

Abstract
Enantioenriched diaryl-, aryl heteroaryl- and diheteroarylmethanols exhibit important biological and medicinal properties. One-pot catalytic asymmetric syntheses of these compounds beginning from readily available aryl bromides are introduced. Thus, lithium-bromide exchange with commercially available aryl bromides and n-BuLi was followed by salt metathesis with ZnCl2 to generate ArZnCl. A second equivalent of n-BuLi was added to form the mixed organozinc, ArZnBu. In the presence of enantioenriched amino alcohol-based catalysts, ArZnBu adds to aldehydes to afford essentially racemic diarylmethanols. The low enantioselectivities were attributed to a LiCl-promoted background reaction. To inhibit this background reaction, the chelating diamine TEEDA (tetraethylethylene diamine) was introduced prior to aldehyde addition. Under these conditions, enantioenriched diarylmethanols were obtained with >90% ee. Arylations of enals generated allylic alcohols with 81–90% ee. This procedure was unsuccessful, however, when applied to heteraryl bromides, which was attributed to decomposition of the heteroaryl lithium under the salt metathesis conditions. To avoid this problem, the metathesis was conducted with EtZnCl, which enabled the salt metathesis to proceed at low temperatures. The resulting EtZn(ArHetero) intermediates (ArHetero=2- and 3-thiophenyl, 2-benzothiophenyl, 3-furyl, and 5-indolyl) were successfully added to aldehydes and heteroaryl aldehydes with enantioselectivities between 81–99%. These are the first examples of catalytic and highly enantioselective syntheses of diheteroarylmethanols. In a similar fashion, ferrocenyl bromide was used to generate FcZnEt and the ferrocenyl group added to benzaldehyde and heteroaromatic aldehydes to form ferrocene-based ligand precursors in 86–95% yield with 96–98% ee. It was also found that the arylation and heteroarylation of enals could be followed by diastereoselective epoxidations to provide epoxy alcohols with high enantio- and diastereoselectivities in a one-pot procedure.
1. Introduction
Enantioenriched diaryl-, aryl heteroaryl-, and diheteroarylmethanols are important intermediates and structural motifs in medicinal chemistry. Diarylmethanols form the core of several biologically active compounds, including (R)-neobenodine, (R)-orphenadrine and (S)-cetrizine.1-6 The 1-benzofuran derivatives 1a-c are intermediates in the synthesis of chiral azoles (2a-c, Figure 1). Compounds 2a-c were initially examined as antifungal agents7 and have been found to be powerful nonsteroidal aromatase inhibitors.8-13 They are indicated in the treatment of hormone-dependent breast cancer.14 Likewise, diheteroarylmethanols have received recent attention. For example, chiral dithienylmethanols have been evaluated as antiallergic and antiischemic agents (3, Figure 1).15 Furthermore, enantioenriched diarylmethanols can be converted into diarylmethane derivatives via SN2 substitution at the C-O bond without loss of ee.1 The diarylmethane motif is found in antimuscarinics,16 antidepressants,17 and endothelin antagonists.18
Figure 1.

Biologically active heteroaryl- and diheteroarylmethanols.
The catalytic enantioselective synthesis of diarylmethanols has been the focus of many studies.19 The most efficient approach to their preparation is the arylation of aromatic aldehydes to generate a C-C bond and stereocenter in a single step. Early studies by Seebach and coworkers employed Ph-Ti(O-iPr)3, generated from Cl-Ti(O-iPr)3 and PhLi, in combination with TADDOL-based titanium catalysts.20-22 To achieve high enantioselectivity, it was necessary to remove the LiCl byproduct formed during salt metathesis by centrifugation.
Pioneering studies by Fu and coworkers23 with a planar-chiral catalyst, diphenylzinc, and 4-chlorobenzaldehyde furnished the diarylmethanol product with 57% ee. A highly enantioselective catalyst was reported by Pu shortly thereafter.24 These works inspired many subsequent investigations using diphenylzinc, as exemplified in Scheme 1A.25-30
Scheme 1.

Asymmetric Arylation of 4-Chlorobenzaldehyde with Ph2Zn (A) and PhZnEt Generated from Ph2Zn and Et2Zn (B) or from PhB(OH)2 and Et2Zn (C).
Despite the large number of studies with diphenylzinc, significant drawbacks remained. Diphenylzinc is prohibitively expensive and limited to phenyl transfer.25,31-34 Furthermore, unlike dialkylzinc additions to aldehydes, which exhibit slow background reactions, the uncatalyzed addition of diphenylzinc to aldehydes is sufficiently rapid to compete with most catalyzed additions.34-36 The latter problem was addressed by Bolm and coworkers, who discovered that the mixed reagent EtZnPh33 exhibited a slower background reaction than diphenylzinc. EtZnPh also resulted in higher enantioselectivities with the same catalysts (Scheme 1B), in part from the reduced contribution of the background reaction.37-42 EtZnPh is easily generated by combining Ph2Zn and Et2Zn (Equation 1).
| Equation 1 |
The development of methods for the enantioselective transfer of substituted aryl groups to aldehydes remained a challenge for several years. Although diarylzinc reagents can be easily prepared from ArLi or ArMgX and zinc halides (ZnX2), the salt byproducts LiX and MgX2 are Lewis acidic and readily promote the background reaction in the presence of enantioenriched catalysts, resulting in diarylmethanols with little or no ee. To bypass this problem, Bolm and coworkers introduced a method whereby aryl boronic acid derivatives underwent transmetallation with dialkylzinc reagents to provide access to salt-free arylzinc reagents, ArZnEt (Scheme 1C).30,42-48 In this procedure,44 2.4 equiv of the aryl boronic acid was heated with 7.2 equiv of diethylzinc for 12 h to generate ArZnEt. Dimethyl (polyethylene glycol) (DiMPEG, 10 mol %, MW∼2,000) was used as an additive to inhibit the background reaction caused by achiral Lewis acidic ZnPh2 or ZnBr2.43,49,50 Further study and optimization by the groups of Pericàs and Magnus significantly reduced the transmetallation time.51,52 Braga demonstrated that the acceleration could be achieved by microwave irradiation.53 Based on Bolm's breakthrough, several aryl boron derivatives, such as triarylboranes42,43,49,54-57 and boroxines,52,58,59 were successfully employed in the asymmetric arylation of aldehydes. It is noteworthy that highly enantioselective late transition metal-based catalysts can also be employed with boronic acid derivatives.60-62
Main group metals other than zinc have been applied to the asymmetric arylation of aldehydes. In 2008 Muramatsu and Harada63 introduced a method wherein Grignard reagents (1.2 equiv) could be added to titanium tetraisopropoxide (3 equiv) in the presence of 2 mol % 3-(3,5-Ph2-C6H3)-H8-BINOL and aldehydes to provide diarylmethanols with high ee. In a similar vein, Gau and coworkers employed salt-free Ar3Al•THF reagents in combination with titanium tetraisopropoxide and either H8-BINOL64 or sulfonamide alcohol-based ligands,65 which led to diarylmethanols with high enantioselectivities. These reactions may proceed via an Ar-Ti intermediate.66
At the outset of our research into the arylation of aldehydes in 2005, two major limitations existed. The first was the necessity for salt-free aryl zinc reagents. The second was the use of costly aryl sources such as Ph2Zn and aryl boronic acids, which are synthesized from aryl halides. To address these problems, we deemed the following criteria essential to a practical, cost effective and scalable protocol: 1) to use readily available aryl bromides, and 2) to avoid filtration or centrifugation20-22,43,67 of metal halide byproducts from the aryl organometallic reagent. Herein we report the full details of the successful development of a method that fulfills these criteria.68 Thus, metallation of an aryl bromide with n-BuLi, transmetallation to zinc, and enantioselective addition to aldehydes in the presence of the MIB-based69,70 catalyst can now be performed in a one-pot procedure (Equation 2).68 To circumvent the need for tedious sublimation, filtration, or centrifugation of the intermediate arylzinc reagents, we introduced a method to sequester the LiCl byproduct, enabling the generation of diarylmethanols with high levels of enantioselectivity in the presence of lithium chloride. Unfortunately, this procedure was unsuccessful when applied to the generation of enantioenriched diheteroarylmethanols. Therefore, an alternative procedure for heteroaryl additions to aldehydes was developed. To our knowledge, these studies represent the first highly enantioselective catalytic asymmetric synthesis of diheteroarylmethanols.
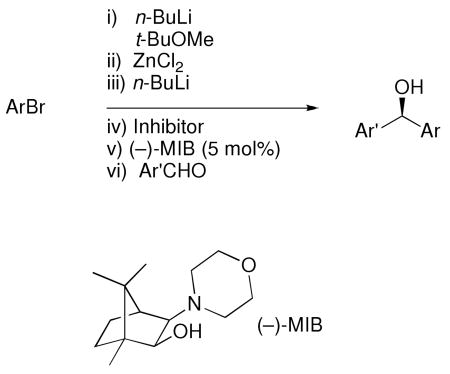 |
Equation 2 |
2. Results and Discussion
The ultimate goal of these investigations was to develop a practical method for the addition of aryl and heteroaryl groups to aldehydes using readily available aryl and heteroaryl bromides. Asymmetric additions with commercial diphenylzinc were used to evaluate catalyst enantioselectivity and for comparison with reactions using bromobenzene.
