

Abstract
We demonstrate the significance of peripheral benzodiazepine receptor (PBR) imaging in living mouse models of Alzheimer's disease (AD) as biomarkers and functional signatures of glial activation. By radiochemically and immunohistochemically analyzing murine models of the two pathological hallmarks of AD, we found that AD-like Aβ deposition is concurrent with astrocyte-dominant PBR expression, in striking contrast with nonastroglial PBR upregulation in accumulations of AD-like phosphorylated tau. Because tau-induced massive neuronal loss was distinct from the marginal neurodegeneration associated with Aβ plaques in these models, cellular localization of PBR reflected deleterious and beneficial glial reactions to tau versus Aβ pathologies, respectively. This notion was subsequently examined in models of various non-AD neuropathologies, revealing the following reactive glial dynamics underlying differential PBR upregulation: (1) PBR(−) astrogliosis uncoupled with microgliosis or coupled with PBR(+) microgliosis associated with irreversible neuronal insults; and (2) PBR(+) astrogliosis coupled with PBR(− or ±) microgliosis associated with minimal or reversible neuronal toxicity. Intracranial transplantation of microglia also indicated that nontoxic microglia drives astroglial PBR expression. Moreover, levels of glial cell line-derived neurotrophic factor (GDNF) in astrocytes were correlated with astroglial PBR, except for increased GDNF in PBR(-) astrocytes in the model of AD-like tau pathology, thereby suggesting that PBR upregulation in astrocytes is an indicator of neurotrophic support. Together, PBR expressions in astrocytes and microglia reflect beneficial and deleterious glial reactions, respectively, in diverse neurodegenerative disorders including AD, pointing to new applications of PBR imaging for monitoring the impact of gliosis on the pathogenesis and treatment of AD.
Keywords: neurodegenerative disorders, microglia, astrocyte, peripheral benzodiazepine receptor, glial cell line-derived neurotrophic factor, Alzheimer's disease
Introduction
A growing body of neuropathological evidence has demonstrated that concurrent microglial and astroglial activation accompanies neurodegeneration in Alzheimer's disease (AD) (Dickson et al., 1993; McGeer and McGeer, 2003) and other related disorders (Arnold et al., 2000; Nelson et al., 2002; Wojtera et al., 2005; Kim and Joh, 2006). In AD, excessive glial responses to the accumulation of Aβ are thought to augment progressive neuronal injury (Combs et al., 2000; Qin et al., 2002). Meanwhile, Aβ immunotherapies have been developed based on a mechanistic concept that induction of microglial responses contribute to the elimination of pathological Aβ aggregates (Schenk et al., 1999; Dodel et al., 2003). Thus, there may be a bifunctionality of reactive gliosis (Zilka et al., 2006; Fiala et al., 2007), but this notion remains elusive without the aid of appropriate biological markers reflecting deleterious and/or beneficial modes of glial responses.
Peripheral benzodiazepine receptor (PBR) (also known as translocator protein) was initially known to be expressed in activated microglia (Myers et al., 1991; Stephenson et al., 1995; Banati, 2002), although it has more recently been revealed that reactive astrocytes also exhibit noticeable levels of PBR (Chen et al., 2004; Maeda et al., 2007a; Rojas et al., 2007). Further, radiolabeled imaging agents have permitted sensitive detection of glial PBR when applied to in vitro autoradiographic and in vivo positron emission tomographic (PET) techniques. For example, [11C]PK11195 was the first to enable PET measurement of PBR in diverse CNS pathologies (Banati et al., 2000; Pappata et al., 2000; Cagnin et al., 2001; Rojas et al., 2007), but other radiolabeled ligands were developed, such as (N-5-fluoro-2-phenoxyphenyl)-N-(2-hydroxy-5-methoxybenzyl)acetamide (or DAA1106) with 11C and 18F resulting in the production of [11C]DAA1106 and [18F]fluoroethyl-DAA1106 ([18F]FE-DAA1106), respectively, to establish PET tracers suitable for imaging PBR in living brains (Zhang et al., 2003, 2004; Fujimura et al., 2006; Ikoma et al., 2007; Venneti et al., 2007).
Based on these technical progresses, the present study was aimed at elucidating the significance of PBR upregulation in microglia and astrocytes. The analyses were initiated by radiochemically and immunohistochemically examining two distinct models of AD, mutant amyloid precursor protein (APP) and tau transgenic (Tg) mice (Sturchler-Pierrat et al., 1997; Yoshiyama et al., 2007). The tau Tg mice show a progressive and substantial loss of neurons (Yoshiyama et al., 2007), in clear contrast to the APP Tg mice that show only minimal or no neuronal death (Van Dam et al., 2005). Moreover, our PBR imaging has demonstrated that gliosis accelerates tau-induced neurodegeneration (Yoshiyama et al., 2007), which contrasts with the amelioration of Aβ deposition by microglial activation in APP Tg mice (Maeda et al., 2007b). We thus presumed that the cellular profiles of PBR expression in tau and APP Tg mice could distinguish the ameliorative versus deleterious responses of microglia and astrocytes to accumulations of pathological Aβ and tau in AD. We also extended these studies by examining the general mechanism by which PBR is differentially upregulated in microglia and astrocytes using other experimental models of CNS injuries.
Materials and Methods
Reagents and antibodies.
The following reagents and all other chemicals were of analytical grade and commercially purchased: methamphetamine (METH), 6-hydroxydopamine (6-OHDA) and kainic acid (KA) from Wako Pure Chemicals; 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), horseradish peroxidase-conjugated isolectin B4 from Griffonia simplicifolia (HRP-ILB4), cuprizone, GBR12909 and PK11195 from Sigma-Aldrich.
We raised a rabbit polyclonal antibody against the C-terminal sequence of PBR using synthetic peptide spanning residues 155–169 of murine PBR (WRDNSGRRGGSRLAE). This antibody (NP155) was affinity-purified and characterized by immunoblotting as well as immunostaining for comparison with commercial anti-PBR antibodies (rabbit polyclonal, R&D Systems; rabbit polyclonal, BioVision; rabbit polyclonal, FL-169, Santa Cruz Biotechnology; goat polyclonal, W-12, Santa Cruz Biotechnology). Other antibodies used in this study are as follows: rabbit polyclonal antibody against ionized calcium binding adapter molecule-1 (Iba-1) (Wako Pure Chemicals), mouse monoclonal antibody against rat CD11b (OX42; AbD Serotec), rat monoclonal antibody against mouse CD11b (M1/70; BMA Biomedicals), rabbit polyclonal (Dako) and rat monoclonal (clone 2.2B10; Zymed/Invitrogen) antibodies against glial fibrillary acidic protein (GFAP), rat monoclonal antibody against myelin basic protein (MBP) (Millipore), rabbit polyclonal antibody against glial cell line-derived neurotrophic factor (GDNF) (Santa Cruz Biotechnology) and rabbit polyclonal antibody against tyrosine hydroxylase (TH) (Millipore).
Western blot analysis.
For evaluation of NP155, the microglial clone termed Ra2 was maintained in Eagle's minimum essential medium (M4655; Sigma-Aldrich) supplemented with 10% fetal bovine serum, 5 μg/ml bovine insulin, 0.2% glucose and 1 ng/ml murine granulocyte-macrophage colony-stimulating factor (G0282; Sigma-Aldrich), as described previously (Sawada et al., 1998). For biochemical analyses, the cells (5 × 106) were scraped and homogenized in 50 mm Tris-HCl, pH 7.4, 4°C, containing 0.1% protease inhibitor mixture (5 mm leupeptin, 1 mg pepstatin and 5 mg aprotinin in 1 ml dimethyl sulfoxide) and 0.5 mm phenylmethylsulfonyl fluoride (PMSF). The suspension was centrifuged at 12,000 rpm (10,000 × g) for 15 min, and the resultant pellet was resuspended in 50 mm Tris-HCl buffer. The protein amounts in the samples were measured according to Lowry's method. For immunocytochemistry, the cells were cultured on cover glasses for 7 d, and washed with PBS, followed by fixation with 4% paraformaldehyde for 20 min.
Hippocampal samples from KA-treated and untreated rats were homogenized in 50 mm Tris-HCl, pH 7.4, 4°C, containing 0.1% protease inhibitor mixture (as in experiment for Ra2 cells) and 0.5 mm PMSF. The suspended homogenates of cultured microglial (Ra2) cells and rat hippocampal tissues, corresponding to 10 and 100 μg protein, respectively, were applied to a 15% SDS polyacrylamide gel. After electrophoresis and transfer of proteins to a polyvinylidene fluoride membrane (Immobilon-P; Millipore), the membrane was immersed in Tris-buffered saline (150 mm NaCl, 10 mm Tris-HCl, pH 8.0) containing 0.05% (v/v) Tween 20 and 3% (w/v) bovine serum albumin (BSA), and then reacted for 1 h with anti-PBR antibodies in TBS containing 0.05% (v/v) Tween 20 and 0.1% (w/v) BSA. The primary antibodies were detected by HRP-conjugated anti-IgG antibodies (GE Healthcare) and enhanced chemiluminescence method (GE Healthcare).
