

Abstract
Lysosome-related organelles (LROs) are a heterogeneous group of vesicles that share various features with lysosomes, but are distinct in function, morphology, and composition. The biogenesis of LROs employs a common machinery, and genetic defects in this machinery can affect all LROs or only an individual LRO, resulting in a variety of clinical features. In this review, we discuss the main components in LRO biogenesis. We also address the function, composition and resident cell type of the major LROs. Finally, we describe the clinical characteristics of the major human LRO disorders.
Keywords: Chediak-Higashi syndrome, Griscelli syndrome, Hermansky-Pudlak syndrome, melanosome, platelet
INTRODUCTION
Lysosomes are membrane-bound cytoplasmic organelles that serve as major degradative compartments in eukaryotic cells (65). Genetic, biochemical, and structural data have demonstrated that certain specialized cell types contain lysosome-related organelles (LROs) that share features with lysosomes but have distinct morphology, composition and/or functions. Such organelles include melanosomes in melanocytes, lytic granules in lymphocytes, delta granules in platelets, lamellar bodies in lung type 2 epithelial cells, and other variants of acidic granules (Table 1). Features that LROs share with lysosomes include a low intra-organellar pH, specific membrane proteins or other components, and a common pathway of formation (20). Recently, cell biologists have studied LROs because of the lessons they impart concerning vesicle formation. Abnormalities in both lysosomes and LROs occur in certain human genetic diseases (Table 2), including the Hermansky-Pudlak (HPS), Griscelli (GS) and Chediak-Higashi syndromes (CHS), further demonstrating the close relationship between such organelles. Cells of patients with these rare disorders (or their mouse, fly, zebrafish, or yeast counterparts) are important tools for investigating membrane trafficking.
TABLE 1.
Function, resident cell types and clinical relevance of the major identified LROs
| LRO | Function | Resident cell type | Major clinical features when defective |
|---|---|---|---|
| Melanosome | Intracellular melanin biosynthesis and storage. Melanin transfer to keratinocytes. | Melanocytes, iris and retinal pigment epithelial cells | Ocular and cutaneous hypopigmentation |
| Delta granule | Storage of small molecules, released for blood coagulation. | Platelets, megakaryocytes | Bleeding diathesis |
| Lytic granule | Intracellular degradation of macromolecules. Upon release, extracellular destruction of virally infected or cancerous target cells. | Cytotoxic T lymphocytes, natural killer cells | Immune deficiency, viral infections |
| Azurophil granule | Storage of hydrolytic enzymes, destruction of phagocytosed bacteria. Upon secretion, support of various pathological processes, including inflammation. | Neutrophils, eosinophils | Neutropenia, immune deficiency, bacterial infections |
| Basophil granule | Storage of histamine, serotonin, heparin, IL-4, and lysosomal proteases. Released for regulation of inflammation. | Basophils, mast cells | Immune deficiency, allergies |
| Lamellar body | Storage and secretion of surfactants for lung function. | Lung type II epithelial cells | Interstitial lung disease, lung inflammation and fibrosis |
| MHC class II compartment | Intracellular processing and incorporation of antigens into cell membranes. | B lymphocytes, macrophages, dendritic cells, other antigen presenting cells | Immune deficiency |
| Neuromelanin granule | Storage of the insoluble pigment neuromelanin which binds iron. | Catecholaminergic neurons of the brainstem | Unknown |
| Ruffled border | Bone resorption and remodeling. Storage, activation and/or release of acid hydrolases. | Osteoclasts | Osteopetrosis |
| Weibel-Palade body | Storage and regulated release of hemostatic and pro-inflammatory factors (von Willebrand Factor, P-selectin) | Endothelial cells | Bleeding diathesis |
TABLE 2.
Protein complexes, genes and their encoded proteins involved in human or animal LRO disorders
| Protein complex | Mutant gene | Human locus | Mutant protein | Human disease | Animal modela |
|---|---|---|---|---|---|
| BLOC-1 | DTNBP1 | 6p22.3 | Dysbindin | HPS-7 | sandy (m) |
| PLDN | 15q21.1 | Pallidin | - | pallid (m) | |
| CNO | 4p16.1 | Cappuccino | - | cappuccino (m) | |
| MUTED | 6p25.1-p24.3 | Muted | - | muted (m) | |
| SNAPAP | 1q21.3 | Snapin | - | - | |
| BLOC1S1 | 12q13-q14 | BLOS1 | - | - | |
| BLOC1S2 | 10q24.31 | BLOS2 | - | - | |
| BLOC1S3/HPS8 | 19q13.32 | BLOS3/HPS8 | HPS-8 | reduced pigmentation (m) | |
| BLOC-2 | HPS3 | 3q24 | HPS3 | HPS-3 | cocoa (m) |
| HPS5 | 11p14 | HPS5 | HPS-5 | ruby-eye 2 (m) pink (f) | |
| HPS6 | 10q24.32 | HPS6 | HPS-6 | ruby-eye (m) | |
| BLOC-3 | HPS1 | 10q23.1–23.3 | HPS1 | HPS-1 | pale ear (m) |
| HPS4 | 22q11.2-q12.2 | HPS4 | HPS-4 | light ear (m) | |
| AP3 | AP3B1 | 5q14.1 | AP3 beta 3A | HPS-2 | pearl (m) orange (f) |
| AP3D1 | 19p13.3 | AP3 delta | - | mocha (m) garnet (f) | |
| AP3M1 | 10q22.2 | AP3 mu 3A | - | carmine (f) | |
| AP3S1/AP3S2 | 5q22/15q26.1 | AP3 sigma 3A/3B | - | ruby (f) | |
| - | CHS1/LYST | 1q42.1-q42.2 | CHS1/LYST | CHS | beige (m,r)b |
| Unnamed complexc | MYO5A | 15q21 | Myosin Va | GS1 | dilute (m) |
| RAB27A | 15q15-q21.1 | Rab27A | GS2 | ashen (m) | |
| MLPH | 2q37.3 | Melanophilin | GS3 | leaden (m) | |
| Unnamed complex | VPS11 | 11q23 | VPS11 | - | pale gray eyes (jm) |
| VPS16 | 20p13-p12 | VPS16A | - | - | |
| VPS18 | 15q14-q15 | VPS18 | - | deep orange (f) vps18(hi2499A) (z) | |
| HOPSd | VPS33A | 12q24.31 | VPS33A | - | buff (m) carnation (f) |
| VPS33B | 15q26.1 | VPS33B | ARC | - | |
| VPS39/Vam6 | 15q15.1 | VPS39/Vam6 | - | leberknödel (z) | |
| VPS41 | 7p14-p13 | VPS41 | - | light (f) | |
| - | RAB38 | 11 q14 | Rab38 | - | chocolate (m) lightoid (f)e,f |
| - | RABGGTA | 14q11.2 | RABGGTA | - | gunmetal (m) |
| - | SLC7A11 | 4q28-q32 | SLC7A11 | - | subtle gray (m) |
| - | p14/MAPBPIP | 1q22 | MAPBPIP/p14 | MIM610798g | - |
Naturally occurring LRO models models; mouse (m), rat (r), fruit fly (f), zebrafish (z), Japanese Medaka ricefish (jm).
Other species with CHS mutants are Aleutian mink, cats, killer whale, and Japanese cattle (58).
These 3 proteins form an unnamed tripartite complex (see Figure 3).
Homotypic vacuolar sorting (HOPS) complex; a conserved protein complex that functions as a tethering factor in membrane fusion events involving lysosomes and/or endosomes (26).
The rat models Ruby (R), Fawn-hooded (FH), and Tester-Moriyama (TM) also carry RAB38 mutations (90).
Unnamed syndrome comprising partial albinism, immunodeficiency, congenital neutropenia, B-cell and cytotoxic T-cell deficiency, and short stature (6).
Here we discuss the machinery of LRO biogenesis, the major recognized LROs, and the known human disorders of LRO biogenesis.
1. BIOGENESIS OF LYSOSOME-RELATED ORGANELLES
Lysosomes and LROs are produced through the interaction of ubiquitous trafficking mechanisms with cell-specific machinery that targets cargo to a particular compartment. Whereas classic secretory granules form directly from the trans-Golgi network (TGN), some or all LRO contents derive from the endosomal system. This group of individual compartments relies on sorting and trafficking of membrane structures, assisted by a network of filaments, tubules, motor proteins, Rabs and other small GTPases, and numerous other components such as BLOCs, SNAREs, syntaxins, and VPS proteins.
A. The endosomal system
Lysosome-related organelles maintain their structure, composition, and function by means of a continuous flow of proteins and membranes/vesicles among endosomal compartments (Figure 1). The endocytic pathway takes up molecules from outside of the cell and internalizes membrane receptors, while the exocytic pathway sorts newly synthesized proteins from the endoplasmic reticulum toward the endosomal system (9). The two pathways connect at the early endosome, a tubulo-vesicular network with a pH of 5.9–6.0 that contains distinct resident proteins, including Early Endosome Associated Protein (EEA)-1, Rab5, and Rabaptin-5. Material is sorted from the early endosome to the cell surface, recycling endosomes, the biosynthetic pathway (of LROs) or late endosomes/lysosomes (9, 36, 99). Components destined for lysosomes are sorted to late endosomes, which have an acidic pH (5–6.0), contain whorls of membranes and vesicles, include multivesicular bodies, and recycle mannose-6-phosphate receptors (MPRs) back to the trans-Golgi membranes. Other late endosome-specific markers are Rab7 and lysobisphosphatidic acid (LBPA). Late endosomes morph into lysosomes, which lack multivesicular structures and MPRs and have a pH of 5.0–5.5. Components destined for specific LROs possess unique, cell-type and LRO-specific sorting and trafficking pathways (99). To support the endosomal system, cell-type specific chaperones exist for vesicle targeting, transport, and fusion events. These include a cytoskeletal system of tubules and filaments, coat associated proteins, SNAREs, syntaxins, rabs, motor proteins, and specific membrane lipids (9, 36, 95, 105).
Figure 1. The endosomal system and LRO biogenesis.

The endosomal system is a collection of highly dynamic compartments defined by their morphology, contingent of marker proteins, function and accessibility to endocytic tracers. Rather than focus on a specific cell type, this model shows the basic endosomal elements involved in generic LRO biogenesis. Solid arrows depict maturation/genesis of a compartment while dashed arrows represent the movement of cargo. The early (sorting) endosome is the major sorting center of the cell and is defined by the presence of EEA1 and Rab5 and by rapid accessibility to endocytic tracers (5–15 min). Sorting of Golgi derived biosynthetic cargo destined for LROs or late endosomes/lysosomes as well as receptors and other molecules internalized from the cell membrane occurs in the early endosome. Some receptors return to the cell membrane by way of recycling endosomes, while others continue on to late endosomes and lysosomes for degradation. As the early endosome matures, intralumenal vesicles containing proteins targeted for lysosomal degradation accumulate and the multivesicular body (MVB)/late endosome is formed. The MVB/late endosome is the last site of sorting and is defined by its morphology and the presence of MPRs and LBPA. MPRs deliver lysosomal hydrolases and are then recycled from the MVB/late endosome back to the TGN. MVBs/late endosomes fuse with (or mature into) lysosomes to deliver their contents for degradation. Lysosomes lack both intralumenal vesicles and MPRs and are considered end-stage degradative compartments. Specialized LROs co-exist in the same cell as non-specialized lysosomes and contents destined for each compartment must be sorted appropriately. In some LRO containing cells, LRO formation involves sequential delivery of LRO-specific proteins. A subset of proteins destined for LROs (e.g. PMEL17 in melanocytes) are sorted from a post-Golgi compartment to an endosomal intermediate and drive the formation of a pre-LRO (e.g. stage I/II melanosome). A second set of LRO specific proteins (e.g. TYR and TYRP1) are sorted from the early endosome to the pre-LRO compartment, resulting in a mature LRO. The mature LRO then acquires specific accessory proteins (Rabs, motor proteins, SNAREs) that assist in its function and/or localization.
B. Membrane budding and fusion
Tight control of membrane flux through the endosomal system occurs through continued vesicular budding and fusion. Budding is largely mediated by coat components, whereas targeting and fusion are generally mediated by SNARE complexes (9, 11, 95). Vesicular structures required for LRO formation arise at the plasma membrane, the trans-Golgi network or other intracellular membranous compartments, with the recruitment of coat components. There exist at least three major vesicle coat proteins, COPI (coatomer) and COPII, functioning in the early endocytic pathway (ER and Golgi) and clathrin, which functions in post-Golgi locations including the TGN, endosomes, and plasma membrane (11). The coat proteins trigger generation of highly curved membrane areas where vesicle-specific cargo recruitment takes place, including recruitment of adaptor proteins and SNAREs. A variety of clathrin-specific “adaptors” exist, including the heterotetrameric adaptor protein (AP) complexes, AP1, AP2, AP3 and AP4 and the monomeric GGA, Hrs, Epsin-1, and ARH proteins. These adaptors bind to transmembrane cargo proteins by recognizing sorting signals, e.g., specific tyrosine or di-leucine motifs or conjugated ubiquitin (9, 11, 73, 99).