2.1. Phenylation with Ph2Zn and MIB
Our first priority was to determine the enantioselectivity of the (−)-MIB-based catalyst in phenyl additions to aldehydes. The substrate selected for these studies was 2-naphthylaldehyde (Table 1), which was used with commercial ZnPh2 and 5 mol % (−)-MIB.69,70 The phenyl addition proceeded with 94% enantioselectivity in toluene and 60% enantioselectivity in diethyl ether (entries 1 and 2). Diethyl ether most likely binds to the MIB-based zinc catalyst, reducing its activity and, therefore, enantioselectivity. When less coordinating tert-butyl methyl ether (TBME) was used, the diarylmethanol was obtained with 88% enantioselectivity (entry 3). In the mixed solvent composed of 1:3 TBME:hexanes the enantioselectivity was 89% at rt (entry 4) and 92% at 0 °C (entry 5). Of the solvents examined, only TBME was suitable for the salt metathesis of PhLi with ZnCl2.
Table 1.
Solvent Screen in the Asymmetric Phenyl Addition to 2-Naphthaldehyde.
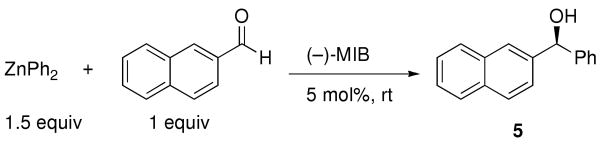 | ||
|---|---|---|
| entry | solvent | ee (%) |
| 1 | toluene | 94 |
| 2 | Et2O | 60 |
| 3 | t-BuOMe | 88 |
| 4 | t-BuOMe/Hex (1:3) | 89 |
| 5 | t-BuOMe/Hex (1:3) | 92a |
| 6 | 2 PhLi + ZnCl2 in t-BuOMe/Hex (1:3) | 2 |
Reaction conducted at 0° C.
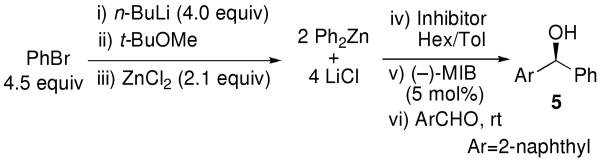
To evaluate the possibility of beginning with aryl bromides, we next generated ZnPh2 by metallation of 4.5 equiv PhBr with 4 equiv n-BuLi in TBME followed by transmetallation with 2 equiv ZnCl2. After addition of hexanes to precipitate additional LiCl, the in situ generated Ph2Zn solution was used in place of the commercial Ph2Zn under otherwise identical conditions (Table 1, entry 6). The expected alcohol product was isolated, but the ee was only 2%. We hypothesized that the Lewis acidic LiCl, generated en route to ZnPh2, promoted the addition to form the racemate faster than the amino alcohol-based Lewis acid catalyst promoted the asymmetric addition. Other researchers have had varying degrees of success employing either filtration or centrifugation of LiCl and MgX2 byproducts.20-22,33,67 These salt byproducts are often produced as a fine particulate and are difficult to remove. Although these procedures are quite useful on laboratory scale, filtration or centrifugation of highly air-sensitive materials is less practical on large scale. To overcome this problem, our strategy was to inhibit the LiCl byproduct rather than remove it. A similar approach was devised by Bolm and coworkers involving the addition of Ph2Zn to aldehydes. These researchers observed a beneficial effect of dimethoxypoly(ethylene glycol) (DiMPEG) on the catalyst enantioselectivity43,49 and proposed that DiMPEG suppressed reactions catalyzed by trace achiral Lewis acids, including ZnBr2 and LiBr, allowing the arylation reaction to proceed via the ligand-accelerated71 pathway.19 Although enantioselectivities reached 93%, yields ranged from 8-31% when Ph2Zn was generated from PhLi and ZnBr2.43 Furthermore, we had difficulties with reproducibility using DiMPEG in combination with the MIB-based catalyst for enantioselective vinylation of aldehydes.72,73
2.2. Development of a Lithium Chloride Selective Inhibitor
The lack of enantioselectivity with Ph2Zn generated from PhBr in Table 1 (entry 6) suggested that the achiral LiCl is a more active Lewis acid than the (−)-MIB-based zinc catalyst. There are three important differences between the lithium and zinc Lewis acids: 1) the lithium is more electropositive and probably the stronger Lewis acid, 2) the lithium center is less sterically saturated than the zinc center in the MIB-based catalyst and 3) the lithium chloride has at least two available coordination sites while the MIB-based zinc catalyst has only one accessible site. Based on this analysis, our strategy was to employ bidentate inhibitors that would chelate lithium and bind tightly, but coordinate in a monodentate fashion to the chiral zinc catalyst. Support for this approach was gained through structures of [TMEDA•LiCl]n, which contain four-coordinate lithium centers with bridging chlorides.74,75
On the basis of this proposal, multidentate amines were screened as LiCl inhibitors in the catalytic enantioselective phenylation of 2-naphthaldehyde with in situ prepared ZnPh2 (Table 2). N,N,N′,N′-Tetraethylethylene diamine (TEEDA) was first examined, because the amino groups are slightly larger than those of the tetramethyl analog TMEDA. Use of 0.2 equiv TEEDA resulted in an increase in the product ee from 2% in the absence of diamine to 55% ee (Table 2, entries 1 and 2). Increasing the amount of TEEDA from 0.2–0.8 and 1.0 equiv resulted in product ee's of up to 83%. A further increase in TEEDA to 1.2 equiv, however, resulted in a slower reaction and a decrease in the product ee to 77%, probably due to inhibition of the MIB-based zinc catalyst by the diamine. It was found that addition of 5 equiv toluene (or hexanes) relative to TBME after transmetallation led to higher enantioselectivity (up to 89%, entry 7). When the temperature of the addition was lowered from rt (entry 7) to 0 °C (entry 8) the enantioselectivity increased to 92%. It is noteworthy that the same enantioselectivity was obtained with commercial Ph2Zn in Table 1 (entry 5), indicating that TEEDA is an excellent inhibitor of LiCl. For comparison, TMEDA was examined in Table 2 (entries 9 and 10) and found to be nearly as effective as TEEDA. Pentamethyldiethylene triamine inhibited LiCl at lower concentrations (0.2 equiv, 80% product ee, entry 12).
Table 2.
Examination of Possible LiCl Inhibitors.
 | ||||
|---|---|---|---|---|
| entry | inhibitor | equiv. | ee (%) | |
| 1 | none | – | 2 | |
| 2 | TEEDA | 0.2 | 55 | |
| 3 | TEEDA | 0.4 | 76 | |
| 4 | TEEDA | 0.8 | 83 | |
| 5 | TEEDA | 1.0 | 83 | |
| 6 | TEEDA | 1.2 | 77 | low conversion |
| 7 | TEEDA | 0.8 | 89 | t-BuOMe/Tol = 1:5 |
| 8 | TEEDA | 0.8 | 92b | t-BuOMe/Tol = 1:5 |
| 9 | TMEDA | 0.8 | 81 | |
| 10 | TMEDA | 1.0 | 81 | |
| 11 | PMDET | 0.1 | 51 | |
| 12 | PMDET | 0.2 | 80 | |
| 13 | PMDET | 0.4 | 71 | |
Solvent = t-BuOMe/Hex = 1:3 unless noted.
Addition conducted at 0 °C.
![]()
2.3. Generation and Application of Mixed Aryl Alkyl Zinc Reagents
As outlined in the Introduction, the background reaction of diarylzinc reagents with aldehydes is often competitive with, or faster than the ligand accelerated pathway71 with amino alcohol-based catalysts. On the basis of the successful application of mixed aryl alkyl zinc reagents by Bolm and co-workers,33 we desired to develop an in situ route to these species to increase enantioselectivities in the aldehyde arylations. To determine the benefit of the mixed organozinc reagents with MIB, our initial experiments involved conproportionation of a 1:1 ratio of commercial Ph2Zn and Et2Zn to generate PhZnEt (Equation 1) followed by addition of (−)-MIB and 2-naphthaldehyde at 0 °C. Under these conditions, the enantioselectivity increased from 92% with Ph2Zn (entry 5, Table 1 and entry 8, Table 2) to 97% with the mixed PhZnEt (Table 3, entry 1).
Table 3.