Animal models.
The mice studied here were maintained and handled in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals and our institutional guidelines. Protocols for the present animal experiments were approved by the Animal Ethics Committees of the National Institute of Radiological Sciences and Central Institute for Experimental Animals (CIEA).
APP Tg mice termed APP23, which overexpress the Swedish doubly mutant APP751 under the control of a neuron-specific Thy-1 promoter element, were developed as described in detail previously (Sturchler-Pierrat et al., 1997), and were maintained on a C57BL/6J background. Tau Tg mice dubbed PS19 were generated by using a cDNA coding a tau isoform containing 1 N-terminal and 1 C-terminal alternatively spliced exons and the P301S MAPT mutation discovered in frontotemporal dementia with parkinsonism linked to chromosome 17 in combination with a murine prion protein promoter (Yoshiyama et al., 2007). The strain was maintained on a B6C3H background. Littermates were used as controls.
Models of KA-induced excitotoxicity were generated by intraperitoneally administering KA (12 mg/kg) to male Sprague Dawley rats (Japan SLC, Hamamatsu) at 8 weeks of age. All rats analyzed here presented visible cramps within 1 h of KA injection. At 1 week after KA challenge, the animals were deeply anesthetized with sodium pentobarbital and transcardially perfused with PBS, and brain tissues were removed. One hemisphere was immediately frozen with dry ice and stored at −80°C pending assays. The other hemisphere was fixed with 4% paraformaldehyde in phosphate buffer.
Selective injury to the nigrostriatal dopaminergic system was caused in mice, rats and marmosets by the use of METH, 6-OHDA and MPTP, respectively. Male in-house C57BL/6J mice (Kito et al., 2003) aged 8 weeks were subcutaneously injected with METH (5 mg/kg) 4 times, 2 h apart, and brain tissues of these mice were obtained at 2 and 7 d after treatment, as in the protocol for KA-injected rats. Lesioning of rat brains was performed as described previously (Inaji et al., 2005). Briefly, male Sprague Dawley rats (Japan SLC) at 8 weeks of age were anesthetized with intraperitoneally administered sodium pentobarbital (60 mg/kg) and placed in a stereotactic frame (Narishige). Four microliters of 2 μg/μl 6-OHDA, freshly dissolved in physiological saline containing 0.1% ascorbic acid, was subsequently injected into the right medial forebrain bundle (stereotactic coordinates: anteroposterior, 4.2 mm from the bregma; mediolateral, 1.8 mm from the midline; and dorsoventral, 7.8 mm below the dura mater) of each rat at a rate of 1.0 μl/min. The animals were lethally anesthetized with sodium pentobarbital at 1 week after treatment, and brain samples were prepared as in the protocol for KA-injected rats. The MPTP challenge was performed using 10 male and 10 female common marmosets (Callithrix jacchus), either in-house bred at CIEA or obtained from CLEA Japan, as in our previous study (Ando et al., 2008). These animals, initially aged 3.8 ± 2.5 years and weighing 316.3 ± 39.6 g, were divided into 2 experimental groups (n = 5 per group) matched for sex, body weight and age. The treatment group consisted of marmosets receiving 3 doses of subcutaneous MPTP administration (2 mg/kg), 24 h apart, and they were killed by deep anesthetization with sodium pentobarbital at 2 weeks. Their brains were immediately extirpated, frozen with dry ice, and stored at −80°C pending assays. Brains of marmosets in the untreated group were also collected for use as controls.
We also generated a mouse model of massive demyelination by continuously feeding male in-house bred C57BL/6J mice aged 8 weeks with a powdered diet containing 0.2% (w/w) cuprizone, as described previously (Chen et al., 2004). Brain tissues were obtained from these animals at 4 weeks of treatment, as in the protocol for KA-injected rats.
Microglial cell cultures and transplantation.
Collected Ra2 cells were labeled with fluorescent dye, PKH26 (MINI26; Sigma-Aldrich), as described previously (Imai et al., 2007), and the final cellular concentration was adjusted to 100,000 cells/μl medium. Male C57BL/6J mice were anesthetized with 1.5% (v/v) isoflurane and were placed in a stereotactic frame (Narishige). Using a 10 μl Hamilton syringe, 2 μl of cell suspension and medium alone were injected into the right and left hippocampi, respectively (stereotactic coordinates: anteroposterior, −2.8 mm; mediolateral, 2.0 mm; dorsoventral 2.0 mm from the bregma), over 4 min. The needle was left in place for 2 min before being withdrawn. These mice were killed 1 week after the procedure, and brain sections were prepared as in the protocol for KA-injected rats.
Immunocytochemical, immunohistochemical and histochemical analyses.
For immunohistochemical assays, the entire brains of animals fixed with 4% paraformaldehyde were cryoprotected using 30% sucrose in phosphate buffer, and 10- or 20-μm-thick frozen sections 40 μm apart were generated in a cryostat (HM560; Carl Zeiss). Alternatively, frozen brain tissues were sliced into 20 μm sections for autoradiographic analysis of dopamine transporter. The brain sections were immunostained based on a standard protocol using either fluorophore-conjugated secondary antibodies (Invitrogen) or a commercial kit for avidin-biotin-diaminobenzidine staining (ABC Staining Systems; Santa Cruz Biotechnology). The sections used for PBR staining were beforehand autoclaved in citric buffer for antigen recovery. Similarly, immunofluorescence staining of Ra2 cells on a cover glass was performed.
Aβ plaques in APP23 mice were visualized by using 0.01% (E,E)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB; Dojindo Laboratories), a fluorescent dye for amyloid fibrils (Maeda et al., 2007b).
Microglial cells in the brain were also histochemically visualized by isolectin labeling. Brain sections were reacted with HRP-ILB4 (10 mg/ml) overnight at 4°C and excess reagent was eliminated by 3 washes in PBS (5 min each). The signals were developed by 3,3′-diaminobenzidine (DAB).
Quantitative analyses were performed using at least 5 slices covering each region of interest (ROI). All stained sections were examined by a confocal laser scanning microscope (FV1000; Olympus) or an all-in-one microscope/digital camera (BZ-9000; Keyence), and photomicrographs captured at 10× (FV1000) or 20× (BZ-9000) were semiautomatically tiled and merged into a large high-resolution image containing the whole ROI. Signal intensities were initially measured for the whole ROI, followed by determination of the overall maximum intensity. A cutoff threshold of the intensity was then assigned for each section as [(background intensity) + (5% of overall maximum intensity)] with additional manual tuning. Areas showing signal intensities above the threshold were calculated for quantification of GFAP-positive astrocytes, and were expressed as ratios against the total area of the ROI. Similarly, areas were estimated for other sections stained with antibodies against PBR, MBP and GDNF. For quantification of Iba-1-immunoreactive microglia, cells containing a hematoxylinophilic nucleus and cytoplasmic Iba-1 signals above the cutoff threshold were counted and expressed as the number per unit area (mm2). Finally, images for which the quantitative value was close to the group average were selected as representative illustrations in the figures. All these analyses were performed with MetaMorph 6.1 (Universal Imaging) and Photoshop 7.0 (Adobe Systems) software.
Radiosynthesis and autoradiography.
Synthesis of 11C-labeled N-(3-iodoprop-2E-enyl)-2β-carbomethoxy-3β-(4-methylphenyl)nortropane ([11C]PE2I), a PET ligand for dopamine transporter, was conducted by O-methylation of its free acid precursor with [11C]methyl triflate according to previously described methods (Halldin et al., 2003). The radiochemical purity of the resultant compound was >99%, and the specific radioactivity was 219.2 ± 38.8 GBq/μmol at the end of synthesis.
[18F]FE-DAA1106 was radiosynthesized using its desmethyl precursor, DAA1123 (generously provided by Taisho Pharmaceutical), based on a previously described protocol (Zhang et al., 2003, 2004). The radiochemical purity of the end product exceeded 95%, and the specific radioactivity was 120 ± 20.5 GBq/μmol at the end of synthesis. [3H]DAA1106 was synthesized by O-methylation of DAA1123 with [3H]methyliodide (specific radioactivity, 3.15 TBq/mmol; GE Healthcare). The reaction product was purified as described previously (Maeda et al., 2007a; Yoshiyama et al., 2007).