Once the vesicle is released by the donor membrane, it is targeted to its acceptor compartment, guided by the cell cytoskeleton, motor proteins, rabs and other “tethering” molecules (9, 11). The final step in a vesicle’s existence is fusion with the acceptor membrane, mediated by SNARE complexes (Soluble N-ethylmaleimide-sensitive factor Association protein REceptors). A SNARE complex assembles when a monomeric v-SNARE on a vesicle binds to an oligomeric t-SNARE on a target membrane, which promotes membrane fusion and cargo transfer from the vesicle to the acceptor compartment. Different v-/t-SNARE complexes form at different steps of intracellular transport, providing specificity of time and place to the membrane fusion process (11, 95). The role of the SNARE and adaptor complexes in LRO biogenesis became apparent from mutant animal and human models, which provided an invaluable resource for elucidating these complex pathways. For example, defects in subunits of adaptor protein complex-3 (AP3) result in human HPS-2 (21, 54) and the HPS mouse mutants pearl and mocha (67). These genetic models demonstrate that AP3 regulates the sorting of proteins specific for the lysosome, melanosome, and platelet granule.
C. Tubules and Filaments
The cytoskeleton, unique to eukaryotic cells, is a dynamic, organized, three-dimensional network of fibers that maintains cell shape and motility. It consists of actin networks, microtubules, and intermediate filaments.
Actin networks, also called microfilaments, consist of fine, thread-like protein fibers, 3–7 nm in diameter and composed of actin monomers. Actin networks serve short-range movements in the cell periphery and in the cell body surrounding the Golgi complex, including muscle contraction, cell motility, cell division and cytokinesis, vesicle and organelle movement, cell signaling, and the establishment and maintenance of cell junctions and cell shape (128). Actin subunits provide polarization to regulate the direction of movement of molecular motors along the filaments. Actin is bound by a variety of proteins assisting in the formation, stability and function of actin networks (128). Myosins are the key motors effecting movement along actin (22, 128).
Microtubules are long polymers of α- and β-tubulin that form cylindrical tubes, 20–25 nm in diameter. Microtubules provide a backbone for long-range (μm to mm) intracellular transport processes such as mitosis, cytokinesis, and vesicular transport (12). Polymerization of microtubules is nucleated in a microtubule organizing center (MTOC), away from which the microtubule grows in the (+) direction. The motor protein kinesin moves toward the (+) end of the microtubule (79), while dynein moves toward the (−) end (96). A variety of microtubule-associated proteins (MAPs) assure proper function, formation and stability of microtubules (12). Actin filaments, microtubules and their molecular motors play critical roles in LRO biogenesis (12). For example, kinesin-2 is a plus end-directed motor for late endosomes and lysosomes (79), and Rab (defective in Charcot-Marie-Tooth disease type 2B) regulates recruitment of dynein/dynactin and movement of late endosomal compartments on microtubules (48, 57). The motor Myosin Va and its interactors Rab27A and Melanophilin are essential for the tethering and actin-based movement of melanosomes in the periphery of the melanocyte (113). Finally, the BLOC-1 complex (Figure 2) (vide infra) associates with actin filaments (27).
Figure 2. Biogenesis of Lysosome-related Organelles Complexes (BLOCs).
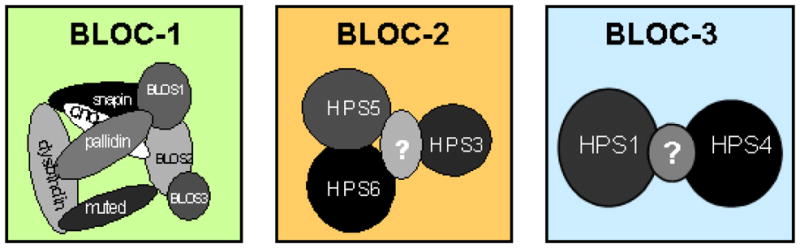
The currently recognized proteins identified as components of each BLOC are indicated. Most of the BLOC subunits are associated with subtypes of Hermansky-Pudlak syndrome. Subunits indicated by “?” are predicted by molecular weight analyses of the entire complex, and may represent a yet-to-be-identified subunit.
Intermediate filaments, approximately 10 nm in diameter, are tissue specific proteins often interconnected with other filamentous systems for mechanical stability. Intermediate filament proteins including keratin, vimentin, desmin and lamin (46). Human disorders of intermediate filaments, which function as mechanical scaffolds (18), number at least 70 (http://www.interfil.org), including diseases of the skin, heart, muscle, liver, brain, adipose tissue and even premature aging. Intermediate filaments assist in endosomal trafficking and LRO biogenesis pathways; defects in specific intermediate filaments affect the endocytic pathway of low-density lipoprotein-cholesterol, the formation of autophagocytic vacuoles, endosome-Golgi recycling of glycosphingolipids, and AP-3 dependent endosomal sorting (17, 18, 33, 114).
D. Motor Proteins
Kinesins, dyneins and myosins are the motors that drive most active transport of proteins and endosomal vesicles (4, 12).
Myosins are actin-associated motor proteins that use ATP hydrolysis to generate mechanical force. They consist of an actin and ATP-binding head domain, a calmodulin-binding neck domain and a cargo-interaction C-terminal tail domain. Myosins regulate organelle and vesicle movement, cytokinesis, muscle contraction, adhesion and migration (4, 129). The dimeric Myosin Va (encoded by MYO5A) achieves actin-dependent transport of LROs, especially melanosomes, by binding of its cargo receptor, Melanophilin (Figure 3) (4, 22, 94, 127, 130).
Figure 3. Functional mechanism of the Rab27a-Melanophilin-MyosinVa tripartite complex.

In melanocytes, Rab27a resides on mature melanosomes which travel to the cell periphery on microtubules via a kinesin motor protein. Once in the periphery, Rab27a recruits the motor protein myosinVa via a direct interaction with Melanophilin. The Rab27a-Melanophilin-MyosinVa tripartite complex is then responsible for the accumulation of mature melanosomes in the actin-rich dendritic tips. This localization is necessary for efficient transfer of melanosomes to keratinocytes for normal pigmentation. If any member of the tripartite complex is defective, melanosomes are not captured in the periphery and return to the cell center on microtubules in a dynein-mediated process. Defects in any member of the tripartite complex result in Griscelli syndrome.
Kinesins are a superfamily of microtubule-associated motor proteins; their catalytic head motor domains bind to microtubules and hydrolize ATP to produce force. Three subfamilies of kinesins differ in position of their motor domain (79). Conventional kinesin is an anterograde motor formed by two heavy chains that possess the ATPase and microtubule binding activity, and two light chains. Their coiled-coil stalk domain is involved in dimer formation and the C-terminal tail participates in cargo binding (4, 79). Kinesins bind LROs, in particular melanosomes, through their binding partner kinectin, which associates with melanosomal membranes (93). Studies in Xenopus melanophores show that an interaction of kinesin II interacts with p150Glued, a subunit of dynactin, to disperse melanosomes (19). Inhibition of kinesin caused bi-directional movement of melanosomes along microtubules, leading to perinuclear accumulation of melanosomes (40).
Dyneins regulate retrograde movement toward microtubule minus-ends. They are large complexes comprised of 2–3 heavy chains, containing a motor domain and variable numbers of intermediate and light chains (4). Cytoplasmic dyneins are expressed in most eukaryotic cells and are implicated in mRNA transport, vesicle trafficking and cell division (96). Cytoplasmic dynein is associated with the protein complex dynactin, which contains ten subunits including p150Glued, p135Glued, dynamitin and Actin-Related Protein 1 (47). The role of dynein in retrograde melanosome transport is well established (4, 47), and its role in melanosome maturation is emerging (57).
E. Rabs and other small GTPases
The Rab family of small GTPases consists of more than 60 members in mammals (105). The unique subcellular distribution of each Rab protein is thought to determine membrane identity (81, 105). Rabs are synthesized as soluble proteins and recognized by a Rab escort protein (REP) that presents them to Rab geranylgeranyl transferase for prenylation (1, 81, 105), which anchors them to a membrane. Rab proteins cycle between GTP-bound “active” and GDP-bound “inactive” forms, assisted by GAP, GDI and GEF proteins (1, 105). This “on/off switch” regulates Rab protein binding to downstream effectors and execution of cellular functions. At least three Rab proteins (Rab27A, Rab38 and Rab32) are involved in LRO biogenesis.
Rab27A assists in the transfer of melanosomes from microtubules to actin filaments (Figure 3). Rab38 is mutated in the mouse chocolate, which displays mild oculocutaneous albinism with small, round hypopigmented melanosomes but no platelet granule deficiency (70). Rab38 is also mutated in three rat models (Table 2) of pigment dilution and platelet granule deficiency (91). Mouse Rab38 and the closely related Rab32 (67% identical) localize to mature melanosomes and to perinuclear vesicles containing melanosome-specific proteins (TYR, TYRP1) (122). In chocolate melanocytes treated with Rab32 siRNA, TYR and TYRP1 fail to exit the TGN, resulting in a complete loss of pigment, suggesting that these rabs are functionally redundant; indeed, only one homologue exists in other species (122). Interestingly, both Rab38 and Rab32 are expressed in melanocytes and bone marrow mast cells but only Rab38 is present in rat basophil leukemia cells and alveolar type II-derived MLE-12 cells (122).
Defects in universal regulators of Rab function such as Rab geranylgeranyl transferase (RabGGTase) and Rab escort protein (REP) can also lead to defects in LRO biogenesis. The gunmetal mouse has reduced RabGGTase activity due to a mutated alpha subunit (RABGGTA) (23). Gunmetal mice have hypopigmentation, macrothrombocytopenia, decreased numbers of platelet alpha and delta granules, and reduced geranylgeranylation and membrane association of Rab27a (105). Deficient REP1 (one of two REP isoforms) in humans leads to choroideremia (CHM, MIM 303100), a form of X-linked retinal degeneration. Rab27a prenylation is reduced in CHM but it is unclear if Rab27a alone or a combination of Rabs is responsible for the disease (105).
F. Biogenesis of Lysosome-related Organelle Complexes (BLOCs)
BLOCs are unique groups of proteins that, together, function in the formation and/or trafficking of lysosome-related endosomal compartments (24, 27, 68, 73, 83, 110) (Figure 2). BLOC-1 consists of the pallidin, muted, cappuccino, HPS7/dysbindin, BLOS1, BLOS2, BLOS3, and Snapin proteins. Hermansky-Pudlak Syndrome subtype 7 (HPS-7, due to defective dysbindin) and HPS-8 (defective BLOC1S3) are BLOC-1 defects (68, 80); other BLOC-1 subunits are associated with a mutant mouse (Table 2) (67).
Potential binding partners of BLOC-1 suggest that this complex regulates SNARE-mediated membrane fusion. Palladin, by interacting with syntaxin 13, may function as a premelanosomal t-SNARE to assist TYRP1 and TYRP2 containing vesicles to fuse with the premelanosome (27, 71). Snapin interacts with SNAP-25/23, SNAREs implicated in membrane fusion events (110). Dysbindin binds alpha and beta dystrobrevins, i.e., dystrophin-related protein components of large dystrophin-associated membrane-spanning complexes (85). However, recent studies indicate that endogenous dysbindin, when assembled into the BLOC-1 complex cannot bind dystrobrevins because of an occupied dystrobrevin binding site (85). BLOC-1 also associates with F-actin (27), supporting a role for the complex in vesicle movement along actin filaments. Moreover, BLOC-1 apparently mediates the egress of cargo from early endosomes toward LROs (106), and interacts with other LRO biogenesis complexes, including AP3 and BLOC-2 (25).
BLOC-2 contains the HPS3, HPS5 and HPS6 proteins, and BLOC-3 consists of the HPS1 and HPS4 proteins. The cell biology of these BLOCs is discussed below, under Hermansky-Pudlak syndrome.
