdw.pdb, 1.85 Å). All waters were removed from the structure along with all occurrences of the co-crystallized peptide ligand (IYT) outside of the active site.11,12,51 The bond orders and charges were then corrected on the co-crystallized protein structure. The copper atoms “atom types” were designated as ‘CuI’, hydrogens were added, and a restrained minimization was performed. The bounding box containing all ligand atoms was set to 14 Å3. Molecular oxygen was removed to prepare the structure using the ProteinPrep wizard for the quantum polarized ligand docking of BIAA (QPLD; Schrödinger First Discovery Suites, www.schrodinger.com). Initially, Glide52 was used to select five top poses using standard precision (SP) mode. These ligand-receptor complexes were analyzed using the Q-site53 module where the bound ligand for each selected pose was treated by ab initio methods to calculate partial atomic charges utilizing electrostatic potential fitting within the receptor. All poses exceeding root mean squared (RMS) and maximum atomic displacement of value 0.5 and 1.3 Ǻ were rejected. Single-point energy determination was treated by the QPLD algorithm to determine the most energetically favorable ligand pose with respect to the receptor. Glide was then used to re-dock the ligand using each of the ligand charge sets previously calculated in Q-site.53 To the QPLD output pose with the BIAA substrate docked into the PHM crystal structure, 3 Na+ ions were added to neutralize (zero net charge) the system along with the molecular oxygen using the coordinates from the original pdb. The system was explicitly solvated using the TIP3P water model to 10 Å from the receptor and equilibrated.37 It should be noted that the O2 coordinates were frozen during the entire system equilibration because it was not formally bound to the CuM domain and would, therefore, traverse the active site during equilibration if its coordinates were not restrained. The final minimized structure of the PHM-ligand central complex was attained through quantum mechanical treatment of the CuM pocket while treating the remainder of the protein structure classically to yield the correct final geometry. The resulting structure was used with Q-Site53 for reaction coordinate calculations. For the QM portion of the calculation a spin unrestricted hybrid density functional theory, with B3LYP hybrid-exchange functional using an LAVCP* basis set was used to more accurately define copper atoms using effective core potentials.54-57 The quantum mechanical region was treated with a spin unrestricted open-shell calculations (UDFT) due to the presence of a radical in the system The molecular mechanics portion of the protein was treated with the OPLS_2001 force field of Jorgensen.58,59
There were two species of interest for study in the PHM oxidation reaction: The copper-hydroxyl (CuII–OH) and the copper-oxyl radical (CuII–O·). The CuII-superoxo species and the bound ligand were set to CuII–OH or CuII–O· and BIAA α-carbon radical in the input file. The quantum mechanical (QM) region was calculated using the CuM atom and domain residues (His240, His242, Met314), the metal-bound oxygen species (either OH or O·), and BIAA Cα radical substrate. On this selection, a Q-site job was run without coordinate constraints to determine the optimized starting point geometry. The calculation was repeated with the oxygen species frozen and the Cα of BIAA frozen to determine the energetic minima starting point. Analysis of the reaction coordinate for substrate oxidation was determined through the difference calculated from incremental increase in the distance between the copper bound oxygen bond and the corresponding decrease in the Cα–O(H) bond. The coordinates determined for each change in reaction coordinate were re-frozen for those two atoms and the energy calculated quantum mechanically. Results were visualized using the Maestro.60
Results
Reaction Stoichiometry and Product Identification for the PHM-catalyzed Oxidation of BIAA
The 13C-NMR of the PHM-generated product generated from BIAA exhibited a spectrum with chemical shifts similar to those of the BIAA starting material, containing signature aromatic and imine carbon shifts (represented A-E, Figure B, supporting information). However, the two carbon shifts found at 171.8 and 69.3 ppm from the O-acetyl moiety in BIAA were not present in the product spectrum. The RP-HPLC retention time of the isolated product was 12.4 min, identical within experimental error to commercially available benzaldoxime, and differed significantly from of the BIAA starting material (6.0 min).
Using BIAA as the limiting substrate, the stoichiometry for the PHM-dependent O2 consumption was 0.97 ± 0.06 O2 consumed/BIAA oxidized. (Figure C and Table C supporting information). The LDH-catalyzed reduction of glyoxylate to glycolate with the concomitant oxidation of NADH to NAD+ provided a convenient spectroscopic method (a) to demonstrate that glyoxylate is a product of the PHM-catalyzed oxidation of BIAA and (b) to measure the glyoxylate concentration generated from BIAA upon PHM treatment. Percent conversion of BIAA to BOX was measured by RP-HPLC. Using the individual methods to measure [BOX] and [glyoxylate], we determined that the [glyoxylate]/[BOX] stoichiometry for the PHM-catalyzed oxidation of BIAA was 1.1 ± 0.1 (Figure 1).
Figure 1.