In vitro autoradiography of dopamine transporters was performed using unfixed frozen brain sections and [11C]PE2I, in accordance with the established procedure (Inaji et al., 2005). The sections were preincubated in 50 mm Tris-HCl buffer, pH 7.4, for 30 min at room temperature, followed by reaction with [11C]PE2I (18.5 MBq/L, ∼20 pm) in 50 mm Tris-HCl buffer, pH 7.4, for 60 min at 25°C. Nonspecific binding of the radioligand was determined by adding a nonradioactive ligand, GBR12909 (10 μm), to the reaction. The samples were then rinsed twice with ice-cold Tris-HCl buffer for 2 min, dipped into ice-cold water for 10 s, warmly blow-dried and contacted to an imaging plate (BAS-MS2025; Fuji Film) for 1 h. Radiolabeling was detected by scanning the imaging plate by means of the BAS5000 system (Fuji Film). Autoradiographic assay for PBR was conducted using paraformaldehyde-fixed frozen sections. The fixation did not affect the binding of this PBR ligand in our preliminary experiments (data not shown). The samples were incubated with [18F]FE-DAA1106 (18.5 MBq/L, ∼0.35 nm) or [3H]DAA1106 (2.4 MBq/L, ∼0.5 nm) in 50 mm Tris-HCl. Nonspecific binding of the radioligand was estimated by adding nonradioactive PK11195 (10 μm) to the reaction. The rest of the protocol was as described above. The samples used for [3H]DAA1106 autoradiography were then coated with warm photographic emulsion (EM-1; GE Healthcare) and exposed, as described in detail previously (Maeda et al., 2007a). The developed emulsion autoradiograms were examined with a microscope (AX-80; Olympus).
Small animal PET imaging.
PET scans were performed using a microPET Focus 220 animal scanner (Siemens Medical Solutions) as described previously (Maeda et al., 2007b). Control and cuprizone-administered mice were anesthetized with 1.5% (v/v) isoflurane, and a 30-G needle connected to a 1 ml polypropylene syringe via a length of polyethylene tubing was inserted into the tail vein. After transmission scans for attenuation correction using a 68Ge-68Ga point source, emission scans were acquired for 60 min in a 3D list mode with an energy window of 350–750 keV, and intravenous injection of [18F]FE-DAA1106 (13.26 ± 14.27 MBq) was performed immediately. Summation images from 0 to 60 min after [18F]FE-DAA1106 injection were reconstructed with maximum a posteriori reconstruction, and dynamic images were reconstructed with filtered back-projection using a 0.5 mm Hanning filter. Volumes of interest (VOIs) were placed on striatal areas including the corpus callosum using PMOD image analysis software (PMOD Group) with reference to the MRI template. Tracer uptake in each VOI was estimated as percentage of injected dose per tissue volume (%ID/ml).
Statistical analysis.
Statistical analyses for group comparisons were performed by Student's t test or ANOVA followed by Bonferroni's post hoc test. Difference between groups was considered significant when the p value was <0.05.
Results
Distinct cellular localizations of PBR in models of Aβ and tau pathologies
Application of [18F]FE-DAA1106 to in vitro autoradiographic imaging of brain slices generated from 20-month-old APP23 mice illustrated intense PBR accumulation primarily in the hippocampus and entorhinal cortex (Fig. 1A), and subsequent immunostaining of the same sections revealed excellent agreement between PBR radiolabeling and Aβ-positive plaque lesions (Fig. 1B,C). PBR upregulation associated with Aβ deposition was also demonstrated at a higher resolution by microautoradiography using [3H]DAA1106 (Fig. 1D) followed by fluorescence staining with FSB (Fig. 1E). High-power photomicrographs for PBR and FSB indicated that the majority of PBR clusters appeared in an annular or semiannular shape outlining the Aβ deposits (Fig. 1F–I). Relative to age-matched WT controls (Fig. 1J), PBR was also markedly upregulated in the hippocampus and entorhinal cortex of 9-month-old PS19 Tg mice (Fig. 1K) in close spatial association with the accumulation of pathologically phosphorylated tau (Fig. 1L). Despite the similarity of regional PBR distributions in the two Tg mouse models, however, the diffuse radiolabeling in PS19 mice concurrent with pronounced hippocampal and entorhinal atrophy and ventricular dilatation contrasted sharply with the patchy and heterogeneous radiographic pattern in the APP23 Tg mice, wherein there was no overt neuronal loss.
Figure 1.

A–C, Glial response to plaque deposition as demonstrated by autoradiography for PBR with [18F]FE-DAA1106 in a brain section of a 20-month-old APP23 mouse (A) followed by Aβ immunolabeling (B; merged image is shown in C). D–I, Pathology in a 20-month-old APP23 mouse detected by emulsion microautoradiography of [3H]DAA1106 (D, F, H) and FSB (E), resulting in double labeling of PBR and plaque amyloid (G, I). Photomicrographs are displayed at low (D, E), middle (F, G) and high (H, I) magnifications. Correspondence between radiolabeled and fluorolabeled lesions in the hippocampus and neocortex is indicated by arrows and arrowheads, respectively. J–L, Brain sections of 9-month-old WT (J) and PS19 (K, L) mice radiolabeled with [18F]FE-DAA1106 (J, K) and immunostained with antibody against phosphorylated tau (L). PS19 mouse exhibited PBR positivity and phospho-tau accumulation in the hippocampus and entorhinal cortex, concurrent with marked atrophy of these structures and ventricular dilatation (asterisks in K, L). High-power view of hippocampal CA1 sector is shown in inset of L. Scale bars: (A–E, J–L) 1 mm, (F, G) 500 μm, (H, I) 100 μm, (inset in L) 50 μm.
Glial subpopulations expressing PBR in these animals were further identified with the aid of NP155, our newly developed anti-PBR antibody, which was shown to detect rodent PBR with a sensitivity and specificity superior to that of the commercially available antibodies (supplemental information; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Double immunofluorescence staining of GFAP and PBR clearly indicated predominant localization of PBR signals to astrocytes in the vicinity of Aβ plaques in APP23 mice (Fig. 2A–C). Although Iba-1 immunostaining clearly captured the accumulation of activated microglia encompassing amyloid plaques in these mice (Fig. 2D), PBR signals were barely detectable in microglia based on double labeling with NP155 and anti-CD11b antibody M1/70 (Fig. 2D, inset). In striking contrast to the glial response to this Aβ pathology, buildup of phospho-tau in the PS19 mice was accompanied by a pronounced nonastroglial PBR expression because PBR was undetectable in most GFAP-labeled astrocytes (Fig. 2E–G). Meanwhile, numerous amoeboid-shaped Iba-1-immunoreactive microglia were present in the corresponding region (Fig. 2H), and double immunofluorescence staining with NP155 and M1/70 demonstrated that a significant subset of CD11b-positive microglia expressed PBR at a detectable level (Fig. 2H, inset) activated microglia are the primary source of PBR signals in PS19 mice showing tau pathology. Unlike these differential profiles of PBR expression triggered by Aβ and tau lesions, GDNF was upregulated in astrocytes of both models of AD-like plaque and tangle pathology, but GDNF upregulation was insufficient to protect neurons from tau-mediated neurotoxicity in the PS19 mice (Fig. 2I–L).
Figure 2.
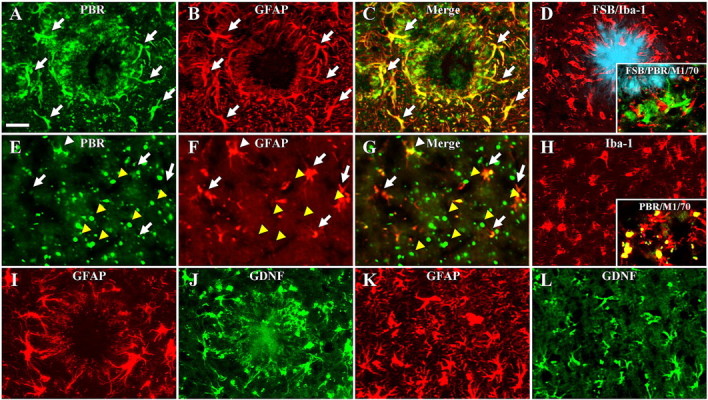
A–D, Glial activation and PBR expression encompassing amyloid plaque in a 20-month-old APP23 mouse. PBR signals (A) were predominantly detected in activated astrocytes (B; 2-channel image in C), although prominent microgliosis (Iba-1; red in D) encircling FSB-positive plaques (blue in D) was observed. Triple staining with FSB (blue in inset of D), NP155 (green in inset of D) and anti-mouse CD11b antibody (M1/70; red in inset of D) also indicated that the majority of plaque-associated microglia did not exhibit intense PBR signals. E–G, Nonastroglial PBR abundantly found in the hippocampus of a 9-month-old PS19 mouse. Double immunostaining for PBR (E) and GFAP (F) indicated that the majority of astrocytes did not express PBR at a detectable level (arrows), and colocalization of PBR and GFAP was observed only in a small subset of PBR-positive astrocytes (white arrowhead; 2-channel image in G), and intense dot-shaped PBR signals (yellow arrowheads) were localized to nonastroglial compartments. H, The same region immunostained for Iba-1, showing numerous microglia with amoeboid shapes distinct from the ramified morphology of resident cells. Double staining with NP155 (green in inset of H) and M1/70 (red in inset of H) revealed that a subset of CD11b-immunoreactive microglia expressed manifest PBRs. I–L, High-level expression of GDNF (J, L) in nearly all GFAP-immunoreactive astrocytes (I, K) reacting to plaque deposition in APP23 mice (I, J) and tau-induced neurotoxicity in PS19 mice (K, L). Scale bars: (A–L) 20 μm, (inset in H) 40 μm.