G. Other endosomal chaperones
Endosomal trafficking and LRO formation requires several additional components and regulatory mechanisms. Human and animal models have helped elucidate the pathways involved. For example, the granule group of Drosophila melanogaster eye color mutants (69) and murine models of hypopigmentation and bleeding (67, 117) have uncovered novel players in LRO biogenesis (Table 2). These include RABGGTA, defective in the gunmetal mouse (23); Slc7a11, a cystine/glutamate exchanger defective in the subtle gray mouse (15); LYST/CHS1, defective in the beige mouse, rat and human Chediak-Higashi syndrome (58); the endosomal adaptor protein p14, defective in a syndrome of albinism and immunodeficiency (6); and VPS proteins (homologs of vacuolar sorting proteins in yeast) such as VPS18, 33, 41, defective in fly models (Table 2). Apart from protein defects, alterations in membrane lipids, rafts, cholesterol and other cellular components can affect LRO biogenesis (36, 105).
2. TYPES OF LYSOSOME RELATED ORGANELLES
LROs share features with lysosomes, but also exhibit cell type-specific components responsible for specialized functions. Table 1 provides a list of the major LROs.
A. Lysosomes
Lysosomes are membrane-bound, acidic, cytoplasmic organelles responsible for intracellular protein degradation (65). They contain mature acid-dependent hydrolases and highly glycosylated integral membrane proteins (LAMPs), but lack mannose 6-phosphate receptors (MPRs) (65). LROs share most or all of these characteristics. In some cell types, LROs such as secretory lysosomes perform both lysosomal functions and specialized LRO functions (99, 112). In other cell types, such as melanocytes and platelets, highly specialized LROs (melanosomes and delta granules, respectively) co-exist with conventional lysosomes, indicating distinct biogenesis and sorting pathways (99).
B. Melanosomes
Melanosomes are specialized LROs responsible for synthesis and storage of melanin in melanocytes (4, 71, 99). The insoluble black to dark-brown eumelanins and the red, brown, and yellow pheomelanins provide color for human skin, eye and hair (71, 98). Morphologically, pheomelanosomes are rich in vesicles but lack an organized structure and will not be further discussed (71). Melanosomes mature through four stages (Figure 4). Stage I pre-melanosomes are clathrin-coated endosomal compartments with intralumenal vesicles (ILV) resembling multivesicular bodies (MVB). Stage II melanosomes are elongated, with intralumenal striations spanning the length of the organelle. Newly synthesized melanin particles are deposited on the striated fibrils, resulting in the black streaks of stage III melanosomes that eventually become fully-melanized stage IV melanosomes (Figure 4) (71, 98). Melanin synthesis requires distinct, non-lysosomal sorting and enrichment of melanosome-specific proteins to different melanosomal stages (98, 100).
Figure 4. Morphological characterization of the four stages of melanosome biogenesis.
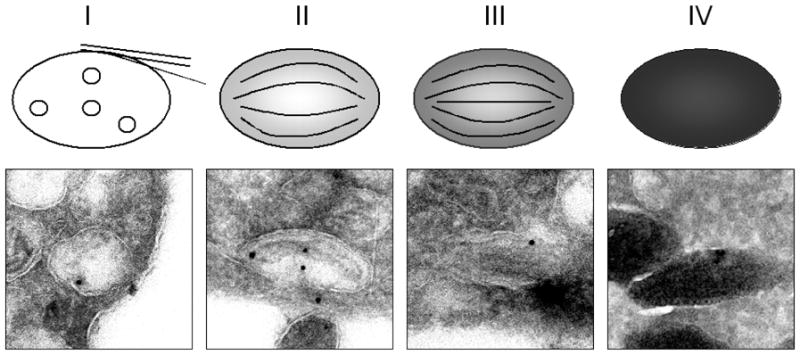
The upper panel represents a cartoon of the four stages of melanosome biogenesis. The lower panel shows an immune-electron microscopy study of human primary epidermal melanocytes. Ultrathin cryosections labeled with anti-NKI beteb, a PMEL17-specific marker, conjugated to 20-nm gold particles. Stage I melanosomes, electron-lucent MVBs without melanin, contain multiple intralumenal vesicles. Stage II melanosomes are more elongated with PMEL17-containing intralumenal striations that run the length of the organelle. Stage III melanosomes exhibit melanin deposits on the striations. These striations are masked in stage IV melanosomes, due to complete melanization.
The melanosome-specific protein PMEL17 is the major component of intralumenal striations, mediated by its N-terminal intralumenal domain (5). For activation, Golgi-glycosylated PMEL17 undergoes proteolytic cleavage by a furin-like proprotein convertase in a post-Golgi compartment. Active PMEL17 is sorted into clathrin-coated MVBs where it becomes enriched in ILVs and initiates striation formation (5). Sorting of melanogenic enzymes (TYR, TYRP1, TYRP2) to melanosomes occurs through a pathway different from that for PMEL17; TYR and TYRP1 are absent from PMEL17-rich clathrin-coated endosomal compartments (99, 100). Melanosomal sorting of TYR and TYRP1 occurs via a distinct population of early endosomes (25, 44, 106). The adaptor protein complex AP3 functions in TYR trafficking from the early endosomes to melanosomes (53). BLOC-1 helps regulate the sorting and budding of TYRP1-containing vesicles from early endosomal compartments, while BLOC-2 is involved in targeting of TYRP1-positive vesicles to melanosomes (25, 44, 106). Human syndromes and animal models continue to shed light on the process of melanosome biogenesis (50, 67, 105).
C. Platelet delta granules
Platelets deliver pro-haemostatic proteins and other mediators to sites of vessel injury. In addition to lysosomes, alpha and delta (also called dense) granules are the major platelet organelles and release mature secretory proteins and small molecules like ADP and serotonin upon stimulation (101). Alpha granules, the most abundant vesicles in platelets, store proteins that promote platelet adhesiveness and wound healing (101). Delta granules, which are platelet LROs, number only 3–8 per platelet and store non-protein small molecules; their membranes contain the lysosomal membrane proteins LAMP2 and CD63 (granulophysin or LAMP3), but not LAMP1 (38, 74). Intralumenal delta granule constituents include calcium, serotonin, ADP, ATP and polyphosphates (38, 74). The high calcium content of delta granules gives them an intrinsic electron density (Figure 6a,b), and their highly osmophilic environment causes them to appear dark on transmission electron microscopy (74).
Figure 6. Clinical features of Hermansky-Pudlak syndrome.
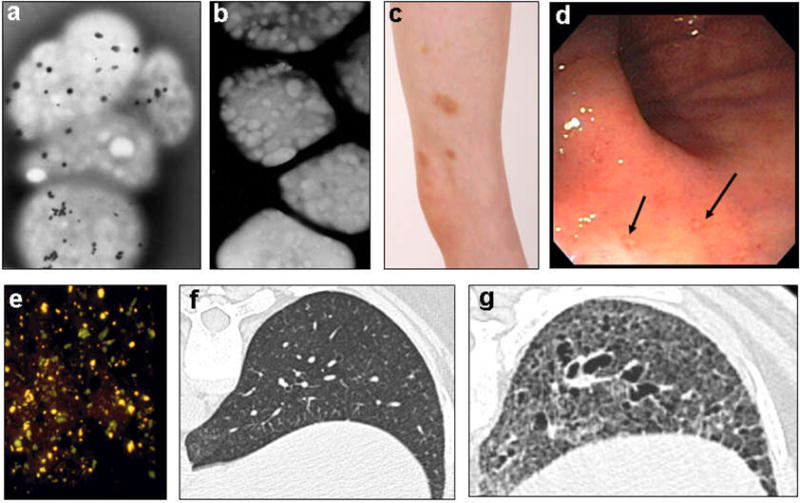
(a) Whole mount electron micrographs of normal platelets, each contain several delta granules (small black dots). (b) HPS platelets, devoid of delta granules. (c) Spontaneous bruising in a boy with HPS. (d) HPS intestine showing mucosal ulcerations (arrows). (e) Autofluorescent ceroid lipofucsin (yellow) in renal tubular cells of an HPS-1 patient sloughed into the urine. (f) High-resolution CT scan of a normal lung. (g) High-resolution CT scan of an HPS-1 lung, showing bullae, fibrosis, and loss of alveoli. [(a) and (b) courtesy of Dr. James G. White, University of Minnesota; (f) and (g) courtesy of Dr. Thomas Markello, NHGRI].
Unlike other secretory cells, circulating platelets do not form their own vesicles (101). Instead, bone marrow megakaryocytes create the granules and transport them into developing proplatelets in the periphery via microtubules and actin networks. During megakaryocytopoiesis, alpha and delta granules arise from the Golgi complex (131), with MVBs as intermediate structures (42, 131). Young megakaryocytes contain numerous MVBs but few alpha and delta granules (42, 131). As megakaryocytes mature, MVBs become fewer, the number of alpha and delta granules increases, a demarcating membrane system develops and the granules enter nascent proplatelets (42, 62, 131). Proplatelets released from the megakaryocyte fragment into individual platelets (62).
D. Lytic granules
Cytotoxic T lymphocytes (CTLs) kill virus-infected cells and tumor cells by releasing their lytic granule contents (28). Lytic granules are specialized secretory LROs that degrade proteins at acidic pH. They also contain proteins such as perforin, which becomes functional upon being secreted into the neutral pH of the extracellular milieu (112). Recognition of MHC peptide complexes on target cells by CTL receptors results in polarization of the secretory machinery and formation of the microtubule organizing center (MTOC) at the contact point between the CTL and target cell. There, lytic granules accumulate and attack the target cell, sparing other cells including the CTL itself (28, 112). Released perforin makes pores in the target cell membrane, allowing granzymes (also lytic granule constituents) to enter the target cells, where they cleave many different substrates and kill the cell. Fas ligand, released from the CTL on small exosome-like vesicles of the lytic granule (103), binds to the Fas receptor on the target cell, beginning a cascade of caspase cleavage events that initiate apoptosis. Lytic granules also contain the lysosomal membrane proteins CD63, LAMP1 and LAMP2 (28, 112).
E. Other LROs
The exact definition and the precise number of LROs has not been established. However, LROs must possess an acidic lumen, a contingent of characteristic lysosomal proteins and a specialized function, usually related to storage or secretion. Accessibility to endocytic markers may be another criterion. Over the past decade, several specialized vesicles, previously considered “orphan” organelles, have been declared LROs. The list may expand as more proteomic studies are performed and additional disease phenotypes are recognized.
Lamellar bodies are secretory granules found in pulmonary type II cells, store and secrete surfactant phospholipids and proteins (123). Thephospholipids form a film at the air-liquid interface, reducing surface tension in the alveoli and preventing alveolar collapse during expiration. Lamellar bodies maintain a pH of ~pH5.5 and contain characteristic lysosomal enzymes (acid phosphatases and cathepsins) and membrane proteins (CD63 and LAMP1). Surfactant protein B (SP-B) is necessary for the formation of lamellar bodies; SP-B (−/−) mice have morphologically and functionally abnormal lamellar bodies (109). Giant lamellar bodies have been reported in disorders of LRO biogenesis both in patients with HPS (82) and in the beige mouse, a model of CHS (13). The relationship between enlarged lamellar bodies and the pulmonary fibrosis of HPS remains speculative.
Azurophilic granules are one of three types of granules found in neutrophils, the most abundant phagocytic cell acting in the innate immune response (37). Like lysosomes, azurophil granules contain proteins (e.g., serine proteases, antibiotic proteins and myeloperoxidase) that undergo proteolytic processing into mature, active proteins. Azurophil granules are affected in at least two disorders of LRO biogenesis. Chediak-Higashi syndrome neutrophils contain giant, CD63-positive azurophil granules that do not properly exocytose myeloperoxidase upon stimulation (63). AP3-deficient individuals with HPS-2 have low intracellular neutrophil elastase, an azurophil granule constituent, resulting in a G-CSF-responsive neutropenia and a diathesis toward bacterial infection (29).
Basophil granules are uniform acidic granules of basophils. Basophils represent 0.5–3% of circulating leukocytes and, together with mast cells, mediate allergic inflammation (72). Upon activation, basophil granules release their contents, which include histamine, serotonin, heparin, and lysosomal proteases (tryptase and chymase). Basophil granules contain lysosomal membrane proteins such as LAMP1, LAMP2, CD63/LAMP3, and LIMPIV/5G10 antigen (20, 115). Basophils are an important source of the cytokine interleukin-4, critical in the development of allergies and the production of IgE antibodies.
Other candidate LROs include Major histocompatibility complex (MHC) class II compartments (MIICs), neuromelanin (NM) granules, ruffled borders and Weibel Palade bodies (WPBs). MIICs are important for antigen processing and are found in B cells, macrophages and dendritic cells. MIICs have multiple internal vesicles and/or membranous whorls that contain lysosomal enzymes and lysosomal membrane markers (41). NM granules, pigmented organelles found in certain catecholaminergic neurons, were recently described as LROs based on proteomics studies (120). Ruffled borders are the site of bone resorption in osteoclasts and may be LRO variants based on their lysosomal proteins and secretory function (111). WPBs are lysosome-related only in that they carry the lysosomal marker CD63; this marker reaches the WPB via an endocytic rather than a direct biosynthetic route (39).