Catalytic Asymmetric Aryl Additions to Aldehydes with ArZnBu Generated from Aryl Bromides
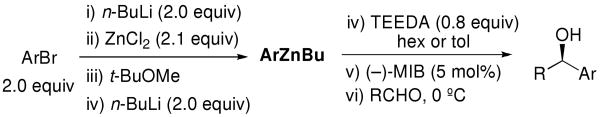 | |||||
|---|---|---|---|---|---|
| entry | ArBr with Ar= | product | # | yield | ee (%) |
| 1 | Ph2Zn/Et2Zn | 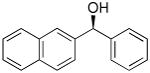 |
5 | - - | 97 |
| 2 | 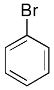 |
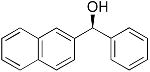 |
5 | 90 | 97 |
| 3 | 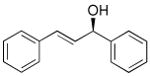 |
6 | 95 | 88 | |
| 4 | 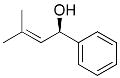 |
7 | 75 | 90 | |
| 5 | 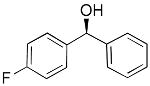 |
8 | 80 | 96 | |
| 6 | 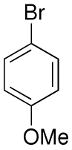 |
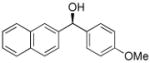 |
9 | 96 | 93 |
| 7 | 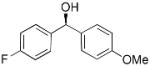 |
10 | 84 | 93 | |
| 8 | 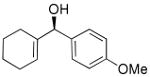 |
11 | 82 | 83 | |
| 9 | 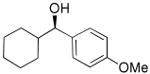 |
12 | 84 | 78 | |
| 10 | 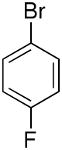 |
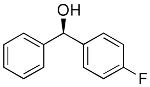 |
13 | 75 | 97 |
| 11 | 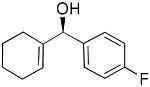 |
14 | 64 | 88 | |
| 12 | 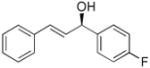 |
15 | 74 | 84 | |
| 13 | 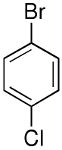 |
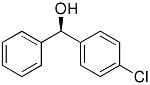 |
4 | 78 | 95 |
| 14 | 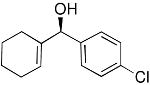 |
16 | 55 | 87 | |
| 15 | 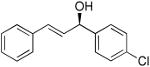 |
17 | 68 | 81 | |
| 16 | 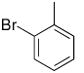 |
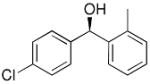 |
18 | 73 | 95 |
| 17 | 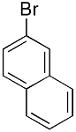 |
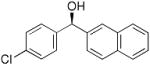 |
19 | 79 | 96 |
| 18 | 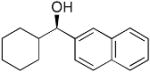 |
20 | 76 | 82 | |
To prepare the mixed aryl alkyl zinc reagents in situ we chose to avoid the use of dialkylzinc reagents, focusing on the more readily available alkyllithiums. Thus, metallation of PhBr with n-BuLi (2 equiv each) and addition of 2.1 equiv ZnCl2 resulted in the generation of PhZnCl. A second dose of n-BuLi (2 equiv) was then added to produce PhZnBu, which was used in combination with 0.8 equiv TEEDA in the asymmetric addition reaction (Table 3, entry 2). Gratifyingly, the enantioselectivity with the in situ generated PhZnBu (97%) was equal to the salt-free PhZnEt, despite the 4 equiv of LiCl in the reaction vessel.
To determine the generality of this method, a series of aryl bromides and aldehydes were employed (Table 3). Bromobenzene and 4-substituted aryl bromides bearing -OMe, -F or -Cl substituents were used in the arylation of benzaldehyde derivatives with 93–97% enantioselectivity. 2-Bromotoluene and 2-bromonaphthalene were added to benzaldehydes with ≥93% enantioselectivity. Aryl additions to α,β-unsaturated aldehydes occurred with 81–90% enantioselectivity (entries 3, 4, 8, 11, 12, 14 and 15). The aliphatic substrate, cyclohexanecarboxaldehyde, underwent aryl addition with 78–82% enantioselectivity (entries 9 and 18). The examples in Table 3 are the first examples of aldehyde arylation beginning with aryl bromides.68
Subsequent to our initial communication,68 a related report appeared by Pu76 employing aryl iodides, and two examples were reported by Harada.63,77 Woodward also developed a method using aryl zinc halides in combination with trimethyl aluminum based on the Schlenk equilibrium.78
Formal Synthesis of (S)-BMS 184394
One example of a biologically active diarylmethanol is BMS 184394 (24, Scheme 2), a RAR γ selective retinoid with activity against skin diseases and cancers, in particular breast cancer and acute promyelocytic leukemia.79-81 Using conditions outlined in Table 3, 3.0 equiv aryl bromide 22 (Scheme 2) was employed to generate the mixed aryl butyl zinc reagent. TEEDA (1.5 equiv) and hexanes were added followed by (+)-MIB (5 mol %) and aldehyde 21. The addition product 23 was produced with 87% enantioselectivity in 88% yield (Scheme 2). Conversion to (S)-BMS 184394 can be accomplished by saponification of the ester.80
Scheme 2.

Synthesis of Enantioenriched 23, the Key Intermediate in the Synthesis of (S)-BMS 184394.
2.4. Attempted Heteroaryl Additions to Aldehydes
General, highly enantioselective additions of heteroaryl groups to aldehydes have not been developed. To our knowledge, the only examples of highly enantioselective heteroaryl additions were published in 2008 by Gau and involved the addition of 2-furyl aluminum reagents to ketones.82 Heteroaryl groups are among the most important pharmacaphors in medicinal chemistry and diheteroarylmethanols have been identified as biologically active structural motifs.15 Thus, not only would methods for heteroaryl additions to aldehydes increase the classes of enantioenriched diarylmethanols accessible, it would enable the catalytic asymmetric synthesis of diheteroarylmethanols that are currently not directly accessible.
With the goal of introducing asymmetric heteroaryl additions to aldehydes, we applied our arylation procedure to metallation of 3-bromothiophene followed by addition to benzaldehyde. The only modification was to maintain the temperature of the heteroaryllithium at −78 °C. Unfortunately, no addition product was observed. When the aryl bromides were used under the conditions outlined in Table 3, the salt metathesis was conducted at room temperature for 4.5 h.68 At this temperature (3-thienyl)Li readily decomposes.
We hypothesized that the absence of product was due to decomposition of the heteroaryllithium in the transmetallation step, which was complicated by the limited solubility of ZnCl2 in TBME at low temperature. To address this problem, we envisaged a more soluble zinc source might undergo transmetallation at lower temperature. Our choice of EtZnCl was based on the large reactivity difference of sp2 hybridized Zn-C bonds over their sp3 counterparts. Another advantage of EtZnCl is that only a single equivalent of LiCl forms during the metathesis, whereas ZnCl2 produces two equivalents (Scheme 3). Lower levels of salt byproduct facilitate inhibition of the LiCl-promoted background reaction.
Scheme 3.

Metathesis with EtZnCl for the Aryl and Heteroaryl Additions to Aldehydes.
The synthesis of EtZnCl was initially performed following the method of Woodward and coworkers by combination of ZnCl2 and ZnEt2 in THF followed by removal of the solvent under reduced pressure.83 Using EtZnCl prepared in this manner, the transmetallation proceeded at −78 °C and the desired heteroaryl addition products were obtained. Unfortunately, product yields and ee's varied greatly from run to run. Our unsuccessful attempts to develop asymmetric heteroaryl additions convinced us to first focus on development of a low temperature transmetallation and then revisit enantioselective heteroaryl additions.
2.5. Development of Low Temperature Transmetallation Conditions and Synthesis of Biologically Active 2a
Momentarily stepping away from the more challenging enantioenriched diheteroarylmethanols, we concentrated on developing low temperature conditions for lithium to zinc transmetallations. We attributed the inconsistencies in the previously described heteroaryl additions to the presence of residual zinc-bound THF in the EtZnCl, which was observed by 1H NMR spectroscopy.83 THF is known to inhibit the MIB-based zinc Lewis acid catalyst. On the basis of this hypothesis, an alternative synthesis of EtZnCl was pursued.84-86 Using toluene in place of THF required heating ZnEt2 and sparingly soluble ZnCl2 at 60 °C for 72 h, after which the solution was filtered to remove any unreacted ZnCl2. The volatiles were then removed under reduced pressure to afford EtZnCl as a white solid that could be stored under nitrogen for months.
The THF-free EtZnCl was first employed with bromobenzene (Equation 3). The transmetallation was conducted at −78 °C to generate PhZnEt, and the addition to 2-benzofurancarbaldehyde was performed at 0 °C. After 12 h, the reaction mixture was quenched with water, worked up, and purified on deactivated silica. We were pleased to isolate the desired addition product 1a in 92% yield with 90% ee (Equation 3). Compound 1a was converted to the promising breast cancer treatment candidate 2a (Figure 1), without loss of ee in 41% unoptimized yield (84% based on recovered 1a of 90% ee) via a Mitsunobu reaction with imidazole.87 SN2 substitutions of this type are known to be very difficult.1 Alternative methods for this transformation also appear potentially useful.89-92
Recently promising diarylmethanes have been examined as possible inhibitors and receptor agonist candidates, but due to limited methods to synthesize the diarylmethanols enantioselectively, most of the studies employed racemic material.11,93,94 Synthesis of 1a with 90% ee suggests that this procedure can be used to prepare diarylmethanols and their derivatives with high ee.
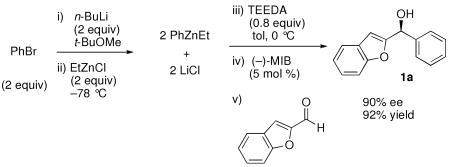 |
Equation 3 |
2.6. Enantioselective Addition of Heteroaryl Groups to Aldehydes
The heteroaryl addition was attempted with 3-bromothiophene under the conditions employed with bromobenzene to generate 1a in Equation 3. Thus, after metallation of 3-bromothiophene with n-BuLi, transmetallation was performed at −78 °C with THF-free EtZnCl. The resulting solution was then warmed to 0 °C and TEEDA, (−)-MIB, and benzaldehyde were added (Table 4). After stirring 12 h, workup, and purification we were pleased to isolate the desired heteroaryl addition product in 68% yield with 90% ee (Table 4, entry 1). It is noteworthy that these revised conditions led to reproducible product ee's and yields.
Table 4.