Astrogliosis without prominent PBR upregulation accompanied by PBR-positive microgliosis in response to excitotoxic insults
We then analyzed diverse experimental models of non-AD neuropathologies to assess PBR expression in glial subtypes after diverse neuronal injuries. As observed in the characterization of NP155 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), marked upregulation of PBR was detected in the midbrain as well as hippocampal/entorhinal areas of KA-treated rats. The midbrain regions of these animals were studied further, because different groups of myelinated and unmyelinated neurons in this anatomical structure are present in a well compartmentalized configuration (Fig. 3A,B). Accumulation of Iba-1-immunoreactive microglia was primarily confined to the necrotic core in the substantia nigra pars reticulata and was encircled by GFAP-positive astrocytes, with only a small spatial overlap between distributions of these two glial subpopulations (Fig. 3C–E). Detailed analysis of this lesion by double immunofluorescence staining with NP155 and anti-GFAP antibody revealed that the majority of astrocytes encompassing the microglia-enriched inflammatory epicenter were PBR-negative (Fig. 3F–J). Meanwhile, prominent PBR expression by activated microglia was demonstrated by double immunohistochemical staining with OX42 and NP155 (Fig. 3F–H,K–M, arrowheads). Double labeling also indicated that activated microglia were concentrated at the site of prominent demyelination (supplemental Fig. 2A,D, available at www.jneurosci.org as supplemental material) associated with astrogliosis in the perimeter surrounding this pathology (supplemental Fig. 2B,E, available at www.jneurosci.org as supplemental material). Interestingly, the loss of TH-positive, unmyelinated dopamine neurons in the substantia nigra pars compacta (supplemental Fig. 2C,F, available at www.jneurosci.org as supplemental material) was accompanied by astrogliosis (supplemental Fig. 2E, available at www.jneurosci.org as supplemental material) minimally immunoreactive for PBR but not by any associated microgliosis (supplemental Fig. 2D, available at www.jneurosci.org as supplemental material). Hence, the excitotoxicity-induced loss of myelinated neurons was tightly linked to PBR-positive microgliosis, whereas damage to unmyelinated neurons did not overtly stimulate microglial activity, both neurotoxic conditions being coexistent with astrogliosis lacking intensified PBR expression.
Figure 3.
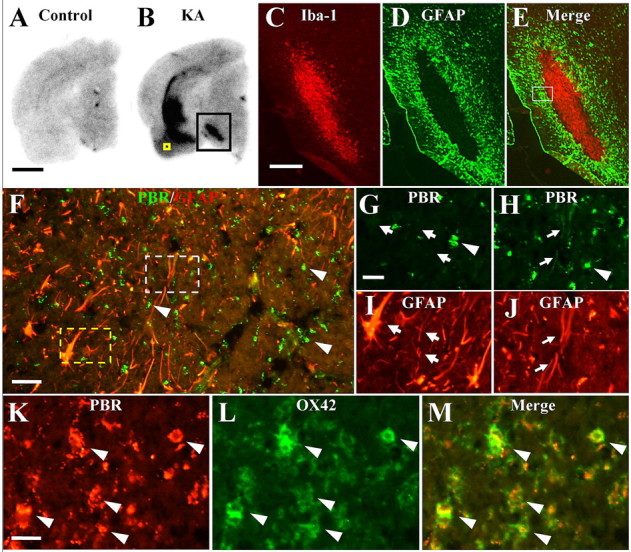
A, B, Autoradiographic labeling of PBR in brain sections of untreated control (A) and KA-treated (B) rats. Black square indicates intense radiolabeling with [18F]FE-DAA1106 in the midbrain, and immunolabeling of gliotic changes in this area is shown in C–E at high magnification. High-power photomicrographs of the entorhinal area indicated by yellow square in B are also shown in K–M for the purpose of clarifying the cellular localization of PBR. C–E, Double immunofluorescence labeling of Iba-1 (C) and GFAP (D) along with two-channel image (E) in the midbrain of KA-treated rat. The section is subadjacent to the sample shown in B. The square indicates a subregion with marked astrogliosis shown in F at high magnification. F–J, Midbrain of KA-treated rat doubly labeled with NP155 (F–H; green) and anti-GFAP (F, I, J; red) antibodies. Broken yellow and white lines delineate areas presented in G and I and H and J, respectively, at high magnification. The majority of astrocytes did not exhibit PBR signals at a visible level (arrows in G, I), whereas a small subset of GFAP labeling overlapped with weak PBR immunoreactivity (arrows in H, J). Round-shaped PBR immunolabeling (arrowheads in G, H) was not colocalized with GFAP staining and was conceived to be microglial. K–M, Double immunolabeling for PBR (K) and CD11b (L) illustrating speckled PBR staining (arrowheads) packed in microglia (2-channel image shown in M). Scale bars: (A, B) 2 mm, (C–E) 500 μm, (F) 40 μm, (G–J) 20 μm, (K–M) 10 μm.
PBR-negative astrogliosis is uncoupled from prominent microgliosis in injuries of nigrostriatal dopaminergic terminals
To determine if gliosis in response to disruption of the nigrostriatal dopaminergic system is characterized by accumulation of PBR-negative astrocytes and lack of pronounced microglial activation, we examined striatal pathologies in rodent models of neurochemical toxicities. The recruitment of PBR-negative astrocytes after the loss of dopaminergic terminals in the striatum was demonstrated in animals showing selective damage of dopamine neurons. Mice undergoing repeated administrations of METH exhibited progressive loss of striatal dopaminergic terminals up to 7 d after the initiation of treatment, as assessed by autoradiography of dopamine transporter using [11C]PE2I (Fig. 4A). Meanwhile, elevation of striatal PBR levels was minimal and insignificant according to autoradiographic assay with [18F]FE-DAA1106 (Fig. 4B). Histological examinations revealed the activity profiles of glial subtypes, indicating prominent GFAP-positive astrogliosis (supplemental Fig. 3A,B,G, available at www.jneurosci.org as supplemental material) with very modest microglial alterations (supplemental Fig. 3C–F,H, available at www.jneurosci.org as supplemental material). Despite marked increase of GFAP immunoreactivity, PBR upregulation was not visible in the striatal astrocytes throughout the observation period (Fig. 4C–H). We also immunohistochemically analyzed gliotic changes in the striatum of 6-OHDA-injected rats, which developed profound degeneration of the nigrostriatal dopaminergic system (Inaji et al., 2005). As seen in the METH challenge, GFAP signals in the lesioned striatum were substantially increased relative to the contralateral control (supplemental Fig. 3I–K, available at www.jneurosci.org as supplemental material) with the absence of overt Iba-1-positive microgliosis (supplemental Fig. 3L–N, available at www.jneurosci.org as supplemental material). This astrogliotic reaction did not coincide with either immunohistochemically (Fig. 4I–L) or autoradiographically (mean ± SE, 100.0 ± 3.6% vs 100.8 ± 6.8% of contralateral mean for contralateral vs ipsilateral striata; p > 0.05 by t test; n = 4 per group) detectable elevation of PBR levels. Similarly, the intensity of PBR radiolabeling in the striatum of MPTP-treated marmosets did not significantly differ from that of untreated controls (mean ± SE, 100.0 ± 4.4% vs 84.8 ± 4.2% of control for control vs MPTP-treated groups; p > 0.05 by t test; n = 5 per group), notwithstanding the severe impairments of striatal dopaminergic terminals (Ando et al., 2008). Together, these findings demonstrate that astroglial PBR in the striatum is not upregulated when the injury provokes no significant activation of striatal microglia.
Figure 4.

A, B, Autoradiographic analyses of dopamine transporter (A) and PBR (B) in striatal regions of mice without treatment and 2 and 7 d after initiation of METH treatment (n = 6 per group). Significant decline of [11C]PE2I binding to dopamine transporter was observed after METH administration (**p < 0.01 by Bonferroni's multiple comparison after ANOVA), whereas changes in binding of [18F]FE-DAA1106 to PBR were insignificant (p > 0.05 by ANOVA). Error bars represent SE. Representative autoradiograms are displayed on top. C–H, Double immunofluorescence staining with anti-GFAP antibody (C–E) and NP155 (F–H) in striatal sections of mice without treatment (left) and 2 (middle) and 7 (right) d of METH challenge. I–L, Double immunofluorescence staining with anti-GFAP antibody (I, J) and NP155 (K, L) in contralateral (left column) and ipsilateral (middle column) striata of 6-OHDA-treated rats. Error bars in graphs represent SE. Scale bars, (C–L) 75 μm.
