

A single replication fork would take more than a year to replicate the genome of Xenopus. By dividing the task among many thousands of replicons, each replicated by forks emanating from individual origins, replication is instead completed in as little as 30 min. While this efficient strategy has been adopted by all eukaryotes, it introduces a complication. To maintain the integrity of the genome, multiple replicons must now be coordinated so that all sequences are replicated exactly once per cell cycle. Because different replicons are often replicated at different times during S phase, replicated regions must be distinguishable from unreplicated regions to avoid problems of rereplication. We suggest that this distinction is based upon a fundamental feature of replication initiation.
Two things happen at origins of replication: proteins are recruited to origins to assemble multiprotein replication machines (Figure 1, left), and these assemblies are triggered to initiate replication forks (Figure 1, right). Replication components accompany the departing forks, leaving behind a spent origin. Consequently, reinitiation should require assembly of new components at the origin. If this assembly is restricted to one part of the cell cycle and the initiation of forks to another, then origin firing would occur only once per cell cycle (Figure 1).
Figure 1. A Model for Limiting DNA Replication to Once per Cell Cycle.
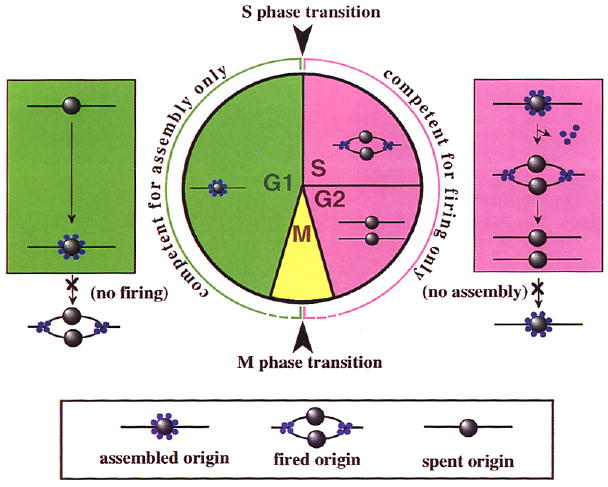
Formation of a functional replication origin (green) involves assembly of essential replication factors (blue spheres) onto DNA at a site marked by an origin recognition complex (ORC) (gray spheres). Origin firing (pink) involves the initiation of replication forks from assembled origins. Some replication factors travel with the fork, while others dissociate or remain at the origin, resulting in a spent origin. Assembly and firing of origins are confined to different parts of the cell cycle (green and pink, respectively), thereby limiting initiation of DNA replication to once per cell cycle.
The transition between replication-competent and replication-incompetent phases of the cell cycle has been explored in a series of early and influential cell fusion experiments (Rao and Johnson, 1970). Upon fusion with an S phase cell, nuclei from G1 cells, but not from G2 cells, replicate their DNA. Thus, even when present in cytoplasm capable of supporting S phase, the G2 nucleus is incompetent to replicate. Since G2 nuclei are converted into G1 nuclei by the passage through mitosis, mitosis must provide replication competence to the G2 nucleus. In the last several years, in vitro experiments using Xenopus egg extracts as well as genetic experiments using fission yeast have given rise to two rather different models for the basis of this mitotic transition. We outline each of these areas of research below and evaluate features of each model in an attempt to bring us closer to a unified understanding of these events.
Licensing and the Minichromosome Maintenance Connection
The Licensing Model
Blow and Laskey (1988) investigated the nature of the mitotic transition by examining the requirements for rereplication in a Xenopus extract system. These extracts can assemble an intact nuclear membrane around added chromatin, and the resulting nuclei can undergo multiple cycles of S phase and mitosis. However, when the accumulation of high levels of cyclin is blocked by inhibition of protein synthesis, the nuclei undergo a single round of DNA replication and arrest in G2 phase. Blow and Laskey found that they could bypass the requirement for mitosis and induce another round of replication by permeabilizing the nuclei with detergents or mechanical disruption. They proposed that the nuclear membrane excludes an essential replication factor that “licenses” the DNA for replication and that this factor is inactivated or destroyed in conjunction with replication. Accordingly, G2 nuclei lack active licensing factor, and replication is thus prohibited until the nuclear envelope breaks down during mitosis (or upon experimental manipulation), allowing entry of new factor. In the context of Figure 1, admission of the essential licensing factor at mitosis allows the assembly of replication proteins at origins, and the destruction of this licensing factor upon origin firing prevents further assembly during the replication phase.