Synthesis of Aryl Heteroaryl- and Diheteroarylmethanols using 5 mol % (−)-MIB (unless noted below).
| entry | ArheteroBr | product | # | yield | ee (%) |
|---|---|---|---|---|---|
| 1 | 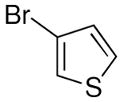 |
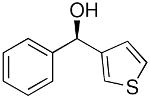 |
25 | 68 | 90 |
| 2a | 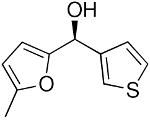 |
26 | 72 | 92 | |
| 3 | 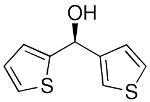 |
27 | 83 | 93 | |
| 4 | 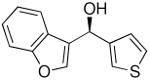 |
28 | 60 | 94 | |
| 5a | 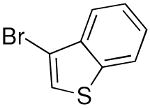 |
 |
29 | 65 | 88 |
| 6a | 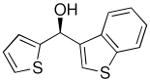 |
30 | 70 | 81 | |
| 7a | 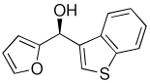 |
31 | 70 | 82 | |
| 8a | 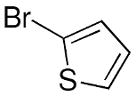 |
 |
32 | 57 | 90 |
| 9 | 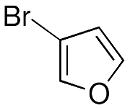 |
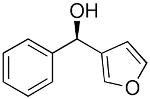 |
33 | 86 | 93 |
| 10a | 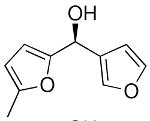 |
34 | 67 | 80 | |
| 11 | 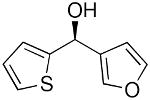 |
35 | 60 | 99 | |
| 12 | 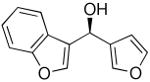 |
36 | 79 | 94 | |
| 13 | 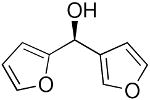 |
37 | 61 | 89 | |
10 mol % MIB. See SI for details.
To explore the enantioselective synthesis of diheteroarylmethanols, the optimized conditions for addition of (3-thienyl)ZnEt to benzaldehyde were employed with heteroaromatic aldehydes. Thus, addition to 5-methyl-2-furan carboxaldehyde, 2-thiophenecarboxaldehyde and 3-benzofurancarboxaldehyde occurred with 92–94% enantioselectivity in 60–83% yield (Table 4, entries 2–4). The differences in yields in entries 1–4 probably arise from a combination of the instability of the heteroaryl organometallic reagents and diminished electrophilicity of the heteroaromatic aldehydes. To develop practical and useful methods, scalability must be demonstrated. Thus, for the synthesis of 27, precursor to potential drug candidate 3 (Figure 1), the asymmetric addition was scaled to produce 820 mg (83% yield and 93% ee, entry 3).
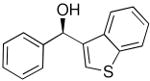
Other heterocycles such as 3-bromobenzothiophene can also be used in the addition with very good enantioselectivities (81–88% ee, entries 5–7). Employing 2-bromothiophene and benzaldehyde afforded diarylmethanol in 90% ee and 57% yield (entry 8). In a similar fashion 3-furanyl ethyl zinc can be added to benzaldehyde (93% ee, 86% yield, entry 9) and heteroaromatic aldehydes with excellent enantioselectivities (89–99% ee, entries 11–13). 5-Methyl-2-furan carboxaldehyde gave addition product of 80% ee (entry 10). Attempts to add 2-furanylzinc reagents to aldehydes, however, resulted in poor enantioselectivities, probably due to the presence of the coordinating oxygen in close proximity to zinc.
To determine if our method for addition of heteroaromatic groups to aldehydes could be extended to other catalysts, we examined the use of Chan's ligand (L2, Figure 2)48,95 with 3-bromothiophene and benzaldehyde under the conditions listed in Table 4, which led to product of 90% ee and 70% yield. These results are virtually identical to those in entry 1 (Table 4) with MIB, indicating that our strategy employing TEEDA to inhibit LiCl is applicable to other amino alcohol-based catalysts.
Figure 2.

Structure of Chan's ligand (L2).
Indoles are regarded as privileged structures in medicinal chemistry and are substructures of an enormous variety of natural products.96,97 We therefore turned our attention toward the synthesis of enantioenriched diarylmethanols containing the indole motif. Metallation of N-silyl-protected 4-bromoindole with n-BuLi was unsuccessful under a variety of conditions, including those in Table 4, most likely due to the electron rich nature of the heterocyclic π-system. More challenging metal-halogen exchange reactions are generally performed with two equiv t-BuLi.98 In these reactions, the first equiv undergoes the metal-halogen exchange with the aryl bromide generating t-BuBr and the second drives the equilibrium by promoting elimination of the liberated t-BuBr to produce isobutylene and LiBr. Unfortunately, diamines that inhibit LiCl had little impact when LiBr was formed. Although we do not understand the intimate differences between LiCl and LiBr at this time, we speculate that weaker bridging Li-Br interactions in [(diamine)LiBr]n facilitate dissociation of the oligomers, opening a coordination site on lithium. To avoid production of LiBr, a 1:1 ratio of 4-bromoindole to t-BuLi was employed, furnishing indole-based diarylmethanols with 90% ee and 60–65% yield (Equation 4).
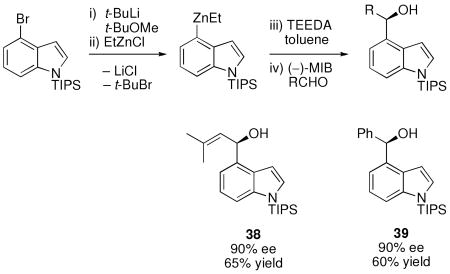 |
Equation 4 |
It is noteworthy that enantioenriched indole 38 is a potential intermediate for the synthesis of (−)-aurantioclavine, which is illustrated in Scheme 4 along with related intermediates in the elegant synthesis of this alkaloid by Stoltz and coworkers.99
Scheme 4.

Structure of (−)-Aurantioclavine and Intermediates in its Synthesis by Stoltz and Coworkers.99
2.7. Tandem Asymmetric Aryl Addition/Diastereoselective Epoxidation
We recently developed a series of tandem reactions involving the asymmetric addition of alkyl,100-102 vinyl,100,103,104 or allyl105 groups to aldehydes and ketones followed by diastereoselective epoxidation to provide epoxy alcohols with three contiguous stereogenic centers.106 These one-pot procedures rapidly increase molecular complexity in a synthetically efficient fashion. To explore the possibility of performing arylation and heteroarylation of enals followed by diastereoselective epoxidation, we examined asymmetric phenyl addition/oxidation with 3-methyl-2-butenal. As shown in Scheme 5A, using the conditions outlined in Table 4 the catalytic asymmetric phenyl addition was performed. The resulting enantioenriched zinc allylic alkoxide was then treated with Et2Zn (1 equiv), TBHP (tert-butylhydroperoxide, 5 equiv), and 20 mol % titanium tetraisopropoxide at 0 °C. The epoxidation reached completion in 3 h, after which the reaction mixture was quenched, worked up, and the product purified by chromatography to afford the epoxy alcohol in 67% yield with 90% ee and >20:1 dr (as determined by 1H NMR). The heteroarylation/epoxidation was examined with the TIPS protected 4-bromoindole (Scheme 5B). Metallation with t-BuLi, transmetallation with EtZnCl, and asymmetric addition as performed in Equation 4 was followed by addition of Et2Zn, TBHP and 20 mol % titanium tetraisopropoxide at 0 °C. Following workup and purification, the enantioenriched indole epoxy alcohol was isolated in 65% yield with 90% ee and >20:1 dr. Interestingly, attempted epoxidation of the isolated indole allylic alcohol product in Equation 4 with m-CPBA resulted in formation of the epoxy alcohol in low yield accompanied by several side products. The examples in Scheme 5 indicate that the asymmetric arylation and heteroarylation are compatible with our tandem diastereoselective epoxidation conditions and could be used to prepare an array of functionalized epoxy alcohols.
Scheme 5.

Tandem Asymmetric Arylation of Aldehydes/Diastereoselective Epoxidation.
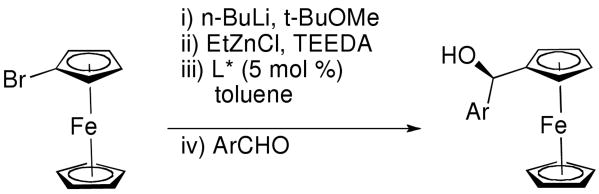
2.8. Synthesis of Ferrocenylzinc and Applications to Asymmetric Additions
Having developed successful methods for the enantioselective addition of aryl and heteroaryl groups to aldehydes, we focused on the generation of the ferrocenylzinc reagent, (Fc)ZnEt. Highly enantioselective additions of ferrocenylzinc reagents to aromatic and heteroaromatic aldehydes would provide rapid access to heteroaryl ferrocenyl methanols. Related motifs107,108 are precursors to important enantioenriched ferrocene-based ligands such as BoPhoz,109 Josiphos,110 FERRIPHOS,111,112 Pigiphos,113,114 PPFA,115 Walphos,116 Taniaphos117 and Trap.109,118,119 The ferrocenyl methanol scaffold is often synthesized by CBS120-122 or Ru/BINAP reduction of ferrocenyl ketones.4,119,123 Asymmetric reduction of heteroaromatic ketone derivatives, however, resulted in only moderate enantioselectivity (X=O, 41% ee; X=S, 68% ee, Equation 5).124
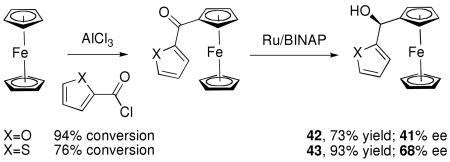 |
Equation 5 |
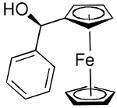
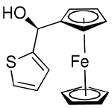
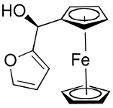
Beginning with ferrocenyl bromide (FcBr) and applying the conditions used in Table 4 to the generation and addition of (Fc)ZnEt to benzaldehyde with (−)-MIB provided product with a disappointing 50% ee (Table 5, entry 1). Inspired by the importance of functionalized ferrocenyl methanols, we screened other amino alcohol ligands. Fortunately, use of Chan's48,95 amino alcohol L2 (Figure 2) with benzaldehyde provided the desired product in 86% yield with 98% ee (Table 5, entry 2). Use of 2-thiophenecarboxaldehyde and 2-furfural in combination with L2 provided the ferrocene-based ligand precursors with enantioselectivities of 96% and yields of 95% (entries 3 and 4). It is known that substitution of furyl groups for phenyl can lead to an increase in catalyst enantioselectivity.125
Table 5.