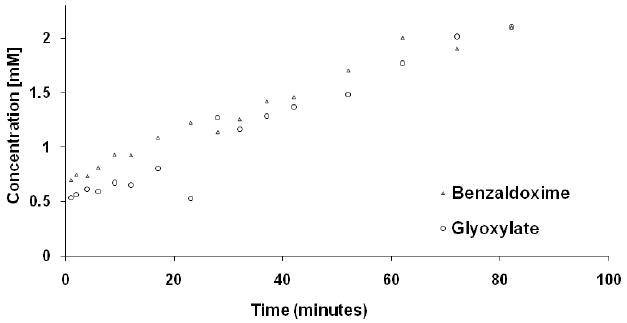
Determination of the PAM dependant ratio of formed products (oxime and glyoxylate) using the imino-oxy acetic acid substrate. The concentrations of oxime and glyoxylate products were independently determined by RP-HPLC and spectrophotometry.
Stability of the BIAA Oxidation Product
Following the PHM-dependent oxidation of BIAA, there are two possible routes for the generation of the observed products, BOX and glyoxylate (Scheme 4a). Path A in Scheme 4 represents a stable α-hydroxylated BIAA which then requires PAL to catalyze BOX and glyoxylate formation while path B requires only PHM for BOX and glyoxylate formation from BIAA. Here, PHM-catalyzed oxidation of the α-carbon would accompany a PAL-independent, chemical dealkylation to yield BOX and glyoxylate.
Scheme 4.


Scheme 4a. Representation of the two possible pathways for BOX and glyoxylate formation following BIAA oxidation in the PHM-domain. Please note that ‘asc’ and ‘sda’ represent single electron contributions from exogenous ascorbate and semi-dehydroascorbate.
Scheme 4b. Experimental design to kinetically differentiate the two possible degradation pathways for BIAA to its corresponding BOX product following PHM-dependent oxidation.
Our approach to differentiating between paths A and B in Scheme 4 was to measure BOX formation in the presence of α-hydroxyhippurate, a known PAL substrate.17,48 If the putative α-hydroxylated BIAA product generated by PHM requires PAL for dealkylation, BOX formation would be inhibited by α-hydroxyhippurate. As a control, benzamide formation via the PAL-catalyzed dealkylation of α-hydroxyhippurate would also be inhibited by BIAA. Of course, if the BIAA product generated by PHM is not a PAL substrate neither BOX formation nor benzamide formation would be affected.
The progress curves for BOX formation from BIAA measured in the presence of either 6 mM or 10 mM α-hydroxyhippurate were unaffected at all BIAA concentrations employed (Fig. 2). Similarly, the progress curves for benzamide formation from α-hydroxyhippurate were unaffected by the presence of 0.5, 2, or 3 mM BIAA (Fig. 3).
Figure 2.

Time course for the PAM-dependent conversion of 3, 2, and 0.5 mM benzaldehyde imino-oxy acetic acid to benzaldoxime in the presence and absence of α-hydroxy-N-benzoylglycine. Results were calculated from RP-HPLC analysis (percent conversion).
Figure 3.

Resolved time course for the conversion of 6mM and 10mM α-hydroxy-N-benzoylglycine to benzamide in the presence and absence of 0.5, 2, and 3 mM BIAA (BenIAA). Please note, time point data was truncated for clarity.
Kinetic Isotope Effects
The D(V/K)APPARENT,BIAA decreased as the concentration of O2 decreased, while the D(V/K)APPARENT,O2 remained constant over the range of BIAA concentrations used (Tables A and B, supporting information). Data fit to the steady-state preferred mechanism (equation 3) had root square values (σ) of 0.237 and 0.140 for α-protiated and α-dideuterated BIAA. Conversely, data fit to the equilibrium preferred mechanism (equation 4) displayed root square values (σ) of 0.236 and 0.156 for the protiated and α-dideuterated substrates. An approximate ten-fold increase in variance was observed for the fit to equations for the equilibrium preferred mechanism, as compared with its steady-state preferred counterpart.49 A large increase in the average residual least square (variance) term was observed showing a greater deviation in error between experimental rates (VEXP) and the predicted/optimized rates (VOPT) for the linear regression model. The variance for [α-1H2]-BIAA and [α-2H2]-BIAA increased from 0.0561(H) and 0.0230(D) from for steady-state preferred to 0.555(H) and 0.242 (D) to for equilibrium preferred. Kinetic data fit to the steady-state preferred model (equation 3) displayed an increased statistical significance based on their variance terms, though the square root (σ) values were statistically very similar.
The graphs of 1/rate vs. 1/[BIAA] and 1/rate vs. 1/[O2] both exhibit intersections in the same quadrant, with a negative abscissa and positive ordinate when fit to the steady-state preferred equation. The extrapolated value of D(V/K)APPARENT, BIAA as [O2] → 0 was determined to be 14.7 ± 1.0 (Figure D, supporting information) and the D(V/K)APPARENT, BIAA as [O2] → ∞ was calculated to be to 1.01 ± 0.03 (Figure E, supporting information). The value for the D(V/K)APPARENT, O2 based on a steady-state preferred mechanism (equation 3), was calculated to be 5.8 ± 0.7 (Table 1). This value is consistent with experimental determinations of D(V/K)APPARENT, O2 obtained at different initial O2 concentrations at one fixed initial BIAA concentration, yielding an average D(V/K)APPARENT, O2 of 4.7 ± 1.1 (Fig. 4, blue circles,  ). D(VMAX) was also calculated to be near unity at 0.8 ± 0.1 while D(KD,BIAA) was found to be 2.1 ± 0.5. The KM,BIAA parameters for both α-protiated and α-dideuterated BIAA were determined to be negative with large standard errors. This suggests that the KM,BIAA was not statistically significant, consistent with a steady-state preferred kinetic mechanism.48,49
). D(VMAX) was also calculated to be near unity at 0.8 ± 0.1 while D(KD,BIAA) was found to be 2.1 ± 0.5. The KM,BIAA parameters for both α-protiated and α-dideuterated BIAA were determined to be negative with large standard errors. This suggests that the KM,BIAA was not statistically significant, consistent with a steady-state preferred kinetic mechanism.48,49
Table 1.