The Minichromosome Maintenance Gene Family
The finding that the protein encoded by the Saccharomyces cerevisiae gene CDC46 is required for DNA replication and is present in the nucleus only from mitosis until S phase led to the suggestion that it might provide licensing function (Hennessy et al., 1990). Since then, CDC46 has been shown to belong to a family of genes, called MCMs (for minichromosome maintenance genes), many of which were identified in screens for mutations that increase the rate of plasmid loss. Several experiments indicate that MCM gene function is required at origins for the initiation of replication. Among these is the finding that CDC46 interacts genetically with ORC-6, which encodes a subunit of a multiprotein complex that binds to yeast replication origins and is required for DNA replication (Li and Herskowitz, 1993).
Isolation of MCM homologs in Schizosaccharomyces pombe (Coxon et al., 1992), Drosophila (Treisman et al., 1995), Xenopus (Coxon et al., 1992), mammals (Hu et al., 1993), and Arabidopsis (Springer et al., 1995) has demonstrated their conservation among eukaryotes. In mammalian cells, MCM gene products are nuclear throughout interphase, but their staining pattern, Triton X-100 extract-ability, and phosphorylation differ between G1 and G2 phases (Kimura et al., 1994). These differences might reflect changes in assembly or activity and thus be consistent with a licensing role for these proteins.
The Licensing Model Meets the MCMs
In an exciting intersection of two previously independent fields, several groups have shown involvement of MCM proteins in the licensing process as assayed in Blow and Laskey–type in vitro systems. Madine et al. (1995) demonstrated that permeabilized HeLa G2 nuclei replicate in a complete Xenopus egg extract, but not in one immunodepleted for a Xenopus MCM3 homolog and its associated proteins (notably, the associated proteins included three additional MCM family members). In contrast, permeabilized HeLa G1 nuclei underwent a complete round of replication in the immunodepleted extract. These results indicate that, while Xenopus MCM3 and associated proteins are required for the replication of G2 nuclei, they have either executed their function prior to G1 phase or are associated with the G1 nuclei.
Chong et al. (1995) and Kubota et al. (1995) took a somewhat different approach to address the role of MCMs in licensing. They used assays based on the previous observation that when chromatin is added to mitotic Xenopus egg extracts that have been treated with kinase inhibitors, it is assembled into a nucleus but fails to undergo DNA replication. Replication can ensue, however, if the chromatin is first exposed to a membrane-depleted interphase extract that is itself incapable of supporting nuclear envelope assembly and chromatin replication. The activity provided by these “impaired” interphase extracts is presumed to be licensing factor. Crude subdivision of the impaired extract produced two fractions (RLF-M and RLF-B) that together, but not alone, supported licensing (Chong et al. 1995). The activity in one of these fractions (RLF-M) was purified to three proteins, including an MCM3 homolog and a second, uncharacterized MCM homolog. Immunodepletion of MCM3 and associated proteins eliminated licensing activity from the impaired interphase extract. Using a similar approach, Kubota et al. (1995) purified three proteins that bound to chromatin during incubation in a similarly impaired interphase extract that is licensing competent, but did not bind in a treated mitotic extract that is licensing incompetent. The cDNA encoding one of the purified proteins, p100, was found to be homologous to yeast MCM3. Immunodepletion of p100 was sufficient to remove licensing activity from impaired interphase extracts.
Although these papers demonstrate that MCM homologs or their associated proteins are required for replication of G2 nuclei, there are several reasons to suspect that the MCM gene products may not be the licensing factors postulated by Blow and Laskey (1988). The observations that the MCMs are required for the replication of G2 nuclei and that they may become stably associated with chromatin upon transition from G2 to G1 are consistent with the predictions of the licensing model, but are not sufficient to define the licensing factor. Any replication protein such as RPA, which is assembled into “prereplication centers” immediately following mitosis (Adachi and Laemmli, 1992), might show such behavior. Furthermore, in contrast with the predictions of the licensing model, two groups (Kimura et al., 1994; Madine et al., 1995) have demonstrated that MCM3 homologs can enter a G1 nucleus in the absence of nuclear membrane breakdown. For this reason, Chong et al. (1995) raise the possibility that their identified MCM fraction (RLF-M) is not itself the licensing factor, but is rather an essential replication function whose action depends upon an additional factor (present in the crude RLF-B fraction) that is regulated by nuclear entry. Thus, although these papers define several molecules that participate in the process whereby a G2 nucleus can acquire the license to replicate, they fail to shed new light on the mechanism by which this is normally prevented.