3. HUMAN DISORDERS OF LRO BIOGENESIS AND TRAFFICKING
Disorders of LRO biogenesis typically affect melanosomes and platelet dense granules, and may or may not affect other LROs. Some LRO disorders have murine (67), fruit fly (26, 66, 69), zebrafish (104), as well as yeast (50) counterparts. The major LRO disorders are listed in Table 2.
A. Hermansky-Pudlak Syndrome (HPS)
There exist eight known human HPS genes, each associated with a murine model (Table 2). The eight HPS gene products operate in distinct complexes, e.g., BLOC-2 (HPS3, HPS5, and HPS6) with relatively mild clinical involvement (3, 49, 52, 124), BLOC-3 (HPS1 and HPS4), with more severe complication (2, 45, 124), and AP3 (HPS-2 gene product), with immune deficiency (54, 124). The pale ear (Hps1 deficient), light ear (Hps4 deficient), and double-homozygous pale ear/light ear mice have identical coat color, and a characteristic hypopigmentation of ears, tail and feet (67, 116), indicating a physical and/or functional interaction between Hps1 and Hps4. In addition, tissues of the light-ear mice (Hps4-deficient) show no Hps1 protein expression (116). Human HPS subtypes involving the same BLOC generally resemble each other.
Clinical manifestations of HPS
All HPS (MIM 203300) patients exhibit some degree of hypopigmentation and a diathesis toward bleeding. The hypopigmentation derives from impaired melanosome formation, trafficking, or transfer to keratinocytes. In contrast, classical albinism is caused by deficiency of melanosomal proteins such as tyrosinase, the P protein, or TYRP1 (61). Patients with albinism have abnormal neuronal crest cell migration during development, with ~90% of their optic nerve fibers decussated (crossed), compared with 55% normally (61). They display horizontal nystagmus, an involuntary, painless lateral movement of the eyes in both directions. The retinal fundus appears pale, and visual acuity is generally stable at 20/200 (legally blind in the United States) or worse. The eyes appear light or blue, and iris transillumination is readily apparent. In this phenomenon, light shone into the pupil is transmitted through the iris due to a decrease in iris pigment. In albinism, the hair is white and the skin is light and sun-sensitive. Solar keratoses and melanocytic nevi are common, and patients are at increased risk of developing squamous cell carcinoma, basal cell carcinoma, and possibly melanoma (119). Sun avoidance is critical.
HPS patients with BLOC-3 defects, i.e., HPS-1 or HPS-4, generally have features of classical albinism, such as light hair (Figure 5a,b), solar keratoses (Figure 5c), significant iris transillumination (Figure 5d), and a hypopigmented retina (Figure 5e). HPS-2 patients have less hair hypopigmentation (Figure 5f). Compared with BLOC-3 patients, individuals with BLOC-2 defects, i.e., HPS-3, HPS-5, or HPS-6, have mild findings, including minimal iris transillumination (Figure 5g) and hypopigmentation of the retina (Figure 5h), skin and hair (Figure 5i). Visual acuity can be as good as 20/50. However, there is enormous variability in each HPS subtype. HPS-7 and HPS-8 patients have not been well characterized with respect to pigmentation (68, 80).
Figure 5. Hair, skin and eye pigmentation in Hermansky-Pudlak syndrome.
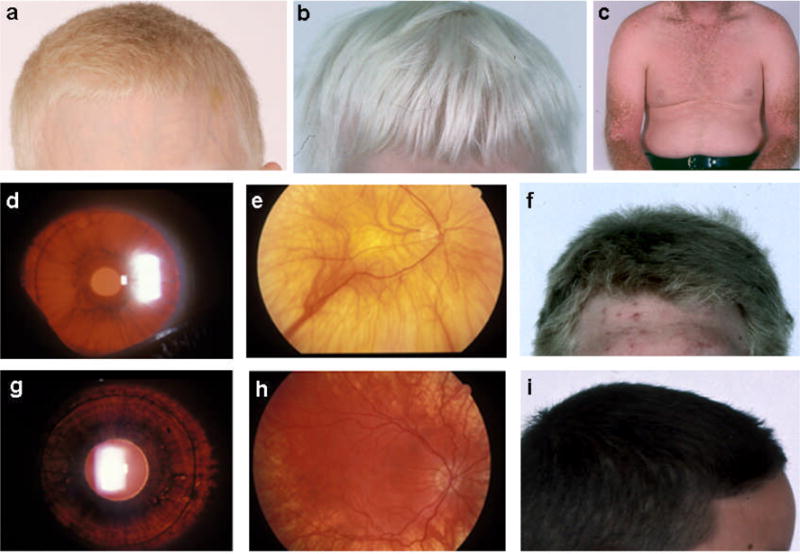
(a) Light colored hair of a boy with HPS-1. (b) White hair of a boy with HPS-4. (c) Keratoses in sun-exposed areas of a Puerto Rican patient with HPS-1. (d) Significant iris transillumination in an HPS-1 patient. Orange light, abnormally present, appears because the iris contains insufficient melanin to block it. (e) Pale retinal fundus in HPS-1, with vessels clearly visible due to lack of retinal pigment epithelium. (f) Tan-blond hair of a patient with HPS-2. (g) Mild iris transillumination in a patient with HPS-3. (h) Mild hypopigmentation of the retina in HPS-3. (i) Dark hair of a Puerto Rican boy with HPS-3. [Figures d, e, g, and h courtesy of Dr. E. Tsilou, National Eye Institute].
The bleeding diathesis of HPS involves defective platelet aggregation due to absence of dense granules, apparent on whole mount electron microscopy (Figure 6a,b). Specifically, the secondary aggregation response of platelets to exogenous stimuli is absent. Bleeding manifestations include spontaneous bruising (Figure 6c), epistaxis, menorrhagia, and prolonged oozing after trauma or minor surgery such as a tooth extraction (30). A platelet transfusion essentially cures this bleeding diathesis, and other interventions such as intravenous 1-desamino-8D-arginine vasopressin and topical thrombin ameliorate the situation. HPS patients vary enormously in their bleeding tendencies.
Besides hypopigmentation and bleeding, the complications of HPS appear somewhat subtype-specific. A colitis resembling Crohn’s disease occurs in up to one-third of patients with BLOC-3 defects, i.e., HPS-1 and HPS-4 (55). The colitis involves granulomas, erosions (Figure 6d), and/or inflammatory cells and generally responds to the anti-TNF-alpha agent, Remicade, as well as to steroids. In HPS-2 lymphocytes, lytic granules are reduced in number and function, leading to infections in childhood. A G-CSF-responsive neutropenia is also specific for HPS-2 (29, 52, 54). Ceroid lipofuscin, an amorphous, autofluorescent lipid-protein material (Figure 6e), has been found in the lysosomes of HPS-1 cells, including renal tubular cells, alveolar macrophages, and cells of the gastrointestinal tract, bone marrow, liver, spleen, lymph nodes, and heart (51). Ceroid lipofuscin may accumulate because cells cannot rapidly degrade mis-targeted vesicle membranes. Pulmonary fibrosis occurs only in HPS patients with subtypes 1 or 4; in HPS-1, upwards of 80% of patients develop this fatal complication in their thirties, forties, or fifties (Figure 6f). The cause of restrictive lung disease in HPS remains unknown; there is no effective therapy, although pirfenidone may have a salutary effect (31). BLOC-2 patients do not develop pulmonary fibrosis (52), and there is insufficient information to know if other HPS subtypes are prone to this complication (124).
Cell biology of HPS
BLOC-2 consists of the HPS3, HPS5 and HPS6 proteins, and its molecular mass (340±64 kDa) is consistent with it being a heterotrimer (324 kDa), though additional subunits cannot be excluded (24). HPS5 and HPS6 proteins interact directly, but no direct interactions with HPS3 have been reported (24). HPS3, 5, and 6 have no homology to known proteins and the only recognizable motif is a clathrin-binding domain (LLDFE) in HPS3 at residues 172–176. HPS3 co-immunoprecipitates with clathrin in melanocytes and GFP-HPS3 co-localizes with clathrin on small vesicles in the perinuclear area only when this motif is intact (43). BLOC-2 interacts with BLOC-1; immunoelectron microscopy localized components of both BLOC-2 (HPS3 and HPS6) and BLOC-1 (pallidin and dysbindin) to tubulovesicular elements of EEA1-positive early endosomes (25). In human HPS-3 and HPS-5 melanocytes, melanosome-targeted cargo (TYRP1, TYR) does not efficiently traffic to melanosomes, but instead resides in small vesicles throughout the cell (8, 44). BLOC-2 deficient melanocytes demonstrate normal early melanosome formation (PMEL17 staining) but later delivery of TYR and TYRP1 to melanosomes is perturbed, causing reduced staining of these markers in the dendritic tips, (Figure 8). Further, increased trafficking of TYRP1 via the cell membrane, demonstrated by increased internalization of anti-TYRP1 antibodies, occurs in BLOC-2, BLOC-1 and AP-3 deficient melanocytes (25, 44). These findings support a role for BLOC-2 in trafficking LRO components (e.g., TYRP1) from an early endosomal compartment to the developing LRO. In the absence of BLOC-2, these components are misdirected to the cell membrane where they are internalized and subsequently degraded. BLOC-2 may also assist in the secretion of lysosomes and related organelles, based on reduced lysosomal enzyme secretion by kidney and platelets from both ruby eye (HPS-6) and ruby eye-2 (HPS-5) mice (117). However, fibroblasts from cocoa (HPS-3) and ruby eye (HPS-6) mice showed normal secretory levels of the enzyme β-hexosaminidase, indicating that BLOC-2 is not critical for secretion of this lysosomal enzyme (24). In addition, an increase in the frequency and duration of transient fusion events in ruby-eye mast cells during stimulation suggests a function for the ruby eye gene product in regulating closure of cell fusion pores (88).
Figure 8. Distribution of melanogenic proteins TYRP1, TYR1 and PMEL17 in melanocytes of patients with LRO disorders.

Confocal immunofluorescence microscopy of primary epidermal melanocytes derived from a normally pigmented individual and patients with LRO biogenesis defects stained for (a–e) TYRP1 (anti-mouse MEL5), (f–j) TYR (anti-mouse Tyrosinase), and (k–o) PMEL17 (anti-mouse HMB45). All melanocytes were co-stained with TO-PRO-3 to visualize the nucleus.
(a,f,k) Normal melanocytes demonstrate punctuate (a) TYRP1, (f) TYR and (k) PMEL17 staining in the perinuclear region and throughout the dendrites with TYRP1 and TYR accumulation in the dendritic tips.
(b,g,l) BLOC-2 deficient melanocytes were derived from an HPS-5 patient compound heterozygous for a nonsense mutation (c.2624C>T; p.R865X) and a one base pair deletion (c.2264delT: P.L875fsX19) in exon 18 of HPS5. BLOC-2 deficient melanocytes display punctate staining of (b) TYRP1 and (g) TYR in the perinuclear region extending into the dendrites, but lack pronounced accumulation of TYRP1 and TYR in the tips. (l) PMEL17 staining in BLOC-2 deficient melanocytes is distributed throughout the cell as in normal melanocytes.
(c,h,m) BLOC-3 deficient melanocytes were derived from an HPS-1 patient homozygous for a 16 base-pair duplication (c.1472_1487dup16) in exon 15 of HPS1. In the absence of BLOC-3 (c) TYRP1 and (h) TYR are concentrated in the perinuclear/TGN region while (m) PMEL17 extends into the dendrites and appears normally distributed.
(d,i,n) GS2 melanocytes were derived from a patient compound heterozygous for two nonsense mutations (c.550C>T; p.R184X and c.598C>T; p.R200X) in Rab27A. In GS2 melanocytes, (d) TYRP1, (i) TYR, and (n) PMEL17 localize to the perinuclear area and do not occupy the dendritic tips.
(e,j,o) CHS melanocytes were derived from a patient carrying a nonsense mutation (c.1540C>T; p.R514X) in exon 5 and a one base pair deletion (c.9893delT; p.F3298fsX3304) in exon 43 in CHS1. In CHS melanocytes, (e) TYRP1 localizes to vesicular structures in the perinuclear area with some dendritic localization, (j) TYR accumulates in large granules that localize to the dendrites but not their tips, and (o) PMEL17 localizes to enlarged vesicular structures that are present in the perinuclear and dendritic area but not in the dendritic tips. All images are 1D projections of confocal z-sections. Scale bars = 10 μm.