Demyelinating but reversible pathology of the striatum leading to synergistic activation of astrocytes with high-level PBR and microglia with low-level PBR
On the assumption that PBR-positive astrocytes and PBR-negative microglia may reflect reversible neuropathological processes, we investigated mice at a subchronic stage of treatment with copper chelator cuprizone (CZ), which is known to induce recoverable loss of myelin (Chen et al., 2004). Oral administration of cuprizone for 4 weeks resulted in concentrations of PBR in the striatum and corpus callosum (Fig. 5A) (mean ± SE, 100.0 ± 13.2% vs 216.6 ± 1.9% of control for control vs CZ-treated striata; p < 0.01 by t test; n = 3 per group), which were spatially consistent with abundant accumulations of GFAP-positive astrocytes and Iba-1-positive microglia (Fig. 5B–E). The Iba-1 immunostaining in the striatum was intensified in patchy clusters (Fig. 5E), and double immunolabeling for MBP and Iba-1 illustrated that these clusters were composed of activated microglia associated with destroyed myelinated fibers (Fig. 5F). Unlike microglial activation, astrogliosis was not confined to the site of demyelination (Fig. 5G). As seen in a low-power image (supplemental Fig. 1G, available at www.jneurosci.org as supplemental material), NP155-labeled PBR signals were diffusely distributed in the striatum of cuprizone-treated mice, which resembled the spatial profile of activated astrocytes and was in clear contrast to the microgliosis locally restricted to demyelinated fiber bundles. At a high magnification, the vast majority of astrocytes exhibited distinct upregulation of PBR, and PBR signals were almost exclusively present in the astroglial compartment (Fig. 5H).
Figure 5.

A, Radiolabeling of PBR in brains of mice without treatment (Control) and treated with CZ. The corpus callosum (CC) and striatum (ST) are outlined by dots. B–E, Immunofluorescence staining of GFAP (B, D) and Iba-1 (C, E) in brain sections of untreated (B, C) and CZ-treated (D, E) mice. Dashed lines define borders of CC and ST. F–H, Double immunofluorescence labeling of MBP and Iba-1 (F), MBP and GFAP (G), and PBR (NP155) and GFAP (H) in striatal sections of a CZ-treated mouse. Arrowheads in H indicate astrocytes doubly positive for PBR and GFAP. I–K, In vivo detection of PBR in CZ-treated mice. Coronal (1 mm anterior to the bregma) PET images in control (I) and CZ-treated (J) mice were generated by summation of dynamic data at 0–60 min after injection of [18F]FE-DAA1106 and were overlaid on the MRI template. Marked increase of radioactivity was found primarily in the corpus callosum and striatum (arrows). Time-radioactivity curves (K) for striatal VOIs in control (open circles) and CZ-fed (closed circles) mice indicate ∼2-fold increase of radioligand accumulation induced by CZ treatment. Scale bars: (A) 2 mm, (B–E) 150 μm, (F–H) 25 μm.
We then conducted in vivo detection of PBR in these mice by using microPET and [18F]FE-DAA1106 to clarify the detectability of astroglial PBR in living brains. CZ-treated mice showed a profoundly increased concentration of radiotracer in the striatal area (Fig. 5I,J), ∼2-fold that in control mice (Fig. 5K), corresponding well with the in vitro autoradiographic data (Fig. 5A). Thus, our findings provide the first clear evidence for the ability of PET and specific radioligand to capture PBR upregulation in astrocytes.
Induction of astroglial PBR by transplantation of immortalized microglia
The mechanistic links between activated microglia and PBR expression in astrocytes were assessed by immunohistochemically assaying PBR levels in the mouse hippocampus transplanted with the PKH26-labeled immortalized microglial cell line, Ra2 (Fig. 6A), which has been shown to be neuroprotective and reactive oxygen species- (ROS) nonproducing (Sawada et al., 2006). Compared with the vehicle-injected side (Fig. 6B–E), the Ra2-injected hippocampus exhibited astrocytes with hypertrophic morphology in close proximity to the grafted microglia, and a high-level expression of PBR was observed in these astrocytes (Fig. 6F–I). Compared with the vehicle-injected side (Fig. 6J), pronounced activation of resident (Iba1-positive, PKH26-negative) microglia was found in the proximity of the transplanted (PHK-positive) Ra2 cells (Fig. 6K–M). As PBR signals showed nearly complete overlap with GFAP immunoreactivity in this area, these activated endogenous microglia were presumed to be PBR-negative. Neuronal damage and loss of myelin were negligible in these areas. Because the hippocampal subregions analyzed here were sufficiently distant from needle tracks (Fig. 6A) and were consequently little affected by traumatic insults, these data demonstrate that activated microglia without evident neurotoxicity can act as the sole initiator of PBR upregulation in nearby astrocytes, and imply the presence of mechanistic links between activations of PBR-negative endogenous microglia and PBR-positive astrocytes.
Figure 6.
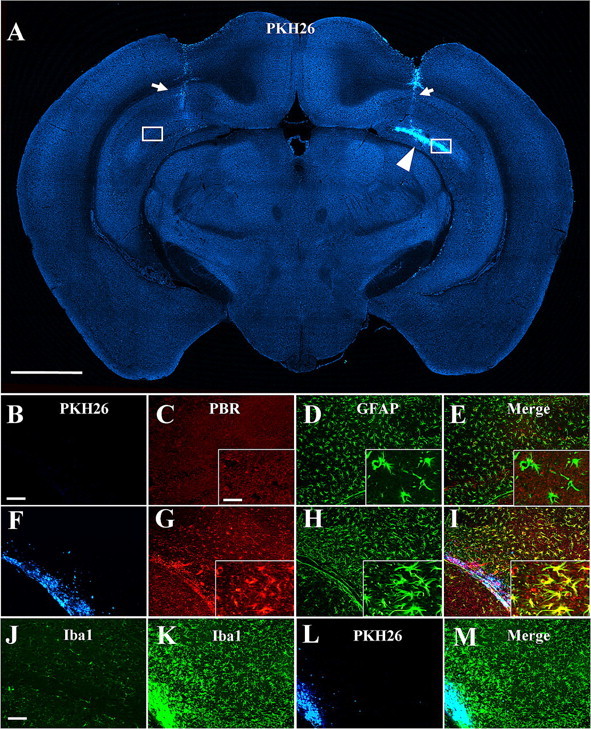
A, Localization of PKH26-labeled Ra2 cells transplanted into unilateral hippocampus of a mouse (arrowhead). Regions of interest (squares) were defined in hippocampal areas sufficiently distant from needle tracts (detectable by weak autofluorescence; arrows) and are shown in B–M at high magnification. Fluorescence of PKH26 is converted to blue for the purpose of the multicolor display. B–I, Fluorescence labeling for exogenous microglia (B, F), PBR (C, G) and GFAP (D, H) along with three-channel images (E, I) in the subregions of the hippocampus injected with vehicle (B–E) and Ra2 (F–I). Transplanted Ra2 cells (F) induced prominent upregulation of PBR (G) as well as hypertrophic changes (H) in neighboring astrocytes. High-power photomicrographs are shown in insets (C–E, G–I). J–M, Fluorescence mapping of microglia positive for Iba1 (J, K) and PHK26 (L) along with a merged image (M) in the subregions of the hippocampus injected with vehicle (J) and Ra2 (K–M). Iba1-positive, PKH26-negative endogenous microglia were manifestly activated in the proximity of the transplantation site containing Iba1/PKH26 double positive exogenous Ra2 cells (K–M), compared with the vehicle-injected contralateral area (J). Scale bars: (A) 1 mm, (B–I) 100 μm, (J–M) 25 μm, (insets) 20 μm.
Augmented GDNF expression in PBR-positive astrocytes
To supplement the evidence for the protective and restorative roles of PBR-expressing astrocytes, we immunohistochemically examined astroglial GDNF in the experimental models beside those of AD. Double fluorescence staining for control rodents illustrated the expression of GDNF in resident astrocytes at a moderate level in the hippocampus (Fig. 7A,B, insets) and at a very low level in the striatum (Fig. 7E,F,I,J) and midbrain (Fig. 7M,N) of mice and rats. A notable increase of astroglial GDNF immunoreactivity was found in the Ra2-transplanted hippocampus (Fig. 7C,D) and CZ-treated striatum (Fig. 7G,H), in which remarkably upregulated PBR in astrocytes was featured. Striatal astrocytes reacting to injuries of dopaminergic terminals exhibited no substantial increase in GDNF levels (Fig. 7K,L), in concordance with the unaltered PBR signals. Activated astrocytes in the midbrain subregion surrounding inflammatory microglia also did not express GDNF at a clearly detectable level (Fig. 7O,P), consistent with the finding that only a tiny subset of these cells was PBR-immunoreactive. Rearrangement of the data for side-by-side comparison of PBR and GDNF immunolabeling in these animal models (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) revealed good agreement between the expression profiles of the two molecules in astrocytes in these models of acute/subacute neurotoxicity.
Figure 7.