Kinetic parameters determined from the steady-state preferred bi-substrate Michaelis-Menten equation 3.
| VMAX (s-1) | KD,BIAA (mM) | KM, O2 (mM) | (VMAX/KM)O2 (M-1s-1) | DKD,BIAA | D(VMAX) | D(VMAX/KM)O2 | |
|---|---|---|---|---|---|---|---|
| Protiated | 4.75 (0.19) | 758.18 (133.42) | 95.37 (12.18) | 4.98E+04 (5.03E+03) | 2.06 (0.52) | 0.84 (.08) | 5.78 (0.71) |
| Deuterated | 5.68 (0.46) | 367.95 (66.58) | 658.08 (95.02) | 8.63E+03 (6.02E+02) |
Figure 4.

The dependence of [oxygen] on D(VMAX/KM)APPARENT, BIAA (BLACK) and the [BIAA] dependence on D(VMAX/KM)APPARENT, O2 (BLUE).
QM/MM Reaction Coordinat
The two species evaluated computationally were the CuII–OH (singlet) and the CuII–O· (quartet). The potential energy of the reaction coordinate for the CuII–OH species, calculated as the difference in bond distance between the Cα–O and Cu–OH bonds, displayed a nearly linear increase in relative energy from the starting point to a ‘saddle point’ value of +45.93 kcal/mol. Following this point, the calculated energy of the reaction coordinate decreased to -25.98 kcal/mol (Fig. 5). The Cα← O bond lengths decreased from 4.60 Å to 2.80 Å at the energetic ‘saddle point’, resting at a final distance of 1.30 Å. The CuII → OH bond lengths increased over the reaction coordinate from 1.82 Å through 2.81 Å at the energetic maxima, to 4.08 Å at the energetic minima. The imino–oxy ↔ α–carbon bond lengths for BIAA increased 0.16 Å over the reaction coordinate, from 1.36 to 1.52 Å (Fig. 5). The sequence shown in scheme B and movie A (supporting information), represent the quantum mechanical aspects of the performed QM/MM study. The displayed poses correspond directly with their respective steps in the Fig. 5. The orientation of the carboxyl of BIAA was unchanged until the step prior to the saddle point (+37.45 kcal/mol). Following this point, a coupled rotation in the hydroxyl moiety and the carboxyl of BIAA was shown which oriented the α-carbon essentially perpendicular to the approaching hydroxyl group (Scheme B, supporting information).
Figure 5.

QM/MM simulated reaction coordinate for the oxidation chemistry observed for the BIAA Cα-radical oxidation with a singlet CuII-OH species (circles). The squares represent dealkylation distances observed for imino-oxy ↔ Cα bond lengths versus reaction coordinate.
The reaction coordinate for the copper-alkoxide (CuII–O·) species (Fig. 6) displayed a more complex relationship with potential energy trend, relative to that of CuII–OH, as an energetic plateau was observed midway to the saddle point at ∼28 kcal/mol. Following this, potential energy at the ‘saddle point’ was +54.75 kcal/mol, +12.82 kcal/mol greater than that observed for the reaction coordinate for the CuII-OH species. Although a higher potential energy was observed for the CuII-O• species, a rapid decline in the potential energy was observed beyond the ‘saddle point’ on the reaction coordinate (Fig. 6), in contrast to slow decline observed for the CuII-OH species (Fig. 5). The ‘saddle points’ for the CuII-OH species occurred with nearly equal bond lengths for the OH between the Cα and CuII (∼2.8 Å at RC = 0.014). Conversely, the ‘saddle point’ of the CuII-O• reaction coordinate was much less symmetrical, reaching its maxima when the Cα-O and CuII-O bond lengths were 1.9 and 3.7 Å, respectively. The CuII-OH potential energy decreased over the reaction coordinate (RC) from +45.93 kcal/mol at RC = 0.014 to -25.97 kcal/mol at RC = 2.78 while the CuII-O• decreased from +58.75 kcal/mol at RC = 1.78 to -9.68 kcal/mol at RC = 3.05. Therefore, the total change in potential energy for each species was similar for each species (within experimental error), 68.43 kcal/mol for CuII–O• and 71.90 kcal/mol for CuII–OH.
Figure 6.