HPS1 and HPS4 are components of BLOC-3, but no direct interaction has been demonstrated. Although the existence of another small subunit cannot be excluded (73, 83, 84), the molecular mass of BLOC-3 (140±30 kDa) is most consistent with BLOC-3 being a heterodimer (156 kDa). HPS1 and HPS4 contain no recognizable homology to each other or to other known proteins nor do they contain structural motifs that would help predict BLOC-3’s function. BLOC-3 is largely cytosolic, with a small fraction associated with membranes (73). In BLOC-3 deficient human melanocytes, TYRP1 and TYR are not properly trafficked to developing, early stage melanosomes and largely remain in the perinuclear/TGN region (Figure 8c,h). The mislocalization of TYR and TYRP1 results in reduced melanin synthesis (102). Formation of early stage melanosomes, marked by PMEL17 staining, appears unaffected in BLOC-3 deficient cells (Figure 8m). Ultrastuctural studies of the choroids of BLOC-3 deficient pale ear (HPS-1) and light ear (HPS-4) mice demonstrated macromelanosomes, suggested to result from abnormal melanosome fusion events (67). BLOC-3 may also be involved in general events effecting lysosome/late endosome biogenesis or movement. Light ear and pale ear mice have enlarged kidney lysosomes, containing increased amounts of hydrolases, with decreased secretion into the urine (75, 117). Pale ear fibroblasts showed reduced perinuclear localization of lysosomal and late endosomal markers compared to control fibroblasts (83). The possible existence of BLOC-4 and BLOC-5 complexes sharing some BLOC-3 subunits has been suggested (14).
AP3 recognizes and sorts proteins with tyrosine-based sorting signals (like LAMPs and TYR) for LRO targeting (89). Consequently, AP3-deficient (i.e., HPS-2) fibroblasts exhibit enhanced abnormal routing of LAMPs through the plasma membrane (21, 54), and AP3 deficient human and mouse melanocytes have an abnormal distribution of TYR (118). HPS-2 is the only HPS subtype that manifests immunodeficiency (21, 53, 54), apparently due to the severely impaired killing ability of HPS-2 CTLs. The secretory lysosomes of HPS-2 CTLs remain in the periphery, and the CTLs fail to polarize upon activation, presumably AP3 fails to sort a protein required forgranule polarization (112).
B. Chediak-Higashi Syndrome (CHS)
CHS (MIM 214500) results from mutations in the CHS1 gene, whose murine analog is the beige gene (56, 58). The clinical phenotype of CHS correlates somewhat with molecular genotype and cell biological phenotype (59, 126).
Clinical manifestations of CHS
Individuals with CHS manifest decreased pigmentation (Figure 7a), giant intracellular granules that are pathognomonic of the disease (Figure 7b), pigment clumping in hair shafts (Figure 7c), and a bleeding diathesis related to platelet dense bodies that are absent or reduced in number (56, 58). The granules, which are azurophilic and contain acid hydrolases and myeloperoxidase (56), are also present in CHS eosinophils, basophils, and monocytes. Children with CHS have life-threatening infections, primarily of the skin and respiratory systems. The pathogens are commonly Staphylococcus aureus and Streptococcus, gram-negative organisms, Candida, or Aspergillus. Immunoglobulins, antibody production, and phagocytosis are normal, but neutropenia is common and leucocytes display impaired migration (56, 58). Natural killer (NK) cells are decreased in function, causing impaired cellular immunity. Platelet counts in CHS are normal (56), but the secondary wave of platelet aggregation is impaired, with an increased ATP-to-ADP ratio and decreased platelet serotonin and calcium. CHS humans, as well as CHS mice (beige) and CHS cattle, have absent or markedly reduced platelet dense bodies (38, 56, 76).
Figure 7. Clinical characteristics of Chediak-Higashi syndrome and Griscelli syndrome.
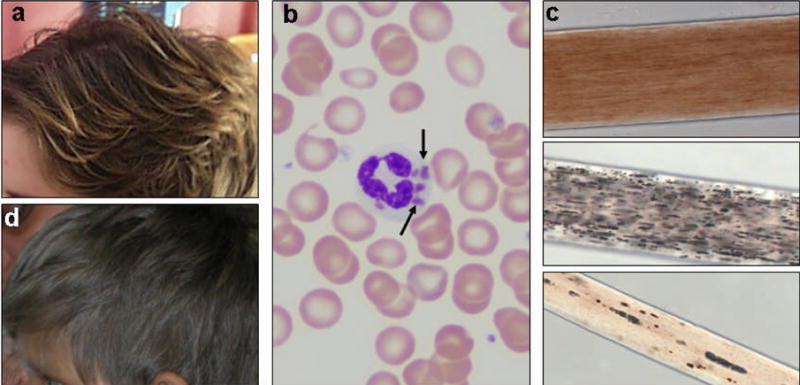
(a) Light brown hair in a girl with CHS, after bone marrow transplantation. (b) Wright stain of a peripheral blood smear from a classic CHS patient, showing abnormal giant granules in a polymorphonuclear leucocyte (arrows). (c) Light microscopy of hair shafts from a normal individual (top), a patient with CHS (middle), and a patient with GS2 (bottom). Uneven granularity characterizes the pigmentation pattern of the bottom two hair shafts. (d) Silvery gray hair of a boy with GS2.
The curious phenomenon of lymphohistiocytosis occurs in approximately 85% of CHS patients. In this fatal complication also called the accelerated phase, the unfettered proliferation of lymphocytes creates a lymphoma-like situation, with fever, anemia, neutropenia, and occasionally thrombocytopenia, hepatosplenomegaly and lymphadenopathy. Liver function tests, serum ferritin, and fibronectin may be elevated, and cellular immunity is decreased (56). Other disorders involving lymphohistiocytosis, such as Autoimmune Lymphoproliferative Syndrome (MIM 601859) or familial hemophagocytic lymphohistiocytosis (FLH2; MIM 603553) (60) have as their cause genetic defects in perforin or in the perforin receptor.
If CHS patients survive until adulthood, they develop signs and symptoms of neurological involvement, including neuropathies, autonomic dysfunction, atrophy, sensory deficits, areflexia, cerebellar signs, seizures, decreased ambulation, and cognitive defects. These findings, which appear independent of lymphohistocytic infiltration of the nervous system, are not prevented by bone marrow transplantation, which essentially cures the diathesis toward infections, lymphohistiocytosis, and immunological disorders of CHS.
Cell biology of CHS
The 429-kDa CHS1 protein is a member of the BEACH protein family and contains several N-terminal ARM/HEAT repeats, a C-terminal WD40 domain and a BEACH motif (56, 58). The exact cell biological function of CHS1 remains unknown. However, studies on other members of the BEACH protein family, and identification of CHS1 interacting proteins such as v-SNAREs, t-SNAREs, signaling protein 14–3-3, casein kinase II, and Hrs, suggest a role in membrane trafficking and/or organelle size-regulation (58). Many cell types in CHS manifest giant lysosomes and LROs. The melanosomes of CHS melanocytes are enlarged (56) and melanosome-specific markers such as PMEL17, TYRP1 and TYR mislocalize to large vesicular structures present in the cell body and occasionally within the dendrites, but not in the dendritic tips (Figure 8e,j,o) (126).
C. Griscelli Syndrome
Griscelli syndrome (GS) is a rare autosomal recessive disorder characterized by mild skin hypopigmentation and by immunological impairment, lymphohistiocytosis, or defects in the central nervous system (77, 78, 94). GS patients do not have an obvious bleeding tendency; whether they have a defect in platelet delta granules is not known. Three types of human GS are recognized, all displaying pigment dilution of the hair and skin (77, 78, 94).
Griscelli syndrome type 1 (GS1; MIM 214450) is caused by mutations in the MYO5A gene on chromosome 15q21, which encodes the actin-associated Myosin Va motor protein (94). Most GS1 patients display primary neurological impairment but no immunological deficiencies (22), resembling the dilute mouse. MYO5A undergoes alternative splicing involving six exons; in melanocytes, the presence of the alternative exon F is required for actin-dependent melanosome transport (127, 130). Since MYO5A exon F transcripts are abundantly expressed in melanocytes but not in brain, the phenotype of patients with mutations in MYO5A exon F is restricted to hypopigmentation (77). Elejalde syndrome (ES) is an autosomal recessive disorder with silver-colored hair, pigment abnormalities of the skin and severe neurological dysfunction, i.e., clinical features comparable to those of GS1 (10). ES patients do not manifest immunological impairment or the hemophagocytic syndrome. ES is thought to correspond to GS1 (10).
Griscelli syndrome type 2 (GS2; MIM 607624) results from mutations in the RAB27A gene on chromosome 15 (78). Clinical features include silver hair (Fig. 6D) with pigment clumping in hair shafts (Fig. 6C), infections, and lymphohistiocytosis. Infiltration of leukocytes in the brain can cause secondary neurological impairment. Primary central nervous system defects have not been observed in GS2 patients (10, 78). The ashen mouse is the murine counterpart of GS2.
Griscelli syndrome type 3 (GS3; MIM 609227) is caused by mutations in Melanophilin (MLPH), a member of the Rab effector family and the human homologue of the murine leaden gene. The only described GS2 patient has a homozygous R35W missense mutation in MLPH (77).
The clinical manifestations of GS result from defects in melanosome transport in melanocytes and lytic granule targeting in CTLs (7, 10, 112). In melanocytes, Rab27A in its GTP-bound form interacts with the membrane of mature melanosomes through its C20 geranylgeranyl lipid tail, added posttranslationally. Within the melanosomal membrane, Rab27A acts as a receptor for Myosin Va through an indirect interaction with the Rab27A effector Melanophilin (127, 130). The Rab27A-Melanophilin-MyosinVa complex results in accumulation of mature melanosomes in the actin-rich dendritic tips, a process necessary for transfer of melanosomes to keratinocytes for normal pigmentation. When Myosin Va, Rab27A or Melanophilin is mutated, the tripartite complex (Figure 3) fails to form and melanosome transport is impaired, resulting in perinuclear accumulation of melanosomes (Figure 8d,i,n). Skin hypopigmentation and silvery-gray hair in GS are not caused by defects in the melanosome biogenesis pathway but by ineffective capturing of melanosomes by the peripheral actin network (127, 130).
D. Platelet-granule specific disorders
“Storage pool deficiency” (SPD; MIM 185050) is the term used to describe the heterogeneous group of disorders characterized by deficiency of granule-bound substances in platelets (38, 62, 87, 125). The newer term “hypogranular platelet disorders” encompasses partial forms of SPD. Combined alpha/delta-SPD defines deficiency of both alpha and delta granules, whereas delta-SPD, which can be isolated or syndromic (as in HPS), refers to patients with delta granule defects.
Non-syndromic delta-SPD, or isolated delta-SPD, is characterized by mild to moderate bleeding due to deficiency of platelet dense granules without other systemic findings (38, 62). Isolated delta-SPD appears to be underdiagnosed. Nieuwenhuis et al. (87) reviewed 390 patients with a bleeding tendency; among the 145 with a prolonged bleeding time and normal platelet counts, 27 (18%) had congenital delta-SPD. Platelet aggregation studies were completely normal in 23% of the delta-SPD patients (87). Definitive diagnosis of delta-SPD requires electron microscopy and/or measurement of adenine nucleotides and serotonin to demonstrate the absence of dense granule-limiting membranes and contents (125). Autosomal dominant inheritance appears to be the primary mode of transmission (125). Mutations in RUNX1 (AML1, CBFA2) are associated with familial predisposition to acute myeloid leukemia, thrombocytopenia and dense granule deficiency (MIM 601399) (32).
Combined alpha/delta-SPD is characterized by decreased numbers of delta granules associated with a variable deficiency of alpha-granules (38, 62). The bleeding tendency in resembles that of delta-SPD. The frequency of alpha/delta-SPD is not known, but it is probably less common than isolated delta-SPD. The mode of inheritance appears to be autosomal dominant. The basic defect in most patients remains unknown. Mutations in the X-linked transcription factor GATA1 result in a form of hypogranular thrombocytopenia (MIM 305371) characterized by a generalized decrease in platelet organelles including alpha and delta granules (86).
Wiskott-Aldrich syndrome (WAS) (MIM 301000) is an X-linked microthrombocytopenia associated with immunodeficiency and eczema, caused by mutations in the WASP gene (121), coding for a protein that regulates signal-mediated actin cytoskeleton rearrangement. WAS platelets have markedly reduced delta granules, alpha-granules and mitochondria.