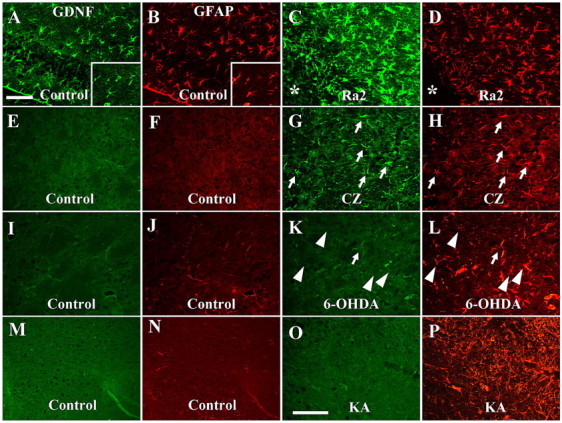
Expression of GDNF in astrocytes assessed by double immunofluorescence staining for GDNF (green) and GFAP (red). A–D, Mouse hippocampi injected with vehicle (A, B) and Ra2 cells (C, D). Relative to astrocytes on the vehicle-injected side, the number, shape and GDNF levels of which were comparable with untreated condition (insets in A, B), astrocytes in the vicinity of the transplantation site (asterisks in C, D) exhibited pronounced activation accompanied by GDNF upregulation. E–H, Striata of mice without treatment (E, F) and at 4 weeks of CZ challenge (G, H). E–H, GDNF- and GFAP-immunoreactivities in the control striatum (E, F) were much less than those in the hippocampus (insets in A, B), whereas markedly increased astrocytes in this region expressed GDNF at a significant level (arrows in G, H). I–L, Striata of control (I, J) and 6-OHDA-treated (K, L) rats. GDNF signals were undetectable in the vast majority of activated astrocytes in the treated rat (arrowheads), and only a limited subset of these cells showed GDNF expression slightly above the detection threshold (arrows). M–P, Midbrain of control (M, N) and KA-treated (O, P) rats. P, O, KA induced prominent astrogliosis (P) with only slight GDNF expression (O) in the region outlining the inflammatory core. Scale bars: (A–L) 50 μm, (M–P) 50 μm.
Discussion
The elevated levels of PBR observed in tau and APP Tg mice that model the core tangle and plaque pathologies of AD by both radiographical and immunohistochemical assays here rationalize the application of PBR imaging for diagnostic and therapeutic assessments of patients with this illness. Significantly, our data also demonstrate distinct mechanisms regulating PBR expression in microglia and astrocytes reacting to tau and Aβ lesions. Of greater interest is the observation that PBR upregulation in astrocytes and microglia may reflect beneficial and deleterious consequences of gliotic changes, respectively, which critically differentiated the modest neurotoxic effects of Aβ versus the more profound neurodegenerative consequences of tau pathologies on hippocampal/entorhinal neurons in APP and tau Tg mice, respectively. Unlike APP Tg mice, patients with AD develop progressive and extensive neuron loss in CNS regions with abundant plaques and tangles, whereas some of the potential restorative processes associated with amyloid plaques, such as axonal sprouting and remodeling of neuritic and synaptic architectures frequently observed in mice (Phinney et al., 1999), are present but less evident in human AD brains (Masliah et al., 1991). Hence, we speculate that “neurotoxic gliosis” characterized by PBR-positive microglia in AD brains may overwhelm the potentially “neutoprotective gliosis” of PBR expressing astrocytes, although this hypothesis will require further testing. For example, this could be examined by developing antibodies that could be used for detecting human PBR in diverse neurodegenerative and other CNS disorders. Moreover, the therapeutic benefit of modulating microglial activity (Dodel et al., 2003; Marx, 2007) could be safely assessed in patients with AD and related tauopathies as well as in other neuroinflammatory CNS conditions by using PBR as a biomarker for microglial neurotoxicity. As a second application, the response to trophic factors could be monitored by detecting increased PBR signals in reactive astrocytes that exert neuroprotective effects. In this context, the implications of PBR changes largely depend on the mechanistic modes of treatments, and changes in PBR expression patterns can be clarified for a given therapeutic approach by preclinical studies using experimental animal models.
As the scope of our research was enlarged from AD pathology to non-AD neurodegenerative or toxic conditions, the common framework of the interplay between microglia and astrocytes after neuronal injury has been delineated with emphasis on PBR as a potentially informative imaging biomarker as schematically illustrated in Figure 8. Disorganization of myelin is a strong inducer of microglial activation, as observed in the midbrain of KA-treated rats and striatum of CZ-exposed mice and documented previously (Smith, 1999), which may involve transient upregulation of microglial PBR (Chen et al., 2004). However, irreversible impairments of neuronal structures are conceivably requisite for long-lasting upregulation of PBR in microglia, in light of our previous and present findings in ethanol-injected rats (Maeda et al., 2007a) and tau Tg mice (Yoshiyama et al., 2007). In contrast, activation of nondeleterious microglia is one of the significant cellular events that result from reversible neuronal injuries, and these glial cells barely express PBR at a high level. This property is exemplified by the CZ challenge in mice, during which spontaneous remyelination occurs at 5–7 weeks (Matsushima and Morell, 2001). As demonstrated by the transplantation of Ra2 cells into the hippocampus of mice, these nontoxic microglia are capable of locally driving PBR expression in astrocytes. Interestingly, our preliminary analysis of transplanted microglia has also indicated that PBR levels in Ra2 cells are notably lower than those in HIV-1 Nef-incorporated Ra2 cells (data not shown), which exhibit dysregulated NADPH oxidase activity and ROS synthesis (Vilhardt et al., 2002), supporting a positive correlation between neurotoxic capability and PBR expression in microglia. This view is additionally supported by a previous demonstration that PK11195, a putative antagonist for PBR, strongly inhibited the lipopolysaccharide-induced production of tumor necrosis factor-α and ROS in primary microglial cells (Wilms et al., 2003). However, PBR-bearing astrocytes present enhanced production of GDNF (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), highlighting the roles of these cells in neuroprotection. This is in line with the previous finding that several PBR ligands could facilitate the survival and regeneration of nerve cells after physical damage (Ferzaz et al., 2002; Mills et al., 2005). The roles of neurotrophins in chronic neurodegeneration may be more complicated, because restorative processes mediated by glial cells are possibly comingled with progressive neuronal injuries. This has been confirmed by the rise of astroglial GDNF expression in PS19 mice. However, GDNF upregulation in these animals was not associated with the emergence of PBR-positive astrocytes, thereby validating the notion that PBR in astrocytes serves as a marker for the repairability of neuronal integrity and sufficiency of neurotrophic support by glial cells.
Figure 8.

Schematic presentation of proposed intercellular network underlying the differential expression of PBR in microglia and astrocytes. Microglial cells are activated by signals from injured neurons, among which myelin components can be highly potent stimulants (1, 1′). Pathologies in which neuronal viability is protected are mutually linked to microgliosis with no or limited PBR expression (1, 2). These microglia trigger activation of PBR-positive astrocytes (3), which may act as neuroprotectors by releasing trophic elements including GDNF (4). Irreversibly damaged neurons have mutual interactions with microglia persistently expressing PBR (1′, 2′). These microglial cells are in close association with activation of astrocytes devoid of significant PBR upregulation (3′), whereas PBR-negative astrogliosis is also inducible by degeneration of unmyelinated neurons without prominent microgliosis (4′).
Glial mediators leading astrocytes to express PBR could be identified by comparatively analyzing signals secreted by microglia in pathologies causing PBR-positive and PBR-negative astrogliosis. Indeed, a subset of secretory molecules, including IL-1 (O'Callaghan et al., 1990), was reported to be undetectable in striatal microglia responding to MPTP lesions of the nigrostriatal dopaminergic projection. As IL-1 receptors are known to be present in astrocytes (Ban et al., 1993), certain IL-1 family cytokines may be key players serving as functional correspondence from microglia to astrocytes, which consequently triggers PBR expression in astrocytes. Neurotrophic components, including IL-10 (Mizuno et al., 1994; Sawada et al., 1998), TGF-β (Constam et al., 1992; Suzumura et al., 1993), plasminogen (Nakajima et al., 1992), nerve growth factors (Elkabes et al., 1996), brain-derived neurotrophic factors (BDNF) and GDNF (Batchelor et al., 1999; Ziv et al., 2006) released from microglia are also plausible candidates for direct or indirect regulators of astroglial PBR enabling PBR-positive astrocytes to promote the protection and repair of injured CNS neurons.