Cyclin-Dependent Kinases and Rereplication
The study of mutations in yeast that allow replication of G2 nuclei has provided insight into how this replication is normally prevented. Broek et al. (1991) attempted to identify genes involved in the prevention of rereplication in S. pombe in a screen for mutations that allowed haploid cells to sporulate; because sporulation can only occur in diploid cells, mutations identified in this screen were expected to have caused endoreplication. They identified two mutations that provoked an additional round of S phase that gave rise to viable diploid cells. Remarkably, both of these mutations were alleles of cdc2, which encodes the cyclin-dependent kinase (CDK) of this organism. Recently, a more direct screen for mutations that allow endoreplication identified three mutations (Hayles et al., 1994). Two of these mutations represent additional alleles of cdc2, and the third is in cdc13, which encodes the G2 cyclin partner of cdc2. These results provide compelling evidence that the cyclin–Cdc2 complex plays a role in preventing rereplication during G2. They also provide a general model for the control of replication within the cell cycle, because cyclin–CDK complexes are present throughout S and G2 phases when licensing is prohibited. The coupling of mitosis to the ability to rereplicate is provided in this model by the cyclin destruction and resulting elimination of CDK activity that universally accompany mitosis. In addition to simply regulating licensing, the presence of this complex appears to define G2 phase itself because when the complex is eliminated, the cells apparently enter G1 phase (Broek et al., 1991).
Because the yeast nuclear envelope remains intact during mitosis, the licensing model does not explain the block to rereplication in yeast. For this reason, perhaps, studies in S. pombe and Xenopus have largely progressed independently of one another. But there are reasons to believe that the role of CDKs in licensing is universal. For example, mutations in Drosophila cyclin A lead to endoreplication in normally mitotic cells (Sauer et al., 1995). In mammals, the addition of a tyrosine kinase inhibitor to cultured cells also induces endoreplication (Usui et al., 1991). This inverse relationship of kinase levels and the ability to rereplicate is also supported by correlations seen in cell cycles that lack the regular alternation of S phase and mitosis. For example, endocycles in Drosophila, which consist of multiple rounds of S phase without intervening mitoses, also lack the G2 cyclins A and B (Lehner and O'Farrell, 1990). A second example is provided by meiosis, which consists of two mitotic-like states without intervening S phase and therefore can be seen as a cycle that lacks licensing. The decline in CDK activity at meiosis I is incomplete. If this residual activity is further reduced by dominant negative CDK expression, then meiosis I is followed by S phase rather than meiosis II (Furuno et al., 1994).
A role for CDKs in preventing rereplication can easily be incorporated into the view represented in Figure 1. We suggest that CDKs couple replication and the cell cycle in two ways. First, they prevent the recruitment of replication proteins to origins in G2, thereby preventing rereplication. This supposition has been supported by the demonstration that CDKs inhibit assembly of “prereplication centers” in Xenopus egg extracts (Adachi and Laemmli, 1994). Second, since CDK activity is required for S phase, the rise in CDK associated with entry into S phase would terminate the period during which replication proteins can assemble at origins.
In several circumstances, the state of a nucleus appears to be determined independently of the state of the cytoplasm. For example, under certain situations, adjacent nuclei in a syncytial Drosophila embryo can be in entirely different phases of the cell cycle (e.g., only one of four products of female meiosis enters interphase, and a local cluster of blastoderm nuclei can undergo an additional cycle to compensate for low nuclear density). And as shown in the work of Rao and Johnson (1970), when G1 and G2 cells are fused to each other, the two nuclei continue to behave differently. These results can be explained in terms of the kinase model if one assumes that nuclei constitute discrete domains of kinase activity. In this way, a nucleus could retain a particular level of kinase even when sharing cytoplasm with a nucleus that has a different level.
The Qualifications for License
The licensing model and the kinase model are at odds over one prediction. In the licensing model, acquisition of the license to replicate is coupled to mitosis by a requirement for nuclear membrane breakdown, while in the kinase model, the coupling is due to a requirement for a collapse in nuclear kinase activity. The genetic finding that removal of kinase during G2 leads to rereplication (in the absence of nuclear membrane breakdown) cannot easily be explained by the licensing model. But we suggest that the observation that nuclear membrane permeabilization allows rereplication in an extract system can be explained by the kinase model. If disruption of the nuclear membrane in the presence of egg extract destroys a nuclear compartment of kinase activity, then it could mimic the normal mitotic collapse of the nuclear kinase activity. We suggest that the licensing factor proposed by Blow and Laskey (1988) responds not to nuclear envelope breakdown but rather to CDK levels.