Asymmetric Addition of (Fc)ZnEt to Aldehydes.
 | ||||||
|---|---|---|---|---|---|---|
| entry | ligand | ArCHO | product | # | yield | ee (%) |
| 1 | (−)-MIB | 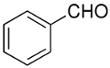 |
 |
44 | 75 | 50 |
| 2 | L2 | 86 | 98 | |||
| 3 | L2 | 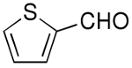 |
 |
43 | 95 | 96 |
| 4 | L2 | 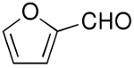 |
 |
42 | 95 | 96 |
The high yields and stereochemical purity of functionalized ferrocenes make them attractive building blocks for the construction of new ferrocene-based ligands for asymmetric catalysis.
3. Summary and Outlook
Herein we described versatile methods for the generation of diaryl- aryl heteroaryl-, and diheteroarylmethanols with high levels of enantioselectivity. The significance of these methods is that asymmetric arylation of aldehydes can now be initiated with aryl bromides, many of which are readily available. Key to the success of our procedures was introduction of a diamine, such as TEEDA. In the absence of TEEDA the addition reaction was promoted by LiCl, generating racemic products. The TEEDA inhibited the LiCl byproduct, allowing the asymmetric addition to proceed via the ligand accelerated pathway.71,126 Importantly, in the presence of the diamine it was not necessary to filter,43 centrifuge,67 or isolate the pyrophoric arylzinc reagents required with previous procedures, making our method desirable for large scale applications.
We also developed the first method for the synthesis of highly enantioenriched diheteroarylmethanols from readily available heteroaryl bromides. A crucial feature of this approach was the use of EtZnCl in the transmetallation step with the heteroaryllithium at −78 °C, at which temperature decomposition of the heteroaryl organometallic species was minimized. Use of EtZnCl in place of ZnCl2 also halves the amount of LiCl byproduct, which was detrimental to the enantioselectivity in the asymmetric addition and must be inhibited by diamine. This method was also shown to be applicable to the tandem asymmetric addition/diastereoselective epoxidation to generate epoxy alcohols with two stereogenic centers in high enantio- and diastereoselectivity.
Finally, we also described the first examples of generation and highly enantioselective addition of ferrocenyl zinc reagents to aldehydes, opening the door to new enantioenriched ferrocene-based ligands. The straightforward methods introduced herein make possible the synthesis of functionalized, previously inaccessible enantioenriched diheteroarylmethanols. We anticipate that these methods will be useful in medicinal chemistry and asymmetric catalysis.
4. Experimental Section
General Considerations
All reactions were performed under a nitrogen atmosphere with oven-dried glassware using standard Schlenk or vacuum line techniques. The progress of reactions was monitored by thin-layer chromatography (TLC) performed on Whatman precoated silica gel 60 Å K6F plates and visualized by ultra-violet light or by staining with cerium-ammonium-molybdate. t-BuOMe was distilled from Na/benzophenone and toluene was dried through alumina columns. TEEDA was distilled and stored under nitrogen. The 1H NMR and 13C{1H} NMR spectra were obtained on a Brüker Fourier transform NMR spectrometer at either 300 or 500 and 75 or 125 MHz, respectively. 1H NMR spectra were referenced to tetramethylsilane in CDCl3 or residual protonated solvent; 13C{1H} NMR spectra were referenced to residual solvent. Analysis of enantiomeric excess was performed using a Hewlett-Packard 1100 Series HPLC and a chiral column. Alternatively, a Berger SFC PioNTo™® was employed when the compounds could not be resolved by HPLC. The optical rotations were recorded using a JASCO DIP-370. Infrared spectra were obtained using a Perkin-Elmer Spectrum 100 Series spectrometer. All reagents were purchased from Aldrich or Acros unless otherwise described. 3-Benzofurancarboxaldehyde was synthesized according to known procedure starting from commercially available 3-methylbenzofuran.127 Binaphthyl amino alcohol ligand was synthesized according to Chan's procedure.48,95 EtZnCl was synthesized following Guerrero's method.84,128 All aldehyde substrates were distilled prior to use. Silica gel (Silicaflash P60 40-63 μm, Silicycle) was used for air-flashed chromatography.
Caution
Dialkylzinc and alkyl lithium reagents are pyrophoric! Care and appropriate laboratory equipment and attire must be used when handling these reagents.
Arylation of Aldehydes
Preparation of (4-Fluoro-phenyl)-(4-methoxy-phenyl)-methanol (10)
A nitrogen purged Schlenk flask was charged with 4-bromoanisole (100.1 μL, 0.8 mmol) and t-BuOMe (1 mL) and cooled to −78 °C. n-BuLi (0.32 mL, 2.5 M in hexanes, 0.8 mmol) was added dropwise and the solution was stirred for 1 h and the temperature raised to 0 °C. ZnCl2 (114.5 mg, 0.84 mmol) was added to the reaction mixture and it was stirred for 30 min. Additional n-BuLi (0.32 mL, 2.5 M in hexanes, 0.8 mmol) was added to the reaction mixture and the resulting solution was allowed to warm to rt and stirred 4.5 h. Toluene (5 mL) and TEEDA (68 μL, 0.32 mmol) were added to the reaction vessel and the solution was stirred. After 1 h (−)-MIB (4.8 mg, 0.02 mmol) was added and the reaction vessel was cooled to 0 °C for 30 min. Finally, 4-fluorobenzaldehyde (43 μL, 0.4 mmol) was added and reaction mixture was stirred at 0 °C and monitored by TLC. After completion (12 h), the reaction mixture was quenched with H2O (20 mL) and extracted with ethyl acetate (3 × 20 mL). The organic layer was dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (hexanes:EtOAc, 95:5) to give 10 (77.7 mg, 84% yield) as a white crystalline solid (m. p. = 52 °C). The enantiomeric excess was determined by HPLC with a Chiralcel AS-H column (hexanes:2-propanol = 95:5, flow rate = 0.5 mL/min), tr (1) = 20.0 min, tr (2) = 22.1 min, = + 13.8(c = 0.195, THF, 93% ee); 1H NMR (C6D6, 300 MHz): δ 2.17 (s, 1H), 3.38 (s, 3H), 5.50 (s, 1H), 6.81-6.95 (m, 4H), 7.17-7.29 (m, 4H); 13C{1H} NMR (C6D6, 75 MHz): δ 55.0, 75.3, 114.3, 115.4 (d, J = 21.2 Hz), 128.4, 128.7 (d, J = 8.0 Hz), 136.9, 141.0 (d, J = 3.0 Hz), 159.7, 162.6 (d, J = 243 Hz); IR (neat): 831, 1033, 1248, 1504, 1609, 2837, 2957, 3422 cm-1; HRMS calcd for C14H13FO2 (M)+: 232.0900, found: 232.0900.
Furan-3-yl(thiophen-2-yl)methanol (35)
A nitrogen purged Schlenk flask was charged with 3-bromofuran (67.0 μL 0.75 mmol) and t-BuOMe (1 mL) and cooled to −78 °C. n-BuLi (0.3 mL, 2.5 M in hexanes, 0.75 mmol) was then added dropwise and the solution was stirred for 1 h at this temperature. During this time a white precipitate formed. Solid EtZnCl (97.0 mg, 0.75 mmol) was added to the reaction mixture at −78 °C followed by toluene (3 mL). The heterogeneous solution was stirred at −78 °C for 30 min and then warmed at 0 °C. TEEDA (64 μL, 0.30 mmol) was added and the solution stirred for an additional 30 min. (−)-MIB (190 μL, 0.1 M solution in hexanes, 0.019 mmol) was added to the reaction flask and the solution was stirred for 5 min before 2-thiophenecarboxaldehyde (35 μL, 0.37 mmol, dissolved in 1.5 mL of toluene) was added over 1.5 h by syringe pump. The reaction mixture was stirred at 0 °C and monitored by TLC until completion (approximately 10 h). The reaction mixture was diluted with 3 mL EtOAc and quenched with water (5 mL). The organic layer was separated and the aqueous solution extracted with EtOAc (3 × 10 mL). The combined organic layer was washed with brine (5 mL), dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on deactivated silica gel (hexanes:EtOAc, 95:5) to give 35 (44.4 mg, 60% yield) as a colorless oil. The enantiomeric excess was determined by HPLC with a Chiralcel OD-H column (hexanes:2-propanol = 97:3, flow rate = 0.5 mL/min), tr (1) = 40.4 min, tr (2) = 47.1 min, = + 18.2(c = 0.032, CHCl3, 99% ee); 1H NMR (CDCl3, 500 MHz): δ 2.27 (d, J = 5.0 Hz, 1H), 6.04 (d, J = 5.0 Hz, 1H), 6.44-6.45 (m, 1H), 6.97-6.99 (m, 1H), 7.01-7.02 (m, 1H), 7.30 (dd, J = 1.5, 5.0 Hz, 1H) 7.41 (t, J = 1.5 Hz, 1H), 7.45-7.46 (m, 1H); 13C{1H} NMR (CDCl3, 125 MHz): δ 65.8, 109.3, 125.0, 125.6, 126.9, 128.6, 140.1, 143,7, 147.4; IR (neat): 3410, 3108, 2924, 2855, 1759, 1672, 1614, 1507, 1416, 1264, 1230, 1156, 1022 cm-1; HRMS calcd for C9H7O2S (M-H)+: 179.0167, found 179.0169.