Thrombocytopenia Absent Radius syndrome (TAR) (MIM 274000) is characterized by thrombocytopenia and bilateral absence of the radii in the presence of thumbs (35). TAR is associated with a microdeletion on chromosome 1q21.1, but the phenotype develops only in the presence of an additional, as-yet-unknown modifier (64). Platelet counts of TAR patients are extremely low (15 to 30×1012/L) in infancy; consequently, 90% of cases are symptomatic within the first 4 months of life (35). However, platelet counts increase with age and may improve to almost normal by adulthood. Defects in platelet granules are reported in a subset of TAR patients.
E. Other LRO disorders
Newly reported LRO-related disorders involve mutations in SNAREs or associated proteins, rabs or rab effectors, VPS proteins or their interactors, microtubules or actin-binding proteins and molecular motors, membrane lipid regulators, and in genes of unknown function (92). For example, mutations in Munc13-4 or in syntaxin 11, encoding SNARE proteins, cause Familial Hemophagocytic Lymphohistocytosis (FHL), i.e., FHL3 (MIM 608898) and FHL4 (MIM 603552), respectively (60, 132). Arthrogryposis-renal dysfunction-cholestasis (ARC; MIM 208085) is caused by mutations in VPS33B, coding for a Sec1/Munc18-related SNARE-interacting protein involved in late endosomal membrane dynamics (34). Mutated SNAP-29 underlies the neurocutaneous CEDNIK syndrome (MIM 609528), in which developmental nervous system and skin lamellar granule defects occur due to impaired SNARE machinery (108). Defective kinesin molecular motors KIF1B and KIF5A cause Charcot-Marie-Tooth disease type 2A1 (MIM 118210) and spastic paraplegia 10 (MIM 604187), respectively (47). Mutated small GTPases or their regulating factors cause: Charcot-Marie-Tooth disease type 2B (MIM 600882), defective in the late endosomal GTPase Rab7 (48); choroideremia (MIM 303100), defective in Rab Escort Protein-1, REP-1, functioning in prenylation of rab GTPases (97); and periventricular heterotopia with microcephaly (MIM 608097), defective in ARFGEF2, a guanine nucleotide exchange factor for ARF (107). Finally, Charcot-Marie-Tooth disease type 4J (MIM 611228) and the pale tremor mutant mouse have late endosome-lysosome defects due to mutaions in FIG 4. FIG 4 codes for a homologue of a yeast SAC (suppressor of actin) domain phosphatidylinositol-3,5-bisphosphate 5-phosphatase located in the vacuolar membrane (16).
Various case reports and genetically unclassified disorders of hypopigmentation and/or bleeding diathesis and/or immunodeficiency have been described, including Cross or Kramer syndrome (MIM 257800), Vici syndrome (MIM 242840), Preus syndrome (MIM 257790), and OOCH (MIM 601220).
CONCLUDING REMARKS
Defining the cellular and molecular events regulating LRO biogenesis and trafficking remains a work in progress, aided enormously by mutant animal and human pigmentation models. The catalog of components involved in LRO biogenesis will expand with evolving experimental tools such as whole genome sequencing, gene silencing, fluorescent imaging, and in vitro expression and transport assays. Insights into mechanisms should translate into rational therapeutic interventions for these intriguing disorders of lysosome-related organelles.
SUMMARY POINTS
LROs share features with lysosomes but have distinct morphology, composition and/or functions.
LROs are maintained by the regulated flow of proteins and membranes/vesicles among a complex endosomal machinery, providing specificity of time and place. Chaperones for this machinery include a cytoskeletal system of tubules and filaments, coat associated proteins, BLOCs, SNAREs, syntaxins, rabs, motor proteins, and membrane lipids.
The biogenesis of LROs employs a common machinery, whose genetic defects can affect all LROs or only an individual LRO. Consequently, a variety of clinical features occur in affected individuals, including hypopigmentation, bleeding and immune deficiency.
Cell biologists study LROs because of the lessons they can impart with respect to vesicle formation in general.
Abnormalities in both lysosomes and LROs have been observed in certain human genetic diseases, including Hermansky-Pudlak syndrome, Griscelli syndrome and Chediak-Higashi syndrome.
Novel players in LRO biogenesis are revealed by human and animal mutations. These models continue to provide invaluable resources for elucidating the complex pathways involved.
FUTURE ISSUES
An exact definition of LROs has not been established, so the precise number of LROs remains unknown. To be considered an LRO, an organelle must possess an acidic lumen, characteristic lysosomal proteins and a specialized function, usually related to storage or secretion. Accesibility to endocytic markers may be included as another criterion.
In the future, newly recognized organelles may be considered LROs.
More human and animal models of albinism/bleeding/immune deficiency/other clinical features will be recognized as LRO disorders. Cells of these patients (or their rodent, fly, zebrafish, or yeast counterparts) will continue to be important tools for analyzing membrane trafficking.
The functions of several proteins in LRO biogenesis, including the BLOC-1, BLOC-2, and BLOC3 subunits and the LYST protein, await elucidation. Understanding these functions will move us toward rational therapeutics for Hermansky-Pudlak syndrome, Chediak-Higashi syndome, and related disorders.
Acknowledgments
This work was supported by the Intramural Research program of the National Human Genome Research Institute (NHGRI), National Institutes of Health, Bethesda, Maryland, USA.
List of important Abbreviations and Acronyms
- LRO
Lysosome-related organelle; An organelle that shares features with the lysosome but has a distinct morphology, composition and/or function
- TGN
Trans-Golgi network; A membranous network of vesicles and tubules located at the trans face of the Golgi apparatus, where it plays a key role in the sorting and targeting of secreted proteins to the correct cellular destinations
- BLOC
Biogenesis of lysosome-related organelles complex
- SNARE
Soluble N-ethylmaleimide-sensitive-factor attachment protein (SNAP) receptor; SNARE proteins that form bridging complexes that are required for intracellular membrane fusion in eukaryotes
- VPS
Vacuolar protein for sorting; Defects in VPS in yeast affect vacuolar biogenesis; their homologues are good candidates for causing LRO disorders in higher organisms
- MPR
Mannose 6-phosphate receptor; A receptor that sorts and delivers newly synthesized lysosomal enzymes at the TGN and assists in their delivery to endosomes, after which the receptor recycles back to the TGN
- TYR/TYRP1
Tyrosinase (TYR) and Tyrosinase-related protein-1 (TYRP1); Melanocyte-specific enzymes that catalyze distinct reactions in melanin synthesis in melanosomes
- PMEL17 (also called gp100)
A melanocyte-specific glycoprotein enriched in the lumen of premelanosomes, where it forms characteristic melanosomal striations, upon which melanin is deposited as the melanosome matures
- MVB
Multiple vesicular body; endosomal structure that contains multiple internal vesicles; MBVs are intermediate structures in the endosomal sorting pathway to lysosomes and/or lysosome-related organelles
- LAMP
Lysosomal associated membrane protein; LAMPs are heavily glycosylated and localized in and targeted to lysosomal membranes.
Footnotes
DISCLOSURE
The authors are not aware of any biases that might be perceived as affecting the objectivity of this review.
Contributor Information
Marjan Huizing, Email: mhuizing@mail.nih.gov.
Amanda Helip-Wooley, Email: ahwooley@mail.nih.gov.
Wendy Westbroek, Email: wwestbro@mail.nih.gov.
Meral Gunay-Aygun, Email: mgaygun@mail.nih.gov.
William A. Gahl, Email: bgahl@helix.nih.gov.
LITERATURE CITED
- 1.Ali B, Seabra M. Targeting of Rab GTPases to cellular membranes. Biochem Soc Trans. 2005;33:652–6. doi: 10.1042/BST0330652. [DOI] [PubMed] [Google Scholar]
- 2.Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet. 2003;113:10–7. doi: 10.1007/s00439-003-0933-5. [DOI] [PubMed] [Google Scholar]
- 3.Anikster Y, Huizing M, White J, Shevchenko YO, Fitzpatrick DL, et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet. 2001;28:376–80. doi: 10.1038/ng576. [DOI] [PubMed] [Google Scholar]
- 4.Barral DC, Seabra MC. The melanosome as a model to study organelle motility in mammals. Pigment Cell Res. 2004;17:111–8. doi: 10.1111/j.1600-0749.2004.00138.x. [DOI] [PubMed] [Google Scholar]
- 5.Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol. 2003;161:521–33. doi: 10.1083/jcb.200302072. This manuscript describes MVBs as intermediates in the generation of stage II melanosomes, and it shows that PMEL17 is the main component of the intralumenal fibrils of stage II melanosomes. These fibrils are generated by cleavage of PMEL17 in a post-Golgi compartment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohn G, Allroth A, Brandes G, Thiel J, Glocker E, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2007;13:38–45. doi: 10.1038/nm1528. [DOI] [PubMed] [Google Scholar]
- 7.Bohn G, Welte K, Klein C. Severe congenital neutropenia: new genes explain an old disease. Curr Opin Rheumatol. 2007;19:644–50. doi: 10.1097/BOR.0b013e3282f05cc2. [DOI] [PubMed] [Google Scholar]
- 8.Boissy RE, Richmond B, Huizing M, Helip-Wooley A, Zhao Y, et al. Melanocyte-specific proteins are aberrantly trafficked in melanocytes of Hermansky-Pudlak syndrome-type 3. Am J Pathol. 2005;166:231–40. doi: 10.1016/S0002-9440(10)62247-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–66. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 10.Cahali JB, Fernandez SA, Oliveira ZN, Machado MC, Valente NS, Sotto MN. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479–82. doi: 10.1111/j.0736-8046.2004.21414.x. [DOI] [PubMed] [Google Scholar]
- 11.Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell. 2007;12:671–82. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Caviston JP, Holzbaur EL. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006;16:530–7. doi: 10.1016/j.tcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Chi E, Pruiett J, Lagunoff D. Abnormal lamellar bodies in type II pneumocytes and increased lung surface active material in the beige mouse. J Histochem Cytochem. 1975;23:863–6. doi: 10.1177/23.11.1242724. [DOI] [PubMed] [Google Scholar]
- 14.Chiang PW, Oiso N, Gautam R, Suzuki T, Swank RT, Spritz RA. The Hermansky-Pudlak syndrome 1 (HPS1) and HPS4 proteins are components of two complexes, BLOC-3 and BLOC-4, involved in the biogenesis of lysosome-related organelles. J Biol Chem. 2003;278:20332–7. doi: 10.1074/jbc.M300090200. [DOI] [PubMed] [Google Scholar]
- 15.Chintala S, Li W, Lamoreux ML, Ito S, Wakamatsu K, et al. Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. Proc Natl Acad Sci U S A. 2005;102:10964–9. doi: 10.1073/pnas.0502856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cordonnier MN, Dauzonne D, Louvard D, Coudrier E. Actin filaments and myosin I alpha cooperate with microtubules for the movement of lysosomes. Mol Biol Cell. 2001;12:4013–29. doi: 10.1091/mbc.12.12.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coulombe PA, Bousquet O, Ma L, Yamada S, Wirtz D. The ‘ins’ and ‘outs’ of intermediate filament organization. Trends Cell Biol. 2000;10:420–8. doi: 10.1016/s0962-8924(00)01828-6. [DOI] [PubMed] [Google Scholar]
- 19.Deacon SW, Serpinskaya AS, Vaughan PS, Lopez Fanarraga M, Vernos I, et al. Dynactin is required for bidirectional organelle transport. J Cell Biol. 2003;160:297–301. doi: 10.1083/jcb.200210066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dell’Angelica EC, Mullins C, Caplan S, Bonifacino JS. Lysosome-related organelles. Faseb J. 2000;14:1265–78. doi: 10.1096/fj.14.10.1265. [DOI] [PubMed] [Google Scholar]