If the elimination of CDKs is what qualifies a nucleus for licensing, what role is played by the MCM proteins? We suggest that these proteins associate with origins and promote the assembly of functional initiation complexes and that their interaction with chromatin is prevented during S and G2 phases by CDK-mediated modifications. This proposal is based on the requirement for MCM proteins for replication initiation in yeast, their genetic interactions with a yeast origin-binding protein, and their differing phosphorylation and chromatin association between G1 and G2 phases in mammals.
If the proposed assembly of proteins at an origin corresponds to the license to replicate, then an analysis of such assembly could provide a physical assay for licensing. Interestingly, the DNA footprint observed at replication origins in yeast changes during the cell cycle (Diffley et al., 1994). During the S and G2 phases, the footprint is identical to that seen in vitro with purified origin recognition complex (ORC). During G1 phase, however, the ORC footprint is extended, indicating the presence of additional proteins. We suggest that this extension of the footprint during G1 corresponds to the MCM-dependent assembly of a licensed origin (Figure 1) and that the analysis of this process will define the events that qualify an origin for licensing.
References
- Adachi Y, Laemmli UK. J Cell Biol. 1992;119:1–15. doi: 10.1083/jcb.119.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi Y, Laemmli UK. EMBO J. 1994;13:4153–4164. doi: 10.1002/j.1460-2075.1994.tb06733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow JJ, Laskey RA. Nature. 1988;332:546–548. doi: 10.1038/332546a0. [DOI] [PubMed] [Google Scholar]
- Broek D, Bartlett R, Crawford K, Nurse P. Nature. 1991;349:388–393. doi: 10.1038/349388a0. [DOI] [PubMed] [Google Scholar]
- Chong JPJ, Mahbubani HM, Khoo CY, Blow JJ. Nature. 1995 doi: 10.1038/375418a0. in press. [DOI] [PubMed] [Google Scholar]
- Coxon A, Maundrell K, Kearsey S. Nucl Acids Res. 1992;20:5571–5577. doi: 10.1093/nar/20.21.5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley JFX, Cocker JH, Dowell SJ, Rowley A. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- Furuno N, Nishizawa M, Okazaki K, Tanaka H, Iwashita J, Nakajo N, Ogowa Y, Sagata N. EMBO J. 1994;13:2399–2410. doi: 10.1002/j.1460-2075.1994.tb06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayles J, Fisher D, Woollard A, Nurse P. Cell. 1994;78:813–822. doi: 10.1016/s0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- Hennessy KM, Clark CD, Botstein D. Genes Dev. 1990;4:2252–2263. doi: 10.1101/gad.4.12b.2252. [DOI] [PubMed] [Google Scholar]
- Hu B, Burkhart R, Schulte D, Musahl C, Knippers R. Nucl Acids Res. 1993;21:5289–5293. doi: 10.1093/nar/21.23.5289-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Nozaki N, Sugimoto K. EMBO J. 1994;13:4311–4320. doi: 10.1002/j.1460-2075.1994.tb06751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Mimura S, Nishimoto Si, Takisawa H, Nojima H. Cell. 1995;81:601–609. doi: 10.1016/0092-8674(95)90081-0. [DOI] [PubMed] [Google Scholar]
- Lehner CF, O'Farrell PH. Cell. 1990;61:535–547. doi: 10.1016/0092-8674(90)90535-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Herskowitz I. Science. 1993;262:1870–1874. doi: 10.1126/science.8266075. [DOI] [PubMed] [Google Scholar]
- Madine MA, Khoo CY, Mills AD, Laskey RA. Nature. 1995 doi: 10.1038/375421a0. in press. [DOI] [PubMed] [Google Scholar]
- Rao PN, Johnson RT. Nature. 1970;225:159–164. doi: 10.1038/225159a0. [DOI] [PubMed] [Google Scholar]
- Sauer K, Knoblich JA, Richardson H, Lehner CF. Genes Dev. 1995 doi: 10.1101/gad.9.11.1327. in press. [DOI] [PubMed] [Google Scholar]
- Springer PS, McCombie WR, Sundaresan V, Martienssen RA. Science. 1995;268:877–880. doi: 10.1126/science.7754372. [DOI] [PubMed] [Google Scholar]
- Treisman JE, Follette PJ, O'Farrell PH, Rubin GM. Genes Dev. 1995 doi: 10.1101/gad.9.14.1709. in press. [DOI] [PubMed] [Google Scholar]
- Usui T, Yoshida M, Abe K, Osada H, Isono K, Beppu T. J Cell Biol. 1991;115:1275–1282. doi: 10.1083/jcb.115.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]