Asymmetric Addition/Diastereoselective Epoxidation Reactions
Preparation of (3,3-Dimethyloxiran-2-yl)(1-(triisopropylsilyl)-1H-indol-4-yl)methanol (41)
A nitrogen purged Schlenk flask was charged with 4-bromo-1-TIPS indole (106.5 mg, 0.3 mmol) and t-BuOMe (1 mL) and cooled to −78 °C. t-BuLi (0.18 mL, 1.7 M in pentane, 0.3 mmol) was added dropwise and the solution was stirred for 1 h. Solid EtZnCl (39.6 mg, 0.3 mmol) was added to the reaction solution at −78 °C. Toluene (3 mL) was next added giving a heterogenous mixture. The solution was warmed at −10 °C and stirred for 3 h. TEEDA (26 μL, 0.12 mmol) was then added and the solution stirred for an additional 30 min. (−)-MIB (150 μL, 0.1 M solution in hexanes, 0.015 mmol) was added to the reaction flask, the solution was stirred for 5 min, and 3-methyl-2-butenal (14.7 μL, 0.15 mmol, dissolved in 1.5 mL of toluene) was added by syringe pump over 1.5 h. The reaction mixture was stirred at 0 °C and monitored by TLC until the starting aldehyde had been consumed. Upon completion of the asymmetric addition, ZnEt2 (0.15 mL, 1 M in hexanes, 0.15 mmol) was added followed by TBHP (0.14 mL, 5.5 M in decane, 0.77 mmol). After stirring for 5 min Ti(O-iPr)4 (30 μL, 1 M in hexanes, 0.03 mmol) was added and the reaction mixture stirred until complete by TLC analysis (approximately 3 h). The reaction mixture was diluted with 3 mL EtOAc and quenched with water (5 mL). The organic layer was next separated and the aqueous solution extracted with EtOAc (3 × 10 mL). The combined organic layer was washed with brine (5 mL), dried over MgSO4, filtered, and the volatile materials were removed under reduced pressure. The crude product was purified by column chromatography on deactivated silica gel (hexanes:EtOAc, 90:10) to give 41 (36.6 mg, 64.9% yield) as a clear oil. The diastereomeric ratio was determined by 1H NMR analysis of the crude product (dr > 20:1); = − 6.1 (c = 0.041, CHCl3, 90% ee); 1H NMR (CDCl3, 500 MHz): δ 1.13 (d, J = 7.6 Hz, 18H), 1.29 (s, 3H), 1.43 (s, 3H), 1.69 (sept, J = 7.6 Hz, 3H), 2.48 (d, J = 3.0 Hz, 1H), 3.31 (d, J = 8.1 Hz, 1H), 4.89 (dd, J = 3.0, 8.1 Hz, 1H), 6.80 (d, J = 3.3 Hz, 1H), 7.10-7.14 (m, 1H), 7.30 (d, J = 3.3 Hz, 1H), 7.47 (dd, J = 2.8, 6.9 Hz, 1H); 13C{1H} NMR (CDCl3, 125 MHz): δ 12.8, 18.0, 19.5, 24.8, 60.1, 67.3, 72.2, 103.3, 113.9, 117.6, 121.1, 129.2, 131.5, 141.3; IR (neat): 3445, 3135, 3081, 3048, 2948, 2892, 2868, 2760, 2729, 2625, 2559, 2361, 2343, 2246, 2150, 2074, 1892, 1824, 1740, 1675, 1599, 1514, 1463, 1428, 1378, 1345, 1323, 1280, 1248, 1209, 1150, 1124, 1096, 1073 cm-1; HRMS calcd for C22H35NO2NaSi (M+Na)+: 396.2335, found 396.2321.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health, National Institute of General Medical Sciences (GM58101) and National Science Foundation (CHE-0615210) for financial support of this work.
Footnotes
Supporting Information Available: Complete experimental procedures, product characterization, and full author list for reference 93. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bolshan Y, Chen Cy, Chilenski JR, Gosselin F, Mathre DJ, O'Shea PD, Roy A, Tillyer RD. Org Lett. 2004;6:111–114. doi: 10.1021/ol0361655. [DOI] [PubMed] [Google Scholar]
- 2.Corey EJ, Helal CJ. Tetrahedron Lett. 1996;37:5675–5678. [Google Scholar]
- 3.Muller P, Nury P, Bernardinelli G. Eur J Org Chem. 2001:4137–4147. [Google Scholar]
- 4.Ohkuma T, Koizumi M, Ikehira H, Yokozawa T, Noyori R. Org Lett. 2000;2:659–662. doi: 10.1021/ol9904139. [DOI] [PubMed] [Google Scholar]
- 5.Rekker RF, Timmerman H, Harms AF, Nauta WT. Arzneim Forsch. 1971;21:688–691. [PubMed] [Google Scholar]
- 6.Van der Stelt C, Heus WJ, Nauta WT. Arzneim Forsch. 1969;19:2010–2012. [PubMed] [Google Scholar]
- 7.Pestellini V, Giannotti D, Giolitti A, Fantò N, Riviera L, Bellotti MG. Chemioterapia. 1987;6:269–271. [PubMed] [Google Scholar]
- 8.Botta M, Corelli F, Gasparrini F, Messina F, Mugnaini C. J Org Chem. 2000;65:4736–4739. doi: 10.1021/jo991937p. [DOI] [PubMed] [Google Scholar]
- 9.Whomsley R, Fernandez E, Nicholls PJ, Smith HJ, Lombardi P, Pestellini V. J Steroid Biochem Mol Biol. 1993;44:675–676. doi: 10.1016/0960-0760(93)90279-6. [DOI] [PubMed] [Google Scholar]
- 10.Saberi MR, Shah K, Simons C. J Enzyme Inhib Med Chem. 2005;20:135–141. doi: 10.1080/14756360400015256. [DOI] [PubMed] [Google Scholar]
- 11.Saberi MR, Vinh TK, Yee SW, Griffiths BJN, Evans PJ, Simons C. J Med Chem. 2006;49:1016–1022. doi: 10.1021/jm0508282. [DOI] [PubMed] [Google Scholar]
- 12.Recanatini M, Cavalli A, Valenti P. Med Res Rev. 2002;22:282–304. doi: 10.1002/med.10010. [DOI] [PubMed] [Google Scholar]
- 13.Botta M, Summa V, Corelli F, Di Pietro G, Lombardi P. Tetrahedron: Asymmetry. 1996;7:1263–1266. [Google Scholar]
- 14.Buzdar AU. Breast Cancer Res Treat. 2002;75:13–17. doi: 10.1023/a:1020305615033. [DOI] [PubMed] [Google Scholar]
- 15.Murai S, Shimano M, Yamamoto H, Koyama T, Nakamura T, Ogawa M, Watanuki M, Okamoto T, Hori T. Eur Pat Appl. Vol. 529365. Kaken Pharmaceutical Co., Ltd.; Japan: Mar 3, 1993. p. A1. [Google Scholar]
- 16.Nilvebrant L, Andersson KE, Gillberg PG, Stahl M, Sparf B. Eur J Pharmacol. 1997;327:195–207. doi: 10.1016/s0014-2999(97)89661-6. [DOI] [PubMed] [Google Scholar]
- 17.Welch WM, Kraska AR, Sarges R, Koe BK. J Med Chem. 1984;27:1508–1515. doi: 10.1021/jm00377a021. [DOI] [PubMed] [Google Scholar]
- 18.Astles PC, Brown TJ, Halley F, Handscombe CM, Harris NV, Majid TN, McCarthy C, McLay IM, Morley A, Porter B, Roach AG, Sargent C, Smith C, Walsh RJA. J Med Chem. 2000;43:900–910. doi: 10.1021/jm990378b. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt F, Stemmler RT, Rudolph J, Bolm C. Chem Soc Rev. 2006;35:454–470. doi: 10.1039/b600091f. [DOI] [PubMed] [Google Scholar]
- 20.Seebach D, Behrendt L, Felix D. Angew Chem Int Ed Engl. 1991;20:1008–1009. [Google Scholar]
- 21.Weber B, Seebach D. Angew Chem Int Ed Engl. 1992;31:84–86. [Google Scholar]
- 22.Weber B, Seebach D. Tetrahedron. 1994;50:7473–7484. [Google Scholar]
- 23.Dosa PI, Ruble JC, Fu GC. J Org Chem. 1997;62:444–445. doi: 10.1021/jo962156g. [DOI] [PubMed] [Google Scholar]
- 24.Huang WS, Hu QS, Pu L. J Org Chem. 1999;64:7940–7956. doi: 10.1021/jo990749w. [DOI] [PubMed] [Google Scholar]
- 25.Bolm C, Muñiz K. Chem Commun. 1999:1295–1296. [Google Scholar]