QM/MM simulated reaction coordinate for the oxidation chemistry observed for the BIAA Cα-radical oxidation with a quartet CuII-O· species (circles). The squares represent dealkylation distances observed for imino-oxy ↔ Cα bond lengths versus reaction coordinate.
Substrate Dealkylation
The increase in imino–oxy ↔ α–carbon bond length for the CuII-O· species (Fig. 6) was observed to be constant over the reaction coordinate until the energetic ‘saddle point’ was reached. It was here that the substrate bond length was dramatically increased as compared to the effect observed studying the CuII–OH species, ranging from 1.42 Å to 2.91 Å. The corresponding poses for the quantum mechanically treated region of the CuII–OH simulation showed very little change in substrate orientation, with major functionalities of the of the BIAA substrates (phenyl ring, imino-oxy, and carboxy termini moieties) remaining constant with respect to starting points. At the ‘saddle point’ of the CuII-O• simulation, an increase in imino–oxy ↔ α–carbon bond length was observed during a small RC window with a 1.47 Å change between RC = 1.78 and 2.09 yielding a final imino–oxy ↔ α–carbon bond of 2.91 Å (scheme C and movie B, supporting information).
Discussion
Dealkylation is Non-Enzymatic
Oxidation of BIAA by PAM yields two products in a 1:1 molar ratio: BOX and glyoxylate. BOX was confirmed by 13C NMR (Figure B, supporting information). In addition, the stoichiometry of [O2]consumed:[BOX]produced was also 1:1 (Figure C and Table C supporting information), consistent with evidence for tight coupling between substrate oxidation and O2 consumption for PHM61 and the related enzyme, DβM.20 PAM-catalyzed oxidation/dealkylation of BIAA is unaffected by α-hydroxyhippurate (a PAL substrate) and the PAL-catalyzed dealkylation of α-hydroxyhippurate is unaffected by BIAA (Figs. 2 and 3). This suggests that the putative α-hydroxylated product generated by PHM, C6H5-CH-N-O-CH(OH)-COOH, is unstable and non-enzymatic decomposes into observed oxime and glyoxylate products. These data provide strong evidence that the oxidative dealkylation of BIAA to BOX and glyoxylate requires only the PHM domain of bifunctional PAM.
Kinetic Mechanism
Previous work with small molecule substrates, N-benzoylglycine48 and N-acetylglycine*, found that D(VMAX/KM) values for both were equal and constant as the O2 concentration was varied. The minimal kinetic mechanism of these small molecule substrates is equilibrium-ordered with O2 binding after N-benzoylglycine or N-acetylglycine. Comparison of equation 3 and 4, shows that KM for the first bound substrate (KM,A[B]) is absent from the denominator for an equilibrium-preferred mechanism. In this case, binding of the first substrate to the free enzyme (E) is in equilibrium. The dissociation constant (KD,A[B]) represents the affinity of the enzyme for the first bound substrate. Analysis of deuterium KIEs for a steady-state preferred mechanism, will include the KM for the first bound substrate (KM,A[B]) in the denominator, yielding a symmetrical mechanism. Therefore, the D(VMAX/KM)APPARENT value cleavage will be dependent on the concentration of the second substrate for a steady-state preferred mechanism, as we observed for Cα-D/H bond cleavage using BIAA as a substrate. The difference between a steady-state ordered and a random mechanisms is that the D(VMAX/KM)APPARENT values approach unity as concentration of the second substrate becomes infinite for ordered substrate addition vs. a non-unity limit for the random substrate addition.62 The values of D(VMAX/KM)APPARENT, BIAA showed a dramatic decrease as the concentration of O2 increased. Extrapolation of the D(VMAX/KM)APPARENT, BIAA to zero [O2] yields a value ∼15 (well beyond the semi-classical limit) while extrapolation of infinite [O2] yields of 1.0 (Figures C and D, supporting information). The D(VMAX/KM)APPARENT, O2 was constant over the range [BIAA] used. This is a clear example of a steady-state ordered mechanism with BIAA binding to reduced PHM prior to O2 (Scheme 5). Using hippurate as the oxidizable substrate, it has been established that PHM exhibits an equilibrium-ordered mechanism with O2 binding after hippurate to reduced enzyme48 while the kinetic mechanism for the Y318F mutant is steady-state-random with either hippurate or O2 binding to reduced enzyme.63 A steady-state random mechanism was observed for the Y318F mutant of PHM as the a non-unity values of D(VMax/KM)APPARENT, HIPPURATE were obtained as [O2] → ∞.63 For the related enzyme, DβM, the absence of the anionic activator (fumarate) at pH 6.0 alters the steady-state mechanism from ordered to random.50,64
Scheme 5.

Minimal kinetic mechanism determined for BIAA oxidation as determined by primary deuterium KIEs.