Our present data also suggest a lack of astroglial PBR and a paucity of microglia-astrocyte coupling in the striata of PD models, presumably in association with a paucity of neuroprotective factors in these tissues. In fact, reduced levels of BDNF and a lack of upregulated GDNF production in the nigrostriatal system of PD patients were reported in previous investigations (Mogi et al., 1999, 2001), despite the presence of prominent GFAP-positive astrogliosis in the same brain region. Feasible explanations for the absence of full-range activation of microglia in these conditions might be that dopaminergic terminals account for <15% of the entire synapses in the striatum (Pickel et al., 1981; Teismann et al., 2003), and that myelinated axons, the loss of which gives strong stimuli to microglia, are absent in the nigrostriatal dopaminergic pathway (Pickel et al., 1981). In line with this contention, MPTP-administered monkeys did not develop microgliosis at a detectable level in the striatum (Hurley et al., 2003). It is also noteworthy that intrastriatal injection of 6-OHDA to rats added a small but not striking enhancement to trauma-induced microgliosis (Depino et al., 2003). In humans, a significant number of studies using postmortem PD brains documented that microglial activation was less robust in the striatum than in the substantia nigra (McGeer et al., 1988; Mirza et al., 2000; Knott et al., 2002). In the context that beneficial coupling of microglia and astrocytes does not emerge in response to PD pathologies, pharmacological augmentation of the dialogue between these glial subtypes in the striatum could have a therapeutic value in PD, conceivably by a mechanism that transforms astrocytes into potent protectors of neuronal structures and functions capable of releasing sufficient neurotrophins. This assertion is supported by studies showing a beneficial outcome of GDNF delivery to the striatum of PD models and patients (Tomac et al., 1995; Gash et al., 1996; Kordower et al., 2000; Gill et al., 2003). Interglial communication to promote neuroprotection against these pathological conditions could also be stimulated by adopting a treatment with either locally or systemically administered exogenous microglia as used for an animal model of transient global brain ischemia (Imai et al., 2007), because this approach provokes neurons and glia to produce BDNF and GDNF (Suzuki et al., 2001; Imai et al., 2007). On the supposition that astroglial PBR is an index of the protective and reconstructive activities of glial cells, the present study highlights the usefulness of PET imaging with radioligands for PBR in monitoring interactions between microglia and astrocytes during the course of therapies to upregulate glial neurotrophins.
In conclusion, cytomolecular and intercellular dynamics governing differential PBR expression between microglia-dominant and astrocytes-dominant states may be relevant to mechanisms underlying major neurodegenerative disorders including AD and PD, and further insights into these phenomena could expand the utility of in vivo PBR imaging as a powerful tool for estimating the therapeutic and adverse effects of inflammatory modulators and glioactive agents in diverse CNS disorders.
Footnotes
This work was supported in part by Grants-in-Aid for the Molecular Imaging Program and Scientific Research on Priority Areas, Research on Pathomechanisms of Brain Disorders (20023036; M.H.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan. We thank Taisho Pharmaceutical (Tokyo, Japan) for providing DAA1123. We thank Takeharu Minamihisamatsu for technical assistance.
The authors declare no competing financial interests.
References
- Ando K, Maeda J, Inaji M, Okauchi T, Obayashi S, Higuchi M, Suhara T, Tanioka Y. Neurobehavioral protection by single dose l-deprenyl against MPTP-induced parkinsonism in common marmosets. Psychopharmacology. 2008;195:509–516. doi: 10.1007/s00213-007-0929-2. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Han LY, Clark CM, Grossman M, Trojanowski JQ. Quantitative neurohistological features of frontotemporal degeneration. Neurobiol Aging. 2000;21:913–919. doi: 10.1016/s0197-4580(00)00173-1. [DOI] [PubMed] [Google Scholar]
- Ban EM, Sarliève LL, Haour FG. Interleukin-1 binding sites on astrocytes. Neuroscience. 1993;52:725–733. doi: 10.1016/0306-4522(93)90421-b. [DOI] [PubMed] [Google Scholar]
- Banati RB. Visualising microglial activation in vivo. Glia. 2002;40:206–217. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, Price G, Wegner F, Giovannoni G, Miller DH, Perkin GD, Smith T, Hewson AK, Bydder G, Kreutzberg GW, Jones T, Cuzner ML, Myers R. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123:2321–2337. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, Howells DW. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci. 1999;19:1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Chen MK, Baidoo K, Verina T, Guilarte TR. Peripheral benzodiazepine receptor imaging in CNS demyelination: functional implications of anatomical and cellular localization. Brain. 2004;127:1379–1392. doi: 10.1093/brain/awh161. [DOI] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol. 1992;148:1404–1410. [PubMed] [Google Scholar]
- Depino AM, Earl C, Kaczmarczyk E, Ferrari C, Besedovsky H, del Rey A, Pitossi FJ, Oertel WH. Microglial activation with atypical proinflammatory cytokine expression in a rat model of Parkinson's disease. Eur J Neurosci. 2003;18:2731–2742. doi: 10.1111/j.1460-9568.2003.03014.x. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- Dodel RC, Hampel H, Du Y. Immunotherapy for Alzheimer's disease. Lancet Neurol. 2003;2:215–220. doi: 10.1016/s1474-4422(03)00349-1. [DOI] [PubMed] [Google Scholar]
- Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16:2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferzaz B, Brault E, Bourliaud G, Robert JP, Poughon G, Claustre Y, Marguet F, Liere P, Schumacher M, Nowicki JP, Fournier J, Marabout B, Sevrin M, George P, Soubrie P, Benavides J, Scatton B. SSR180575 (7-chloro-N,N,5-trimethyl-4-oxo-3-phenyl-3,5-dihydro-4H-pyridazino[4,5-b]indole-1-acetamide), a peripheral benzodiazepine receptor ligand, promotes neuronal survival and repair. J Pharmacol Exp Ther. 2002;301:1067–1078. doi: 10.1124/jpet.301.3.1067. [DOI] [PubMed] [Google Scholar]
- Fiala M, Cribbs DH, Rosenthal M, Bernard G. Phagocytosis of amyloid-beta and inflammation: two faces of innate immunity in Alzheimer's disease. J Alzheimers Dis. 2007;11:457–463. doi: 10.3233/jad-2007-11406. [DOI] [PubMed] [Google Scholar]
- Fujimura Y, Ikoma Y, Yasuno F, Suhara T, Ota M, Matsumoto R, Nozaki S, Takano A, Kosaka J, Zhang MR, Nakao R, Suzuki K, Kato N, Ito H. Quantitative analyses of 18F-FEDAA1106 binding to peripheral benzodiazepine receptors in living human brain. J Nucl Med. 2006;47:43–50. [PubMed] [Google Scholar]
- Gash DM, Zhang Z, Ovadia A, Cass WA, Yi A, Simmerman L, Russell D, Martin D, Lapchak PA, Collins F, Hoffer BJ, Gerhardt GA. Functional recovery in parkinsonian monkeys treated with GDNF. Nature. 1996;380:252–255. doi: 10.1038/380252a0. [DOI] [PubMed] [Google Scholar]
- Gill SS, Patel NK, Hotton GR, O'Sullivan K, McCarter R, Bunnage M, Brooks DJ, Svendsen CN, Heywood P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9:589–595. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- Halldin C, Erixon-Lindroth N, Pauli S, Chou YH, Okubo Y, Karlsson P, Lundkvist C, Olsson H, Guilloteau D, Emond P, Farde L. [(11)C]PE2I: a highly selective radioligand for PET examination of the dopamine transporter in monkey and human brain. Eur J Nucl Med Mol Imaging. 2003;30:1220–1230. doi: 10.1007/s00259-003-1212-3. [DOI] [PubMed] [Google Scholar]
- Hurley SD, O'Banion MK, Song DD, Arana FS, Olschowka JA, Haber SN. Microglial response is poorly correlated with neurodegeneration following chronic, low-dose MPTP administration in monkeys. Exp Neurol. 2003;184:659–668. doi: 10.1016/S0014-4886(03)00273-5. [DOI] [PubMed] [Google Scholar]
- Ikoma Y, Yasuno F, Ito H, Suhara T, Ota M, Toyama H, Fujimura Y, Takano A, Maeda J, Zhang MR, Nakao R, Suzuki K. Quantitative analysis for estimating binding potential of the peripheral benzodiazepine receptor with [(11)C]DAA1106. J Cereb Blood Flow Metab. 2007;27:173–184. doi: 10.1038/sj.jcbfm.9600325. [DOI] [PubMed] [Google Scholar]
- Imai F, Suzuki H, Oda J, Ninomiya T, Ono K, Sano H, Sawada M. Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab. 2007;27:488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- Inaji M, Okauchi T, Ando K, Maeda J, Nagai Y, Yoshizaki T, Okano H, Nariai T, Ohno K, Obayashi S, Higuchi M, Suhara T. Correlation between quantitative imaging and behavior in unilaterally 6-OHDA-lesioned rats. Brain Res. 2005;1064:136–145. doi: 10.1016/j.brainres.2005.09.055. [DOI] [PubMed] [Google Scholar]
- Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson's disease. Exp Mol Med. 2006;38:333–347. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
- Kito S, Noguchi Y, Ohta Y. Developmental responses of two substrains of in vitro fertilized C57BL/6J mouse embryos to oxygen and amino acids. Exp Anim. 2003;52:63–66. doi: 10.1538/expanim.52.63. [DOI] [PubMed] [Google Scholar]