- 21.Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. This paper demonstrates that mutations of an AP3 subunit cause Hermansky-Pudlak syndrome type 2. It shows for the first time that defects in intracellular vesicle transport pathways underlie Hermansky-Pudlak syndrome. [DOI] [PubMed] [Google Scholar]
- 22.Desnos C, Huet S, Darchen F. ‘Should I stay or should I go?’: myosin V function in organelle trafficking. Biol Cell. 2007;99:411–23. doi: 10.1042/BC20070021. [DOI] [PubMed] [Google Scholar]
- 23.Detter JC, Zhang Q, Mules EH, Novak EK, Mishra VS, et al. Rab geranylgeranyl transferase alpha mutation in the gunmetal mouse reduces Rab prenylation and platelet synthesis. Proc Natl Acad Sci U S A. 2000;97:4144–9. doi: 10.1073/pnas.080517697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Pietro SM, Falcon-Perez JM, Dell’Angelica EC. Characterization of BLOC-2, a complex containing the Hermansky-Pudlak syndrome proteins HPS3, HPS5 and HPS6. Traffic. 2004;5:276–83. doi: 10.1111/j.1600-0854.2004.0171.x. [DOI] [PubMed] [Google Scholar]
- 25.Di Pietro SM, Falcon-Perez JM, Tenza D, Setty SR, Marks MS, et al. BLOC-1 interacts with BLOC-2 and the AP-3 complex to facilitate protein trafficking on endosomes. Mol Biol Cell. 2006;17:4027–38. doi: 10.1091/mbc.E06-05-0379. This study applies biochemical, morphological and genetic techniques to demonstrate that BLOC-1 physically and functionally interacts with AP-3 and with BLOC-2. It also shows that BLOC-1 and BLOC-2 localize mainly to early endosome-associated tubules. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falcon-Perez JM, Romero-Calderon R, Brooks ES, Krantz DE, Dell’Angelica EC. The Drosophila pigmentation gene pink (p) encodes a homologue of human Hermansky-Pudlak syndrome 5 (HPS5) Traffic. 2007;8:154–68. doi: 10.1111/j.1600-0854.2006.00514.x. [DOI] [PubMed] [Google Scholar]
- 27.Falcon-Perez JM, Starcevic M, Gautam R, Dell’Angelica EC. BLOC-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet-dense granules. J Biol Chem. 2002;277:28191–9. doi: 10.1074/jbc.M204011200. [DOI] [PubMed] [Google Scholar]
- 28.Fischer A, Latour S, de Saint Basile G. Genetic defects affecting lymphocyte cytotoxicity. Curr Opin Immunol. 2007;19:348–53. doi: 10.1016/j.coi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Fontana S, Parolini S, Vermi W, Booth S, Gallo F, et al. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood. 2006;107:4857–64. doi: 10.1182/blood-2005-11-4398. [DOI] [PubMed] [Google Scholar]
- 30.Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) N Engl J Med. 1998;338:1258–64. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 31.Gahl WA, Brantly M, Troendle J, Avila NA, Padua A, et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2002;76:234–42. doi: 10.1016/s1096-7192(02)00044-6. This study describes a clinical treatment study of the small molecule pirfenidone, which was found to slow the progression of pulmonary fibrosis in a subset of Hermansky-Pudlak syndrome patients. [DOI] [PubMed] [Google Scholar]
- 32.Ganly P, Walker LC, Morris CM. Familial mutations of the transcription factor RUNX1 (AML1, CBFA2) predispose to acute myeloid leukemia. Leuk Lymphoma. 2004;45:1–10. doi: 10.1080/1042819031000139611. [DOI] [PubMed] [Google Scholar]
- 33.Gillard BK, Clement R, Colucci-Guyon E, Babinet C, Schwarzmann G, et al. Decreased synthesis of glycosphingolipids in cells lacking vimentin intermediate filaments. Exp Cell Res. 1998;242:561–72. doi: 10.1006/excr.1998.4126. [DOI] [PubMed] [Google Scholar]
- 34.Gissen P, Johnson CA, Morgan NV, Stapelbroek JM, Forshew T, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400–4. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 35.Greenhalgh KL, Howell RT, Bottani A, Ancliff PJ, Brunner HG, et al. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet. 2002;39:876–81. doi: 10.1136/jmg.39.12.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gruenberg J. The endocytic pathway: a mosaic of domains. Nat Rev Mol Cell Biol. 2001;2:721–30. doi: 10.1038/35096054. [DOI] [PubMed] [Google Scholar]
- 37.Gullberg U, Bengtsson N, Bulow E, Garwicz D, Lindmark A, Olsson I. Processing and targeting of granule proteins in human neutrophils. J Immunol Methods. 1999;232:201–10. doi: 10.1016/s0022-1759(99)00177-5. [DOI] [PubMed] [Google Scholar]
- 38.Gunay-Aygun M, Huizing M, Gahl WA. Molecular defects that affect platelet dense granules. Semin Thromb Hemost. 2004;30:537–47. doi: 10.1055/s-2004-835674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hannah MJ, Williams R, Kaur J, Hewlett LJ, Cutler DF. Biogenesis of Weibel-Palade bodies. Semin Cell Dev Biol. 2002;13:313–24. doi: 10.1016/s1084-9521(02)00061-7. [DOI] [PubMed] [Google Scholar]
- 40.Hara M, Yaar M, Byers HR, Goukassian D, Fine RE, et al. Kinesin participates in melanosomal movement along melanocyte dendrites. J Invest Dermatol. 2000;114:438–43. doi: 10.1046/j.1523-1747.2000.00894.x. [DOI] [PubMed] [Google Scholar]
- 41.Harding C. Intracellular organelles involved in antigen processing and the binding of peptides to class II MHC molecules. Semin Immun. 1995;7:355–60. doi: 10.1006/smim.1995.0040. [DOI] [PubMed] [Google Scholar]
- 42.Heijnen HF, Debili N, Vainchencker W, Breton-Gorius J, Geuze HJ, Sixma JJ. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood. 1998;91:2313–25. [PubMed] [Google Scholar]
- 43.Helip-Wooley A, Westbroek W, Dorward H, Mommaas M, Boissy RE, et al. Association of the Hermansky-Pudlak syndrome type-3 protein with clathrin. BMC Cell Biol. 2005;6:33. doi: 10.1186/1471-2121-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helip-Wooley A, Westbroek W, Dorward HM, Koshoffer A, Huizing M, et al. Improper trafficking of melanocyte-specific proteins in Hermansky-Pudlak syndrome type-5. J Invest Dermatol. 2007;127:1471–8. doi: 10.1038/sj.jid.5700737. This article employs BLOC-2 deficient human melanocytes to demonstrate that early stage melanosome formation and PMEL17 trafficking are preserved in these cells. Other melanosome-targeted cargo (TYRP1, TYR) are mistrafficked and fail to be efficiently delivered to melanosomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hermos CR, Huizing M, Kaiser-Kupfer MI, Gahl WA. Hermansky-Pudlak syndrome type 1: gene organization, novel mutations, and clinical-molecular review of non-Puerto Rican cases. Hum Mutat. 2002;20:482. doi: 10.1002/humu.9097. [DOI] [PubMed] [Google Scholar]
- 46.Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8:562–73. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- 47.Hirokawa N, Takemura R. Biochemical and molecular characterization of diseases linked to motor proteins. Trends Biochem Sci. 2003;28:558–65. doi: 10.1016/j.tibs.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 48.Houlden H, King RH, Muddle JR, Warner TT, Reilly MM, et al. A novel RAB7 mutation associated with ulcero-mutilating neuropathy. Ann Neurol. 2004;56:586–90. doi: 10.1002/ana.20281. [DOI] [PubMed] [Google Scholar]
- 49.Huizing M, Anikster Y, Fitzpatrick DL, Jeong AB, D’Souza M, et al. Hermansky-Pudlak syndrome type 3 in Ashkenazi Jews and other non-Puerto Rican patients with hypopigmentation and platelet storage-pool deficiency. Am J Hum Genet. 2001;69:1022–32. doi: 10.1086/324168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huizing M, Boissy RE, Gahl WA. Hermansky-Pudlak syndrome: vesicle formation from yeast to man. Pigment Cell Res. 2002;15:405–19. doi: 10.1034/j.1600-0749.2002.02074.x. [DOI] [PubMed] [Google Scholar]
- 51.Huizing M, Gahl WA. Disorders of vesicles of lysosomal lineage: the Hermansky-Pudlak syndromes. Curr Mol Med. 2002;2:451–67. doi: 10.2174/1566524023362357. [DOI] [PubMed] [Google Scholar]
- 52.Huizing M, Hess R, Dorward H, Claassen DA, Helip-Wooley A, et al. Cellular, molecular and clinical characterization of patients with Hermansky-Pudlak syndrome type 5. Traffic. 2004;5:711–22. doi: 10.1111/j.1600-0854.2004.00208.x. [DOI] [PubMed] [Google Scholar]
- 53.Huizing M, Sarangarajan R, Strovel E, Zhao Y, Gahl WA, Boissy RE. AP-3 mediates tyrosinase but not TRP-1 trafficking in human melanocytes. Mol Biol Cell. 2001;12:2075–85. doi: 10.1091/mbc.12.7.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huizing M, Scher C, Strovel E, Fitzpatrick D, Hartnell L, et al. Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome type 2. Pediatr Res. 2002;51:150–8. doi: 10.1203/00006450-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 55.Hussain N, Quezado M, Huizing M, Geho D, White JG, et al. Intestinal disease in Hermansky-Pudlak syndrome: occurrence of colitis and relation to genotype. Clin Gastroenterol Hepatol. 2006;4:73–80. doi: 10.1016/s1542-3565(05)00858-x. [DOI] [PubMed] [Google Scholar]
- 56.Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999;68:283–303. doi: 10.1006/mgme.1999.2927. [DOI] [PubMed] [Google Scholar]
- 57.Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, et al. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol. 2001;11:1680–5. doi: 10.1016/s0960-9822(01)00531-0. [DOI] [PubMed] [Google Scholar]
- 58.Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15:22–9. doi: 10.1097/MOH.0b013e3282f2bcce. [DOI] [PubMed] [Google Scholar]
- 59.Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet. 2002;108:16–22. [PubMed] [Google Scholar]
- 60.Katano H, Cohen JI. Perforin and lymphohistiocytic proliferative disorders. Br J Haematol. 2005;128:739–50. doi: 10.1111/j.1365-2141.2004.05305.x. [DOI] [PubMed] [Google Scholar]
- 61.King RAHV, Creel DJ, Oetting WS. Albinism. In: Scriver BACR, Sly WS, Valle B, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 5587–627. [Google Scholar]
- 62.King SM, Reed GL. Development of platelet secretory granules. Semin Cell Dev Biol. 2002;13:293–302. doi: 10.1016/s1084952102000599. [DOI] [PubMed] [Google Scholar]
- 63.Kjeldsen L, Calafat J, Borregaard N. Giant granules of neutrophils in Chediak-Higashi syndrome are derived from azurophil granules but not from specific and gelatinase granules. J Leukoc Biol. 1998;64:72–7. doi: 10.1002/jlb.64.1.72. [DOI] [PubMed] [Google Scholar]
- 64.Klopocki E, Schulze H, Strauss G, Ott CE, Hall J, et al. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet. 2007;80:232–40. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- 66.Kramer H. Sorting out signals in fly endosomes. Traffic. 2002;3:87–91. doi: 10.1034/j.1600-0854.2002.030201.x. [DOI] [PubMed] [Google Scholar]
- 67.Li W, Rusiniak ME, Chintala S, Gautam R, Novak EK, Swank RT. Murine Hermansky-Pudlak syndrome genes: regulators of lysosome-related organelles. Bioessays. 2004;26:616–28. doi: 10.1002/bies.20042. [DOI] [PubMed] [Google Scholar]
- 68.Li W, Zhang Q, Oiso N, Novak EK, Gautam R, et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) Nat Genet. 2003;35:84–9. doi: 10.1038/ng1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lloyd V, Ramaswami M, Kramer H. Not just pretty eyes: Drosophila eye-colour mutations and lysosomal delivery. Trends Cell Biol. 1998;8:257–9. doi: 10.1016/s0962-8924(98)01270-7. [DOI] [PubMed] [Google Scholar]
- 70.Loftus S, Larson DM, Baxter LL, Antonellis A, Chen Y, Wu X, Jiang Y, Bittner M, Hammer JA, 3rd, Pavan WJ. Mutation of melanosome protein RAB38 in chocolate mice. Proc Natl Acad Sci USA. 2002;99:4471–6. doi: 10.1073/pnas.072087599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marks MS, Seabra MC. The melanosome: membrane dynamics in black and white. Nat Rev Mol Cell Biol. 2001;2:738–48. doi: 10.1038/35096009. [DOI] [PubMed] [Google Scholar]
- 72.Marone G, Casolaro V, Patella V, Florio G, Triggiani M. Molecular and cellular biology of mast cells and basophils. Int Arch Allergy Immunol. 1997;114:207–17. doi: 10.1159/000237670. [DOI] [PubMed] [Google Scholar]
- 73.Martina J, Moriyama K, Bonifacino J. BLOC-3, a protein complex containing the Hermansky-Pudlak syndrome gene products HPS1 and HPS4. J Biol Chem. 2003;278:29376–84. doi: 10.1074/jbc.M301294200. [DOI] [PubMed] [Google Scholar]