- 26.Huang WS, Pu L. J Org Chem. 1999;64:4222–4223. [Google Scholar]
- 27.Huang WS, Pu L. Tetrahedron Lett. 2000;41:145–149. [Google Scholar]
- 28.Zhao G, Li XZ, Wang XR. Tetrahedron: Asymmetry. 2001;12:399–403. [Google Scholar]
- 29.Ko DH, Kim KH, Ha DC. Org Lett. 2002;4:3759–3762. doi: 10.1021/ol026761j. [DOI] [PubMed] [Google Scholar]
- 30.Rudolph J, Schmidt F, Bolm C. Synthesis. 2005:840–842. [Google Scholar]
- 31.Hermanns N, Dahmen S, Bolm C, Brase S. Angew Chem Int Ed. 2002;41:3692–3694. doi: 10.1002/1521-3773(20021004)41:19<3692::AID-ANIE3692>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 32.Bolm C, Hildebrand JP, Muñiz K, Hermanns N. Angew Chem Int Ed. 2001;40:3284–3308. doi: 10.1002/1521-3773(20010917)40:18<3284::aid-anie3284>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 33.Bolm C, Hermanns N, Hildebrand JP, Muñiz K. Angew Chem Int Ed. 2000;39:3465–3467. doi: 10.1002/1521-3773(20001002)39:19<3465::aid-anie3465>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 34.Fontes M, Verdaguer X, Solà L, Pericàs MA, Riera A. J Org Chem. 2004;69:2532–2543. doi: 10.1021/jo035824o. [DOI] [PubMed] [Google Scholar]
- 35.Rudolph J, Bolm C, Norrby PO. J Am Chem Soc. 2005;127:1548–1552. doi: 10.1021/ja046254s. [DOI] [PubMed] [Google Scholar]
- 36.Rudolph J, Rasmussen T, Bolm C, Norrby PO. Angew Chem Int Ed. 2003;42:3002–3005. doi: 10.1002/anie.200351150. [DOI] [PubMed] [Google Scholar]
- 37.Bolm C, Kesselgruber M, Grenz A, Hermanns N, Hildebrand JP. New J Chem. 2001;25:13–15. [Google Scholar]
- 38.Bolm C, Kesselgruber M, Hermanns N, Hildebrand JP, Raabe G. Angew Chem Int Ed. 2001;40:1488–1490. [PubMed] [Google Scholar]
- 39.Bolm C, Hermanns N, Kesselgruber M, Hildebrand JP. J Organomet Chem. 2001;624:157–161. [Google Scholar]
- 40.Bolm C, Hermanns N, Classen A, Muniz K. Bioorg Med Chem Lett. 2002;12:1795–1798. doi: 10.1016/s0960-894x(02)00271-8. [DOI] [PubMed] [Google Scholar]
- 41.Pizzuti MG, Superchi S. Tetrahedron: Asymmetry. 2005;16:2263–2269. [Google Scholar]
- 42.Özçubukçu S, Schmidt F, Bolm C. Org Lett. 2005;7:1407–1409. doi: 10.1021/ol050242+. [DOI] [PubMed] [Google Scholar]
- 43.Rudolph J, Hermanns N, Bolm C. J Org Chem. 2004;69:3997–4000. doi: 10.1021/jo0495079. [DOI] [PubMed] [Google Scholar]
- 44.Bolm C, Rudolph J. J Am Chem Soc. 2002;124:14850–14851. doi: 10.1021/ja028518l. [DOI] [PubMed] [Google Scholar]
- 45.Braga AL, Ludtke DS, Vargas F, Paixao MW. Chem Commun. 2005:2512–2514. doi: 10.1039/b501485a. [DOI] [PubMed] [Google Scholar]
- 46.Ito K, Tomita Y, Katsuki T. Tetrahedron Lett. 2005;46:6083–6086. [Google Scholar]
- 47.Ji JX, Wu J, Au-Yeung TTL, Yip CW, Haynes RK, Chan ASC. J Org Chem. 2005;70:1093–1095. doi: 10.1021/jo048395i. [DOI] [PubMed] [Google Scholar]
- 48.Lu G, Kwong FY, Ruan JW, Li YM, Chan ASC. Chem Eur J. 2006;12:4115–4120. doi: 10.1002/chem.200501048. [DOI] [PubMed] [Google Scholar]
- 49.Rudolph J, Lormann M, Bolm C, Dahmen S. Adv Synth Catal. 2005;347:1361–1368. [Google Scholar]
- 50.Wu PY, Wu HL, Uang BJ. J Org Chem. 2006;71:833–835. doi: 10.1021/jo052017b. [DOI] [PubMed] [Google Scholar]
- 51.Jimeno C, Sayalero S, Fjermestad T, Colet G, Maseras F, Pericàs MA. Angew Chem Int Ed. 2008;47:1098–1101. doi: 10.1002/anie.200703103. [DOI] [PubMed] [Google Scholar]
- 52.Magnus NA, Anzeveno PB, Coffey DS, Hay DA, Laurila ME, Schkeryantz JM, Shaw BW, Staszak MA. Org Process Res Dev. 2007;11:560–567. [Google Scholar]
- 53.Braga AL, Paixao MW, Westermann B, Schneider PH, Wessjohann LA. J Org Chem. 2008;73:2879–2882. doi: 10.1021/jo702413n. [DOI] [PubMed] [Google Scholar]
- 54.Rudolph J, Schmidt F, Bolm C. Adv Synth Catal. 2004;346:867–872. [Google Scholar]
- 55.Bolm C, Zani L, Rudolph J, Schiffers I. Synthesis. 2004:2173–2180. [Google Scholar]
- 56.Bolm C, Schmidt F, Zani L. Tetrahedron: Asymmetry. 2005;16:1367–1376. [Google Scholar]
- 57.Dahmen S, Lormann M. Org Lett. 2005;7:4597–4600. doi: 10.1021/ol0515659. [DOI] [PubMed] [Google Scholar]
- 58.Wu X, Liu X, Zhao G. Tetrahedron: Asymmetry. 2005;16:2299–2305. [Google Scholar]
- 59.Liu XY, Wu XY, Chai Z, Wu Y, Zhao G, Zhu SZ. J Org Chem. 2005;70:7432–7435. doi: 10.1021/jo050994h. [DOI] [PubMed] [Google Scholar]
- 60.Sakai M, Ueda M, Miyaura N. Angew Chem Int Ed. 1998;37:3279–3281. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3279::AID-ANIE3279>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 61.Duan HF, Xie JH, Shi WJ, Zhang Q, Zhou QL. Org Lett. 2006;8:1479–1481. doi: 10.1021/ol060360c. [DOI] [PubMed] [Google Scholar]
- 62.Yamamoto Y, Kurihara K, Miyaura N. Angew Chem Int Ed. 2009;48:4414–4416. doi: 10.1002/anie.200901395. [DOI] [PubMed] [Google Scholar]
- 63.Muramatsu Y, Harada T. Chem Eur J. 2008;14:10560–10563. doi: 10.1002/chem.200801612. [DOI] [PubMed] [Google Scholar]
- 64.Wu KH, Gau HM. J Am Chem Soc. 2006;128:14808–14809. doi: 10.1021/ja062080y. [DOI] [PubMed] [Google Scholar]
- 65.Hsieh SH, Chen CA, Chuang DW, Yang MC, Yang HT, Gau HM. Chirality. 2008;20:924–929. doi: 10.1002/chir.20572. [DOI] [PubMed] [Google Scholar]
- 66.Balsells J, Davis TJ, Carroll PJ, Walsh PJ. J Am Chem Soc. 2002;124:10336–10348. doi: 10.1021/ja0171658. [DOI] [PubMed] [Google Scholar]
- 67.Cote A, Charette AB. J Am Chem Soc. 2008;130:2771–2773. doi: 10.1021/ja710864p. [DOI] [PubMed] [Google Scholar]
- 68.Kim JG, Walsh PJ. Angew Chem Int Ed. 2006;45:4175–4178. doi: 10.1002/anie.200600741. For our preliminary communication see: [DOI] [PubMed] [Google Scholar]
- 69.Chen YK, Jeon SJ, Walsh PJ, Nugent WA. Org Synth. 2005;82:87–92. [Google Scholar]
- 70.Nugent WA. Chem Commun. 1999:1369–1370. [Google Scholar]
- 71.Berrisford DJ, Bolm C, Sharpless KB. Angew Chem Int Ed Engl. 1995;34:1059–1070. [Google Scholar]
- 72.Kerrigan MH, Jeon SJ, Chen Y, Walsh PJ. J Am Chem Soc. 2009;131:8434–8445. doi: 10.1021/ja809821x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salvi L, Jeon SJ, Fisher EL, Carroll PJ, Walsh PJ. J Am Chem Soc. 2007;129:16119–16125. doi: 10.1021/ja0762285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoffmann D, Dorigo A, Schleyer PvR, Reif H, Stalke D, Sheldrick GM, Weiss E, Geissler M. Inorg Chem. 1995;34:262–269. [Google Scholar]
- 75.Chitsaz S, Pauls J, Neumuller B. Z Naturforsch B: J Chem Sci. 2001;56:245–248. [Google Scholar]
- 76.DeBerardinis AM, Turlington M, Pu L. Org Lett. 2008;10:2709–2712. doi: 10.1021/ol8008478. [DOI] [PubMed] [Google Scholar]