- Knott C, Stern G, Kingsbury A, Welcher AA, Wilkin GP. Elevated glial brain-derived neurotrophic factor in Parkinson's diseased nigra. Parkinsonism Relat Disord. 2002;8:329–341. doi: 10.1016/s1353-8020(02)00008-1. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, Brown WD, Holden JE, Pyzalski R, Taylor MD, Carvey P, Ling Z, Trono D, Hantraye P, Déglon N, Aebischer P. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science. 2000;290:767–773. doi: 10.1126/science.290.5492.767. [DOI] [PubMed] [Google Scholar]
- Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, Zhang MR, Suzuki K, Suhara T. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007a;1157:100–111. doi: 10.1016/j.brainres.2007.04.054. [DOI] [PubMed] [Google Scholar]
- Maeda J, Ji B, Irie T, Tomiyama T, Maruyama M, Okauchi T, Staufenbiel M, Iwata N, Ono M, Saido TC, Suzuki K, Mori H, Higuchi M, Suhara T. Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer's disease enabled by positron emission tomography. J Neurosci. 2007b;27:10957–10968. doi: 10.1523/JNEUROSCI.0673-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx J. Alzheimer's disease. A new take on tau. Science. 2007;316:1416–1417. doi: 10.1126/science.316.5830.1416. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, Terry R, Baudier J, Saitoh T. Patterns of aberrant sprouting in Alzheimer's disease. Neuron. 1991;6:729–739. doi: 10.1016/0896-6273(91)90170-5. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Inflammatory processes in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:741–749. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Mills CD, Bitler JL, Woolf CJ. Role of the peripheral benzodiazepine receptor in sensory neuron regeneration. Mol Cell Neurosci. 2005;30:228–237. doi: 10.1016/j.mcn.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Mirza B, Hadberg H, Thomsen P, Moos T. The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson's disease. Neuroscience. 2000;95:425–432. doi: 10.1016/s0306-4522(99)00455-8. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Sawada M, Marunouchi T, Suzumura A. Production of interleukin-10 by mouse glial cells in culture. Biochem Biophys Res Commun. 1994;205:1907–1915. doi: 10.1006/bbrc.1994.2893. [DOI] [PubMed] [Google Scholar]
- Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, Ichinose H, Nagatsu T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson's disease. Neurosci Lett. 1999;270:45–48. doi: 10.1016/s0304-3940(99)00463-2. [DOI] [PubMed] [Google Scholar]
- Mogi M, Togari A, Kondo T, Mizuno Y, Kogure O, Kuno S, Ichinose H, Nagatsu T. Glial cell line-derived neurotrophic factor in the substantia nigra from control and parkinsonian brains. Neurosci Lett. 2001;300:179–181. doi: 10.1016/s0304-3940(01)01577-4. [DOI] [PubMed] [Google Scholar]
- Myers R, Manjil LG, Cullen BM, Price GW, Frackowiak RS, Cremer JE. Macrophage and astrocyte populations in relation to [3H]PK 11195 binding in rat cerebral cortex following a local ischaemic lesion. J Cereb Blood Flow Metab. 1991;11:314–322. doi: 10.1038/jcbfm.1991.64. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Tsuzaki N, Nagata K, Takemoto N, Kohsaka S. Production and secretion of plasminogen in cultured rat brain microglia. FEBS Lett. 1992;308:179–182. doi: 10.1016/0014-5793(92)81270-v. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Soma LA, Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002;34:491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller DB, Reinhard JF., Jr Characterization of the origins of astrocyte response to injury using the dopaminergic neurotoxicant, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 1990;521:73–80. doi: 10.1016/0006-8993(90)91526-m. [DOI] [PubMed] [Google Scholar]
- Pappata S, Levasseur M, Gunn RN, Myers R, Crouzel C, Syrota A, Jones T, Kreutzberg GW, Banati RB. Thalamic microglial activation in ischemic stroke detected in vivo by PET and [11C]PK1195. Neurology. 2000;55:1052–1054. doi: 10.1212/wnl.55.7.1052. [DOI] [PubMed] [Google Scholar]
- Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–8559. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickel VM, Beckley SC, Joh TH, Reis DJ. Ultrastructural immunocytochemical localization of tyrosine hydroxylase in the neostriatum. Brain Res. 1981;225:373–385. doi: 10.1016/0006-8993(81)90843-x. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS. Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem. 2002;83:973–983. doi: 10.1046/j.1471-4159.2002.01210.x. [DOI] [PubMed] [Google Scholar]
- Rojas S, Martín A, Arranz MJ, Pareto D, Purroy J, Verdaguer E, Llop J, Gómez V, Gispert JD, Millán O, Chamorro A, Planas AM. Imaging brain inflammation with [(11)C]PK11195 by PET and induction of the peripheral-type benzodiazepine receptor after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2007;27:1975–1986. doi: 10.1038/sj.jcbfm.9600500. [DOI] [PubMed] [Google Scholar]
- Sawada M, Imai F, Suzuki H, Hayakawa M, Kanno T, Nagatsu T. Brain-specific gene expression by immortalized microglial cell-mediated gene transfer in the mammalian brain. FEBS Lett. 1998;433:37–40. doi: 10.1016/s0014-5793(98)00879-5. [DOI] [PubMed] [Google Scholar]
- Sawada M, Imamura K, Nagatsu T. Role of cytokines in inflammatory process in Parkinson's disease. J Neural Transm. 2006;(Suppl):373–381. doi: 10.1007/978-3-211-45295-0_57. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Smith ME. Phagocytosis of myelin in demyelinative disease: a review. Neurochem Res. 1999;24:261–268. doi: 10.1023/a:1022566121967. [DOI] [PubMed] [Google Scholar]
- Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995;15:5263–5274. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Imai F, Kanno T, Sawada M. Preservation of neurotrophin expression in microglia that migrate into the gerbil's brain across the blood-brain barrier. Neurosci Lett. 2001;312:95–98. doi: 10.1016/s0304-3940(01)02198-x. [DOI] [PubMed] [Google Scholar]
- Suzumura A, Sawada M, Yamamoto H, Marunouchi T. Transforming growth factor-beta suppresses activation and proliferation of microglia in vitro. J Immunol. 1993;151:2150–2158. [PubMed] [Google Scholar]
- Teismann P, Tieu K, Cohen O, Choi DK, Wu DC, Marks D, Vila M, Jackson-Lewis V, Przedborski S. Pathogenic role of glial cells in Parkinson's disease. Mov Disord. 2003;18:121–129. doi: 10.1002/mds.10332. [DOI] [PubMed] [Google Scholar]
- Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, Olson L. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. 1995;373:335–339. doi: 10.1038/373335a0. [DOI] [PubMed] [Google Scholar]
- Van Dam D, Vloeberghs E, Abramowski D, Staufenbiel M, De Deyn PP. APP23 mice as a model of Alzheimer's disease: an example of a transgenic approach to modeling a CNS disorder. CNS Spectr. 2005;10:207–222. doi: 10.1017/s1092852900010051. [DOI] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wang G, Slagel SL, Mason NS, Mathis CA, Fischer ML, Larsen NJ, Mortimer AD, Hastings TG, Smith AD, Zigmond MJ, Suhara T, Higuchi M, Wiley CA. A comparison of the high-affinity peripheral benzodiazepine receptor ligands DAA1106 and (R)-PK11195 in rat models of neuroinflammation: implications for PET imaging of microglial activation. J Neurochem. 2007;102:2118–2131. doi: 10.1111/j.1471-4159.2007.04690.x. [DOI] [PubMed] [Google Scholar]
- Vilhardt F, Plastre O, Sawada M, Suzuki K, Wiznerowicz M, Kiyokawa E, Trono D, Krause KH. The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J Biol Chem. 2002;277:42136–42143. doi: 10.1074/jbc.M200862200. [DOI] [PubMed] [Google Scholar]
- Wilms H, Claasen J, Röhl C, Sievers J, Deuschl G, Lucius R. Involvement of benzodiazepine receptors in neuroinflammatory and neurodegenerative diseases: evidence from activated microglial cells in vitro. Neurobiol Dis. 2003;14:417–424. doi: 10.1016/j.nbd.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Wojtera M, Sikorska B, Sobow T, Liberski PP. Microglial cells in neurodegenerative disorders. Folia Neuropathol. 2005;43:311–321. [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang MR, Kida T, Noguchi J, Furutsuka K, Maeda J, Suhara T, Suzuki K. [(11)C]DAA1106: radiosynthesis and in vivo binding to peripheral benzodiazepine receptors in mouse brain. Nucl Med Biol. 2003;30:513–519. doi: 10.1016/s0969-8051(03)00016-7. [DOI] [PubMed] [Google Scholar]
- Zhang MR, Maeda J, Ogawa M, Noguchi J, Ito T, Yoshida Y, Okauchi T, Obayashi S, Suhara T, Suzuki K. Development of a new radioligand, N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoroethyl-5-methoxybenzyl)acetamide, for PET imaging of peripheral benzodiazepine receptor in primate brain. J Med Chem. 2004;47:2228–2235. doi: 10.1021/jm0304919. [DOI] [PubMed] [Google Scholar]
- Zilka N, Ferencik M, Hulin I. Neuroinflammation in Alzheimer's disease: protector or promoter? Bratisl Lek Listy. 2006;107:374–383. [PubMed] [Google Scholar]
- Ziv Y, Avidan H, Pluchino S, Martino G, Schwartz M. Synergy between immune cells and adult neural stem/progenitor cells promotes functional recovery from spinal cord injury. Proc Natl Acad Sci U S A. 2006;103:13174–13179. doi: 10.1073/pnas.0603747103. [DOI] [PMC free article] [PubMed] [Google Scholar]