The steady-state mechanism is attained for BIAA through a decrease in the rate constant responsible for dissociation of the binary complex (E-S), k2.48 Unlike an equilibrium preferred mechanism, the binary complex (E-S) becomes the predominant enzyme state. The reduction in D(VMAX/KM)APPARENT,BIAA with increasing [O2] was due to an increase in the “commitment to catalysis”.65-67 Under steady-state ordered conditions, as [O2] → ∞, dissociation of the reactant (RCα-H) from the central complex does not occur as the commitment to the forward reaction also becomes infinite (D(VMAX/KM)APPARENT,BIAA = ∼1). The magnitude of D(VMAX/KM)BIAA as [O2] → 0, has a commitment to the forward reaction which is dependent on the ratio of the chemical step vs. the dissociation of O2 from the central complex (k5/k4, Scheme 5). Therefore, the predominant enzyme form would be E-RCα-H due to the decrease in the rate constant for O2 insertion to achieve the central complex, k3[O2].
The dissociation constant for BIAA showed a normal isotope effect of DKD,BIAA = 2.06 ± 0.52 (Table 1). Conversely, the binding isotope effect for hippurate is inverse for both PHM and PAM, DKD,hippurate <1.0.43 Often the effect of isotopic labeling on binding is dismissed as negligible, but binding isotope effects have been reported for a number of enzymes, including lactate dehydrogenase,68 hexokinase,69 thymidine phosphorylase,70 and purine nucleoside phosphorylase.71 The DKD,BIAA suggests that deuteration of the BIAA α-carbon reduces stringent vibrational contributions associated with ground state destabilization during formation of the E•BIAA complex formation, potentially decreasing k2. A decrease in k2 increases the forward commitment to catalysis, k3/k2, and may be a contributing factor in the kinetic mechanism for BIAA being steady-state ordered vs. equilibrium ordered. The forward commitment for an equilibrium order mechanism will be relatively low.
While neither hippurate nor BIAA showed an appreciable DVMAX, the turnover number decreased approximately six-fold for BIAA relative to hippurate. This difference in product release (VMAX) for BIAA cannot be attributed to an uncoupling of O2 consumption from BOX and glyoxylate formation (Figures 1, B and Table B), as the reaction stoichiometries for BIAA oxidation match those reported for glycine extended substrates.72 One possible reason for the relatively low VMAX for BIAA is that the imino-oxy moiety is complicating Cα–O insertion into its respective Cα-radical species. This conclusion would mean that BIAA dealkylation would be included in the minimal kinetic mechanism and would require no other rate constant for product release other than k7. This rate constant describes the slow step responsible for the irreversible conversion of the E•P complex to free enzyme (E) and product (P) for all PHM substrates. Therefore, the absence of DVMAX may best be explained as a requisite rate limiting conformational change necessary for product release with glycine-extended substrates, though an additional energetic penalty associated with BIAA oxidation (dealkylation) appears to further decrease the magnitude of k7 for this substrate.
Substrate Oxidation and Product Release are Uncoupled
QM/MM simulation of both the oxygen insertion into the BIAA Cα–radical and the resulting dealkylation process provided an experimental constraint used to probe the nature of the Cu/O oxidant species. The CuII–OH (singlet) species was postulated to affect the direct OH transfer and product release steps of the ‘side-on/η2’ and ‘copper-oxo pathway B’ mechanisms. The effect of direct product formation on the bond length of our dealkylation probe was negligible, suggesting that imino–oxy ↔ Cα bond dissociation would likely not occur through this mechanism. Formation of the copperII–alkoxide species (quartet) had a dramatic effect on the structure of the dealkylation probe, suggesting that the process for substrate oxidation and product release were completely uncoupled, and likely to pass through a copper-alkoxide intermediate (red coordinates, Figure 7). The doublet copper-oxo species (‘copper-oxo pathway A’) could not be used for this analysis as H-abstraction and oxidation steps were concerted resulting in an absence of a substrate intermediate (radical).37
Figure 7.

Combined plot of QM/MM reaction coordinate simulation comparing the CuII-OH (singlet) and the CuII-O· (quartet) species again bond distance for the BIAA dealkylation event. Please note, squares represent the distance change between the NO←Cα bond distance while the circles signify Cu→O bond distance changes over the reaction coordinate. For more detail, please see methods section.
As the separation of H-abstraction from substrate oxidation was a necessary caveat for our theoretical treatment of dealkylation, calculations using the double species could not be performed.
Conclusion
Due to the complexities of PHM, theoretical models lacking experimental constraint often lead to ambiguous conclusions in thermodynamic preference between chemical models. Here, the catalytic power of the enzyme is determined at its upper limit and neglects that the reaction regime only requires the necessary energy for catalysis. The work of Karlin (supra vide) infers that the nature of the Cu/O oxidant cannot definitively characterized in PHM, as the reaction coordinate may proceed through several possible catalytic paths. Therefore, the dealkylation event was designed to describe the nature of substrate oxidation chemistry in PHM. BIAA was a superb probe for the glycine extended substrate functionalization as it retained all reaction stoichiometries important to PHM. When docked to PHM, non-Lewis hyperconjugative stabilization of the oxime moiety is electronically analogous to an amide (in preparation). For the QM/MM study, Cα-H bond cleavage was not part of the reaction coordinate; instead the dealkylation of BIAA was used to provide the system with an independent variable to constrain the theoretical model. The mixture of both in vitro and in silico data suggest that direct hydroxyl insertion into the Cα-radical of BIAA is unlikely as the NO ↔ Cα distance did not significantly increase. Interestingly, the singlet species reaction coordinate was thermodynamically favored as the oxidant with high symmetry over the simulation. This mechanism would likely lead to a complete uncoupling of the BIAA radical and a reduced oxygen species from the PHM active site, as the substrate radical would resist conversion into a thermodynamically unstable hemiacetal derivative. PHM would not be expected to stabilize the putative HO-Cα BIAA intermediate as the active site is highly solvated during catalysis.73 Thus, if this hemiacetal formed, its instability would not be suppressed through protein docking.