- 77.Knochel P, Dohle W, Gommermann N, Kneisel FF, Kopp F, Korn T, Sapountzis I, Vu VA. Angew Chem Int Ed. 2003;42:4302–4320. doi: 10.1002/anie.200300579. [DOI] [PubMed] [Google Scholar]
- 78.Shannon J, Bernier D, Rawson D, Woodward S. Chem Commun. 2007:3945–3947. doi: 10.1039/b710681e. [DOI] [PubMed] [Google Scholar]
- 79.Thacher SM, Vasudevan J, Chandraratna RAS. Curr Pharm Des. 2000;6:25–58. doi: 10.2174/1381612003401415. [DOI] [PubMed] [Google Scholar]
- 80.Yu KL, Spinazze P, Ostrowski J, Currier SJ, Pack EJ, Hammer L, Roalsvig T, Honeyman JA, Tortolani DR, Reczek PR, Mansuri MM, Starrett JE. J Med Chem. 1996;39:2411–2421. doi: 10.1021/jm9502293. [DOI] [PubMed] [Google Scholar]
- 81.Klaholz BP, Mitschler A, Moras D. J Mol Biol. 2000;302:155–170. doi: 10.1006/jmbi.2000.4032. [DOI] [PubMed] [Google Scholar]
- 82.Wu KH, Chuang DW, Chen CA, Gau HM. Chem Commun. 2008:2343–2345. doi: 10.1039/b802441c. [DOI] [PubMed] [Google Scholar]
- 83.Blake AJ, Shannon J, Stephens JC, Woodward S. Chem Eur J. 2007;13:2642–2672. doi: 10.1002/chem.200601739. [DOI] [PubMed] [Google Scholar]
- 84.Guerrero A, Martin E, Hughes DL, Kaltsoyannis N, Bochmann M. Organometallics. 2006;25:3311–3313. [Google Scholar]
- 85.Boersma J, Noltes JG. Tetrahedron Lett. 1966:1521–1525. [Google Scholar]
- 86.Fabicon RM, Richey HG., Jr Organometallics. 2001;20:4018–4023. [Google Scholar]
- 87.Mitsunobu O. Synthesis. 1981:1–28. [Google Scholar]
- 88.Botta M, Summa V, Trapassi G, Monteagudo E, Corelli F. Tetrahedron: Asymmetry. 1994;5:181–184. [Google Scholar]
- 89.Tang Y, Dong Y, Vennerstrom JL. Synthesis. 2004:2540–2544. [Google Scholar]
- 90.Njar VCO. Synthesis. 2000:2019–2028. [Google Scholar]
- 91.Álvarez C, Álvarez R, Corchete P, Pérez-Melero C, Peláez R, Medarde M. Bioorg Med Chem. 2008;16:8999–9008. doi: 10.1016/j.bmc.2008.08.040. [DOI] [PubMed] [Google Scholar]
- 92.Lipshutz BH, Chung DW, Rich B, Corral R. Org Lett. 2006;8:5069–5072. doi: 10.1021/ol0618757. [DOI] [PubMed] [Google Scholar]
- 93.Plobeck N, et al. J Med Chem. 2000;43:3878–3894. doi: 10.1021/jm000228x. [DOI] [PubMed] [Google Scholar]
- 94.Wood PM, Woo LWL, Labrosse JR, Trusselle MN, Abbate S, Longhi G, Castiglioni E, Lebon F, Purohit A, Reed MJ, Potter BVL. J Med Chem. 2008;51:4226–4238. doi: 10.1021/jm800168s. [DOI] [PubMed] [Google Scholar]
- 95.Lu G, Li X, Zhou Z, Chan WL, Chan ASC. Tetrahedron: Asymmetry. 2001;12:2147–2152. [Google Scholar]
- 96.Sundberg RJ. Indoles. Academic Press; San Diego: 1996. [Google Scholar]
- 97.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. 2nd. John Wiley; New York: 2002. [Google Scholar]
- 98.Moyer MP, Shiurba JF, Rapoport H. J Org Chem. 1986;51:5106–5110. [Google Scholar]
- 99.Krishnan S, Bagdanoff JT, Ebner DC, Ramtohul YK, Tambar UK, Stoltz BM. J Am Chem Soc. 2008;130:13745–13754. doi: 10.1021/ja804738b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lurain AE, Maestri A, Kelly AR, Carroll PJ, Walsh PJ. J Am Chem Soc. 2004;126:13608–13609. doi: 10.1021/ja046750g. [DOI] [PubMed] [Google Scholar]
- 101.Jeon SJ, Walsh PJ. J Am Chem Soc. 2003;125:9544–9545. doi: 10.1021/ja036302t. [DOI] [PubMed] [Google Scholar]
- 102.Jeon SJ, Li H, Walsh PJ. J Am Chem Soc. 2005;127:16416–16425. doi: 10.1021/ja052200m. [DOI] [PubMed] [Google Scholar]
- 103.Lurain AE, Carroll PJ, Walsh PJ. J Org Chem. 2005;70:1262–1268. doi: 10.1021/jo048345d. [DOI] [PubMed] [Google Scholar]
- 104.Kelly RA, Lurain AE, Walsh PJ. J Am Chem Soc. 2005;127:14668–14674. doi: 10.1021/ja051291k. [DOI] [PubMed] [Google Scholar]
- 105.Kim JG, García IF, Kwiatkowski D, Walsh PJ. J Am Chem Soc. 2004;126:12580–12585. doi: 10.1021/ja047758t. [DOI] [PubMed] [Google Scholar]
- 106.Hussain MH, Walsh PJ. Acc Chem Res. 2008;41:883–893. doi: 10.1021/ar800006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marquarding D, Klusacek H, Gokel G, Hoffmann P, Ugi I. J Am Chem Soc. 1970;92:5389–5393. [Google Scholar]
- 108.Ireland T, Perea JJA, Knochel P. Angew Chem Int Ed. 1999;38:1457–1460. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1457::AID-ANIE1457>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 109.Boaz NW, Debenham SD, Mackenzie EB, Large SE. Org Lett. 2002;4:2421–2424. doi: 10.1021/ol0261736. [DOI] [PubMed] [Google Scholar]
- 110.Togni A, Breutel C, Schnyder A, Spindler F, Landert H, Tijani A. J Am Chem Soc. 1994;116:4062–4066. [Google Scholar]
- 111.Perea JJA, Börner A, Knochel P. Tetrahedron Lett. 1998;39:8073–8076. [Google Scholar]
- 112.Lotz M, Ireland T, Perea JJA, Knochel P. Tetrahedron: Asymmetry. 1999;10:1839–1842. [Google Scholar]
- 113.Barbaro P, Currao A, Herrmann J, Nesper R, Pregosin PS, Salzmann R. Organometallics. 1996;15:1879–1888. [Google Scholar]
- 114.Fadini L, Togni A. Chem Commun. 2003:30–31. doi: 10.1039/b210680a. [DOI] [PubMed] [Google Scholar]
- 115.Hayashi T, Mise T, Fukushima M, Kagotani M, Nagashima N, Hamada Y, Matsumoto A, Kawakami S, Konishi M. Bull Chem Soc Jap. 1980;53:1138–1151. [Google Scholar]
- 116.Sturm T, Weissensteiner W, Spindler F. Adv Synth Catal. 2003;345:160–164. [Google Scholar]
- 117.Ireland T, Grossheimann G, Wieser-Jeunesse C, Knochel P. Angew Chem Int Ed. 1999;38:3212–3215. [PubMed] [Google Scholar]
- 118.Sawamura M, Hamashima H, Ito Y. Tetrahedron: Asymmetry. 1991;2:593–596. [Google Scholar]
- 119.Arrayás RG, Adrio J, Carretero JC. Angew Chem Int Ed. 2006;45:7674–7715. doi: 10.1002/anie.200602482. [DOI] [PubMed] [Google Scholar]
- 120.Boaz NW. Tetrahedron Lett. 1989;30:2061–2064. [Google Scholar]
- 121.Corey EJ, Bakshi BK, Shibata S. J Am Chem Soc. 1987;109:5551–5553. [Google Scholar]
- 122.Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 123.Ohkuma T, Ooka H, Hashiguchi S, Ikariya T, Noyori R. J Am Chem Soc. 1995;117:2675–2676. [Google Scholar]
- 124.Lam WS, Kok SHL, Au-Yeung TTL, Wu J, Cheung HY, Lam FL, Yeung CH, Chan ASC. Adv Synth Catal. 2006;348:370–374. [Google Scholar]
- 125.Northrup AB, MacMillan DWC. J Am Chem Soc. 2002;124:2458–2460. doi: 10.1021/ja017641u. [DOI] [PubMed] [Google Scholar]
- 126.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito: 2008. [Google Scholar]
- 127.Zaidlewicz M, Chechlowska A, Prewysz-Kwinto A, Wojtczak A. Heterocycles. 2001;55:569–577. [Google Scholar]
- 128.Guerrero A, Hughes DL, Bochmann M. Organometallics. 2006;25:1525–1527. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.