Using the natural glycine-extended substrate, hydrolysis of the ‘copper-alkoxide/substrate’ intermediate complex yields a stable α-hydroxylated glycine product. The BIAA model displayed changes in the bond lengths for the oxygen within the CuII↔O• complex approaching the carbon radical (a decrease) as NO↔Cα (an increase) over the reaction coordinate (Fig. 7). The steady-state data suggests that this process is irreversible and tightly coupled with respect to both oxygen and BIAA, similar to the PHM natural substrate.20 Taken together, the reason for the O-dealkylation of BIAA occurring independent of PAL is likely that the oxidation step becomes increasingly rate limiting versus the subsequent hydrolysis step in BIAA compared to a glycine-extended substrate. This irreversible event was calculated to be an endothermic process (Fig. 7) in both singlet and quartet copper-oxo complexes, though O-dealkylation of BIAA was only observed for the singlet state species calculation. By comparison, glycine-extended substrates, this event has been calculated to be an exothermic process.19,35
Our proposal was that the ‘CuII-O·’ species was the oxidant in PHM is based on the dealkylation characteristics unique to BIAA. Formation of the ‘inner-sphere alcohol’ complex during BIAA oxidation was the only mechanism which could account for the tightly coupled reaction stoichiometries while still displaying the ability to dealkylate independent of PAL. The copperII-alkoxide was thermodynamically unfavored compared to the singlet CuII-OH species, though it was the increase in NO ↔ Cα bond length that pointed strongly to an ‘inner sphere alcohol’ intermediate. Therefore, production of an oxidized copperII–alkoxide intermediate complex would be favored to facilitate the reaction characteristics observed during the BIAA O–de–alkylation process (Scheme 6). In conclusion, this study provides compelling evidence that PHM-dependent oxidation of glycine-extended substrates occurs through a covalently linked copperII–alkoxide substrate complex.
Scheme 6.

Dealkylation reaction predicted for the CuII-alkoxide intermediate formation with BIAA as the substrate.
Supplementary Material
Scheme 3.

Degenerate electronic ground state structures of the CuII-hydroperoxo and CuII-hydroxyl oxidant species proposed for direct substrate oxidation/product release. Please note, copper domain residues were omitted for clarity. Refer to text for more detail.
Acknowledgments
This work was supported, in part, by grants from the National Institutes of Health - General Medical Sciences (R15-GM067257 and R15-GM073659), the Shirley W. & William L. Griffin Foundation, the Alpha Research Foundation, Inc., the Eppley Foundation for Research, the Gustavus and Louise Pfieffer Research Foundation, the Milheim Foundation for Cancer Research, the Shin Foundation for Medical Research, the Wendy Will Case Cancer Fund, and the University of South Florida - Established Researcher Grant Program, to D.J.M, and a predoctoral fellowship to E.W.L. from the American Heart Association (0415259B).
Footnotes
N. R. McIntyre and Merkler, D. J., manuscript in preparation.
Supporting Information Available: Experimental figures, tables, schemes and movies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Prigge ST, Mains RE, Eipper BA, Amzel LM. Cell Mol Life Sci. 2000;57:1236–59. doi: 10.1007/PL00000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merkler DJ, Chew GH, Gee AJ, Merkler KA, Sorondo JP, Johnson ME. Biochemistry. 2004;43:12667–74. doi: 10.1021/bi049529p. [DOI] [PubMed] [Google Scholar]
- 3.Kolhekar AS, Roberts MS, Jiang N, Johnson RC, Mains RE, Eipper BA, Taghert PH. J Neurosci. 1997;17:1363–76. doi: 10.1523/JNEUROSCI.17-04-01363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williamson M, Hauser F, Grimmelikhuijzen CJ. Biochem Biophys Res Commun. 2000;277:7–12. doi: 10.1006/bbrc.2000.3629. [DOI] [PubMed] [Google Scholar]
- 5.Hauser F, Williamson M, Grimmelikhuijzen CJ. Biochem Biophys Res Commun. 1997;241:509–12. doi: 10.1006/bbrc.1997.7854. [DOI] [PubMed] [Google Scholar]
- 6.Spijker S, Smit AB, Eipper BA, Malik A, Mains RE, Geraerts WP. Faseb J. 1999;13:735–48. doi: 10.1096/fasebj.13.6.735. [DOI] [PubMed] [Google Scholar]
- 7.Klinman JP. J Biol Chem. 2006;281:3013–6. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 8.Klinman JP, Krueger M, Brenner M, Edmondson DE. J Biol Chem. 1984;259:3399–402. [PubMed] [Google Scholar]
- 9.Ash DE, Papadopoulos NJ, Colombo G, Villafranca JJ. J Biol Chem. 1984;259:3395–8. [PubMed] [Google Scholar]
- 10.Kulathila R, Consalvo AP, Fitzpatrick PF, Freeman JC, Snyder LM, Villafranca JJ, Merkler DJ. Arch Biochem Biophys. 1994;311:191–5. doi: 10.1006/abbi.1994.1225. [DOI] [PubMed] [Google Scholar]
- 11.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864–7. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 12.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Nat Struct Biol. 1999;6:976–83. doi: 10.1038/13351. [DOI] [PubMed] [Google Scholar]
- 13.Kolhekar AS, Bell J, Shiozaki EN, Jin L, Keutmann HT, Hand TA, Mains RE, Eipper BA. Biochemistry. 2002;41:12384–94. doi: 10.1021/bi0260280. [DOI] [PubMed] [Google Scholar]
- 14.De M, Bell J, Blackburn NJ, Mains RE, Eipper BA. J Biol Chem. 2006;281:20873–82. doi: 10.1074/jbc.M513886200. [DOI] [PubMed] [Google Scholar]
- 15.Padgette SR, Wimalasena K, Herman HH, Sirimanne SR, May SW. Biochemistry. 1985;24:5826–39. doi: 10.1021/bi00342a021. [DOI] [PubMed] [Google Scholar]
- 16.Katopodis AG, May SW. Biochemistry. 1990;29:4541–8. doi: 10.1021/bi00471a006. [DOI] [PubMed] [Google Scholar]
- 17.Katopodis AG, Ping D, May SW. Biochemistry. 1990;29:6115–20. doi: 10.1021/bi00478a001. [DOI] [PubMed] [Google Scholar]
- 18.Gherman BF, Heppner DE, Tolman WB, Cramer CJ. J Biol Inorg Chem. 2006;11:197–205. doi: 10.1007/s00775-005-0066-5. [DOI] [PubMed] [Google Scholar]
- 19.Chen P, Solomon EI. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 20.Evans JP, Ahn K, Klinman JP. J Biol Chem. 2003;278:49691–8. doi: 10.1074/jbc.M300797200. [DOI] [PubMed] [Google Scholar]
- 21.Itoh S. Curr Opin Chem Biol. 2006;10:115–22. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 22.Hatcher LQ, Karlin KD. J Biol Inorg Chem. 2004;9:669–83. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 23.Bollinger JM, Jr, Krebs C. Curr Opin Chem Biol. 2007;11:151–8. doi: 10.1016/j.cbpa.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 24.Fujisawa K, Tanaka M, Moro-oka Y, Kitajima N. J Am Chem Soc. 1994;116:12079–12080. [Google Scholar]
- 25.Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. J Am Chem Soc. 2003;125:466–74. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- 26.Karlin KD, Wei N, Jung B, Kaderli S, Zuberbuhler AD. J Am Chem Soc. 1991;113:5868–5870. [Google Scholar]
- 27.Karlin KD, Wei N, Jung B, Kaderli S, Niklaus P, Zuberbuhler AD. J Am Chem Soc. 1993;115:9506–9514. [Google Scholar]
- 28.Karlin KD, Kaderli S, Zuberbuhler AD. Acc Chem Res. 1997;30:139–147. [Google Scholar]
- 29.Schatz M, Raab V, Foxon SP, Brehm G, Schneider S, Reiher S, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem Int Ed. 2004;43:4306–4363. doi: 10.1002/anie.200454125. [DOI] [PubMed] [Google Scholar]
- 30.Maiti D, Sarjeant AA, Karlin KD. J Am Chem Soc. 2007;129:6720–1. doi: 10.1021/ja0719024. [DOI] [PubMed] [Google Scholar]
- 31.Maiti D, Lucas HR, Sarjeant AA, Karlin KD. J Am Chem Soc. 2007;129:6998–9. doi: 10.1021/ja071704c. [DOI] [PubMed] [Google Scholar]
- 32.Lee DH, Hatcher LQ, Vance MA, Sarangi R, Milligan AE, Sarjeant AA, Incarvito CD, Rheingold AL, Hodgson KO, Hedman B, Solomon EI, Karlin KD. Inorg Chem. 2007 doi: 10.1021/ic700541k. [DOI] [PubMed] [Google Scholar]
- 33.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2007;129:264–5. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 34.Chen P, Solomon EI. J Inorg Biochem. 2002;88:368–74. doi: 10.1016/s0162-0134(01)00349-x. [DOI] [PubMed] [Google Scholar]
- 35.Chen P, Solomon EI. Proc Natl Acad Sci U S A. 2004;101:13105–10. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bauman AT, Yukl ET, Alkevich K, McCormack AL, Blackburn NJ. J Biol Chem. 2006;281:4190–8. doi: 10.1074/jbc.M511199200. [DOI] [PubMed] [Google Scholar]
- 37.Crespo A, Marti MA, Roitberg AE, Amzel LM, Estrin DA. J Am Chem Soc. 2006;128:12817–28. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]
- 38.Kamachi T, Kihara N, Shiota Y, Yoshizawa K. Inorg Chem. 2005;44:4226–36. doi: 10.1021/ic048477p. [DOI] [PubMed] [Google Scholar]
- 39.Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Inorg Chem. 2006;45:3034–41. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]
- 40.Roth JP. Curr Opin Chem Biol. 2007 doi: 10.1016/j.cbpa.2007.01.683. [DOI] [PubMed] [Google Scholar]
- 41.Chen P, Bell J, Eipper BA, Solomon EI. Biochemistry. 2004;43:5735–47. doi: 10.1021/bi0362830. [DOI] [PubMed] [Google Scholar]
- 42.Merkler DJ, Young SD. Arch Biochem Biophys. 1991;289:192–6. doi: 10.1016/0003-9861(91)90461-q. [DOI] [PubMed] [Google Scholar]
- 43.Liu KC, Shelton BR, Howe RK. Journal of Organic Chemistry. 1980;45:3917–1919. [Google Scholar]
- 44.Morrison TJ, Billett F. J Chem Soc. 1948:2033–5. [Google Scholar]
- 45.McIntyre NR, Lowe EW, Jr, Chew GH, Owen TC, Merkler DJ. FEBS Lett. 2006;580:521–32. doi: 10.1016/j.febslet.2005.12.058. [DOI] [PubMed] [Google Scholar]
- 46.Buettner GR, Jurkiewicz BA. Radiat Res. 1996;145:532–41. [PubMed] [Google Scholar]
- 47.Samuni A, Aronovitch J, Godinger D, Chevion M, Czapski G. Eur J Biochem. 1983;137:119–24. doi: 10.1111/j.1432-1033.1983.tb07804.x. [DOI] [PubMed] [Google Scholar]
- 48.Francisco WA, Merkler DJ, Blackburn NJ, Klinman JP. Biochemistry. 1998;37:8244–52. doi: 10.1021/bi973004y. [DOI] [PubMed] [Google Scholar]
- 49.Cleland WW. Methods Enzymol. 1979;63:103–38. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 50.Klinman JP, Humphries H, Voet JG. J Biol Chem. 1980;255:11648–51. [PubMed] [Google Scholar]
- 51.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Science. 1997;278:1300–5. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 52.GLIDE Schrodinger, LLC Portland, OR, 2000.
- 53.QSITE Schrodinger, LLC Portland, OR, 2000.
- 54.Becke AD. J Chem Phys. 1993;98:1372–1377. [Google Scholar]
- 55.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 56.Hay JP, Wadt WR. J Chem Phys. 1985;82:299–310. [Google Scholar]
- 57.Cui Q, Elstner M, Kaxiras E, Frauenheim T, Karplus M. J Phys Chem B. 2001;105:569–585. [Google Scholar]
- 58.Jorgensen WL, Tirado-Rives J. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 59.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 60.MAESTRO Schrodinger, LLC Portland, OR, 2002.
- 61.Evans JP, Blackburn NJ, Klinman JP. Biochemistry. 2006;45:15419–15429. doi: 10.1021/bi061734c. [DOI] [PubMed] [Google Scholar]
- 62.Cook PF, Cleland WW. Biochemistry. 1981;20:1790–6. doi: 10.1021/bi00510a013. [DOI] [PubMed] [Google Scholar]
- 63.Francisco WA, Blackburn NJ, Klinman JP. Biochemistry. 2003;42:1813–9. doi: 10.1021/bi020592t. [DOI] [PubMed] [Google Scholar]
- 64.Miller SM, Klinman JP. Biochemistry. 1985;24:2114–27. doi: 10.1021/bi00330a004. [DOI] [PubMed] [Google Scholar]
- 65.Northrop DB, Cho YK. Biophys J. 2000;79:1621–8. doi: 10.1016/S0006-3495(00)76412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cleland WW. CRC Crit Rev Biochem. 1982;13:385–428. doi: 10.3109/10409238209108715. [DOI] [PubMed] [Google Scholar]
- 67.Blanchard JS, Cleland WW. Biochemistry. 1980;19:4506–13. doi: 10.1021/bi00560a019. [DOI] [PubMed] [Google Scholar]
- 68.LaReau R, Wah W, Anderson V. Biochemistry. 1989;34:6050–6058. [Google Scholar]
- 69.Lewis BE, Schramm VL. J Am Chem Soc. 2003;125:4785–4798. doi: 10.1021/ja0298242. [DOI] [PubMed] [Google Scholar]
- 70.Birck MR, Schramm VL. J Am Chem Soc. 2004;126:6882–6883. doi: 10.1021/ja0492642. [DOI] [PubMed] [Google Scholar]
- 71.Murkin AS, Birck MR, Rinaldo-Matthis A, Shi W, Taylor EA. Biochemistry. 2007;46:5038–5049. doi: 10.1021/bi700147b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kulathila R, Merkler KA, Merkler DJ. Nat Prod Rep. 1999;16:145–54. doi: 10.1039/a801346b. [DOI] [PubMed] [Google Scholar]
- 73.Owen TC, Merkler DJ. Med Hypotheses. 2004;62:392–400. doi: 10.1016/j.mehy.2003.11.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.