- 74.McNicol A, Israels SJ. Platelet dense granules: structure, function and implications for haemostasis. Thromb Res. 1999;95:1–18. doi: 10.1016/s0049-3848(99)00015-8. [DOI] [PubMed] [Google Scholar]
- 75.Meisler M, Levy J, Sansone F, Gordon M. Morphologic and biochemical abnormalities of kidney lysosomes in mice with an inherited albinism. Am J Pathol. 1980;101:581–93. [PMC free article] [PubMed] [Google Scholar]
- 76.Menard M, Meyers KM. Storage pool deficiency in cattle with the Chediak-Higashi syndrome results from an absence of dense granule precursors in their megakaryocytes. Blood. 1988;72:1726–34. [PubMed] [Google Scholar]
- 77.Menasche G, Ho CH, Sanal O, Feldmann J, Tezcan I, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1) J Clin Invest. 2003;112:450–6. doi: 10.1172/JCI18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25:173–6. doi: 10.1038/76024. This paper is the first demonstration that Griscelli syndrome is heterogeneous, and that the majority of patients with the classic syndrome are affected by mutations in RAB27A. [DOI] [PubMed] [Google Scholar]
- 79.Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005;15:467–76. doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 80.Morgan NV, Pasha S, Johnson CA, Ainsworth JR, Eady RAJ, et al. A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8) Am J Hum Genet. 2006;78:160–6. doi: 10.1086/499338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Munro S. Organelle identity and the targeting of peripheral membrane proteins. Curr Opin Cell Biol. 2002;14:506–14. doi: 10.1016/s0955-0674(02)00350-2. [DOI] [PubMed] [Google Scholar]
- 82.Nakatani Y, Nakamura N, Sano J, Inayama Y, Kawano N, Yamanaka S, Miyagi Y, Nagashima Y, Ohbayashi C, Mizushima M, Manabe T, Kuroda M, Yokoi T, Matsubara O. Interstitial pneumonia in Hermansky-Pudlak syndrome: significance of florid foamy swelling/degeneration (giant lamellar body degeneration) of type-2 pneumocytes. Virchos Arch. 2000;437:304–13. doi: 10.1007/s004280000241. [DOI] [PubMed] [Google Scholar]
- 83.Nazarian R, Falcon-Perez JM, Dell’Angelica EC. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): a complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc Natl Acad Sci U S A. 2003;100:8770–5. doi: 10.1073/pnas.1532040100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nazarian R, Huizing M, Helip-Wooley A, Starcevic M, Gahl W, Dell’angelica E. An immunoblotting assay to facilitate the molecular diagnosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2007 doi: 10.1016/j.ymgme.2007.09.001. online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nazarian R, Starcevic M, Spencer MJ, Dell’Angelica EC. Reinvestigation of the dysbindin subunit of BLOC-1 (biogenesis of lysosome-related organelles complex-1) as a dystrobrevin-binding protein. Biochem J. 2006;395:587–98. doi: 10.1042/BJ20051965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24:266–70. doi: 10.1038/73480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nieuwenhuis HK, Akkerman JW, Sixma JJ. Patients with a prolonged bleeding time and normal aggregation tests may have storage pool deficiency: studies on one hundred six patients. Blood. 1987;70:620–3. [PubMed] [Google Scholar]
- 88.Oberhauser A, Fernandez J. A fusion pore phenotype in mast cells of the ruby-eye mouse. Proc Natl Acad Sci USA. 1996;93:14349–54. doi: 10.1073/pnas.93.25.14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ohno H, Aguilar RC, Yeh D, Taura D, Saito T, Bonifacino JS. The medium subunits of adaptor complexes recognize distinct but overlapping sets of tyrosine-based sorting signals. J Biol Chem. 1998;273:25915–21. doi: 10.1074/jbc.273.40.25915. [DOI] [PubMed] [Google Scholar]
- 90.Oiso N, Riddle SR, Serikawa T, Kuramoto T, Spritz RA. The rat Ruby (R) locus is Rab38: identical mutations in Fawn-hooded and Tester-Moriyama rats derived from an ancestral Long Evans rat sub-strain. Mamm Genome. 2004;15:307–14. doi: 10.1007/s00335-004-2337-9. [DOI] [PubMed] [Google Scholar]
- 91.Oiso N, Riddle SR, Serikawa T, Kuramoto T, Spritz RA. The rat Ruby (R) locus is Rab38: identical mutations in Fawn-hooded and Tester-Moriyama rats derived from an ancestral Long Evans rat sub-strain. Mamm Genome. 2004;15:307–14. doi: 10.1007/s00335-004-2337-9. [DOI] [PubMed] [Google Scholar]
- 92.Olkkonen VM, Ikonen E. When intracellular logistics fails--genetic defects in membrane trafficking. J Cell Sci. 2006;119:5031–45. doi: 10.1242/jcs.03303. [DOI] [PubMed] [Google Scholar]
- 93.Ong LL, Lim AP, Er CP, Kuznetsov SA, Yu H. Kinectin-kinesin binding domains and their effects on organelle motility. J Biol Chem. 2000;275:32854–60. doi: 10.1074/jbc.M005650200. [DOI] [PubMed] [Google Scholar]
- 94.Pastural E, Barrat FJ, Dufourcq-Lagelouse R, Certain S, Sanal O, et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet. 1997;16:289–92. doi: 10.1038/ng0797-289. [DOI] [PubMed] [Google Scholar]
- 95.Pelham HR. SNAREs and the specificity of membrane fusion. Trends Cell Biol. 2001;11:99–101. doi: 10.1016/s0962-8924(01)01929-8. [DOI] [PubMed] [Google Scholar]
- 96.Pfister KK, Shah PR, Hummerich H, Russ A, Cotton J, et al. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2006;2:e1. doi: 10.1371/journal.pgen.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rak A, Pylypenko O, Niculae A, Pyatkov K, Goody RS, Alexandrov K. Structure of the Rab7:REP-1 complex: insights into the mechanism of Rab prenylation and choroideremia disease. Cell. 2004;117:749–60. doi: 10.1016/j.cell.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 98.Raposo G, Marks M. Melanosomes--dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 2007;8:786–97. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Raposo G, Marks MS, Cutler DF. Lysosome-related organelles: driving post-Golgi compartments into specialisation. Curr Opin Cell Biol. 2007;19:394–401. doi: 10.1016/j.ceb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raposo G, Tenza D, Murphy D, Berson J, Marks M. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol. 2001;152:809–24. doi: 10.1083/jcb.152.4.809. This article describes the model for melanosome biogenesis discussed in the current review. It is the first paper to demonstrate that melanosomes are distinct from lysosomes, with a common precursor at the stage I melanosome/vacuolar early endosome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rendu F, Brohard-Bohn B. The platelet release reaction: granules’ constituents, secretion and functions. Platelets. 2001;12:261–73. doi: 10.1080/09537100120068170. [DOI] [PubMed] [Google Scholar]
- 102.Sarangarajan R, Budev A, Zhao Y, Gahl W, Boissy R. Abnormal translocation of tyrosinase and tyrosinase-related protein 1 in cutaneous melanocytes of Hermansky-Pudlak Syndrome and in melanoma cells transfected with anti-sense HPS1 cDNA. J Invest Dermatol. 2001;117:641–6. doi: 10.1046/j.0022-202x.2001.01435.x. [DOI] [PubMed] [Google Scholar]
- 103.Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, et al. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med. 1998;187:1205–13. doi: 10.1084/jem.187.8.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schonthaler HB, Fleisch VC, Biehlmaier O, Makhankov Y, Rinner O, et al. The zebrafish mutant lbk/vam6 resembles human multisystemic disorders caused by aberrant trafficking of endosomal vesicles. Development. 2008;135:387–99. doi: 10.1242/dev.006098. [DOI] [PubMed] [Google Scholar]
- 105.Seabra MC, Mules EH, Hume AN. Rab GTPases, intracellular traffic and disease. Trends Mol Med. 2002;8:23–30. doi: 10.1016/s1471-4914(01)02227-4. [DOI] [PubMed] [Google Scholar]
- 106.Setty SR, Tenza D, Truschel ST, Chou E, Sviderskaya EV, et al. BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol Biol Cell. 2007;18:768–80. doi: 10.1091/mbc.E06-12-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36:69–76. doi: 10.1038/ng1276. [DOI] [PubMed] [Google Scholar]
- 108.Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, Rapaport D, Goldsher D, et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet. 2005;77:242–51. doi: 10.1086/432556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stahlman M, Gray M, Falconieri M, Whitsett J, Weaver T. Lamellar body formation in normal and surfactant B-deficient mice. Lab Invest. 2000;80:395–402. doi: 10.1038/labinvest.3780044. [DOI] [PubMed] [Google Scholar]
- 110.Starcevic M, Dell’Angelica EC. Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1) J Biol Chem. 2004;279:28393–401. doi: 10.1074/jbc.M402513200. [DOI] [PubMed] [Google Scholar]
- 111.Stenbeck G. Formation and function of the ruffled border in osteoclasts. Semin Cell Dev Biol. 2002;13:285–92. doi: 10.1016/s1084952102000587. [DOI] [PubMed] [Google Scholar]
- 112.Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: the secrets of secretory lysosomes. Science. 2004;305:55–9. doi: 10.1126/science.1095291. [DOI] [PubMed] [Google Scholar]
- 113.Strom M, Hume AN, Tarafder AK, Barkagianni E, Seabra MC. A family of Rab27-binding proteins. Melanophilin links Rab27a and myosin Va function in melanosome transport. J Biol Chem. 2002;277:25423–30. doi: 10.1074/jbc.M202574200. [DOI] [PubMed] [Google Scholar]
- 114.Styers ML, Salazar G, Love R, Peden AA, Kowalczyk AP, Faundez V. The endo-lysosomal sorting machinery interacts with the intermediate filament cytoskeleton. Mol Biol Cell. 2004;15:5369–82. doi: 10.1091/mbc.E04-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Suarez-Quian CA. The distribution of four lysosomal integral membrane proteins (LIMPs) in rat basophilic leukemia cells. Tissue Cell. 1987;19:495–504. doi: 10.1016/0040-8166(87)90043-7. [DOI] [PubMed] [Google Scholar]
- 116.Suzuki T, Li W, Zhang Q, Karim A, Novak EK, et al. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet. 2002;30:321–4. doi: 10.1038/ng835. [DOI] [PubMed] [Google Scholar]
- 117.Swank RT, Novak EK, McGarry MP, Rusiniak ME, Feng L. Mouse models of Hermansky Pudlak syndrome: a review. Pigment Cell Res. 1998;11:60–80. doi: 10.1111/j.1600-0749.1998.tb00713.x. This review (together with its recent update, ref. 71) thoroughly describes and classifies the mouse models of hypopigmentation and bleeding. These models provide the bases for identifying novel genes and for investigating the cell biology of the homologous human disorders, including Hermansky-Pudlak and Griscelli syndrome. [DOI] [PubMed] [Google Scholar]
- 118.Theos AC, Tenza D, Martina JA, Hurbain I, Peden AA, et al. Functions of adaptor protein (AP)-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol Biol Cell. 2005;16:5356–72. doi: 10.1091/mbc.E05-07-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Toro J, Turner M, Gahl WA. Dermatologic manifestations of Hermansky-Pudlak syndrome in patients with and without a 16-base pair duplication in the HPS1 gene. Arch Dermatol. 1999;135:774–80. doi: 10.1001/archderm.135.7.774. [DOI] [PubMed] [Google Scholar]
- 120.Tribl F, Marcus K, Meyer HE, Bringmann G, Gerlach M, Riederer P. Subcellular proteomics reveals neuromelanin granules to be a lysosome-related organelle. J Neural Transm. 2006;113:741–9. doi: 10.1007/s00702-006-0452-3. [DOI] [PubMed] [Google Scholar]
- 121.Villa A, Notarangelo L, Macchi P, Mantuano E, Cavagni G, et al. X-linked thrombocytopenia and Wiskott-Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat Genet. 1995;9:414–7. doi: 10.1038/ng0495-414. [DOI] [PubMed] [Google Scholar]
- 122.Wasmeier C, Romao M, Plowright L, Bennett DC, Raposo G, Seabra MC. Rab38 and Rab32 control post-Golgi trafficking of melanogenic enzymes. J Cell Biol. 2006;175:271–81. doi: 10.1083/jcb.200606050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weaver T, Na C, Stahlman M. Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin Cell Dev Biol. 2002;13:263–70. doi: 10.1016/s1084952102000551. [DOI] [PubMed] [Google Scholar]
- 124.Wei ML. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. This extensive review of the recent clinical, molecular, biochemical and cell biologic aspects of each Hermansky-Pudlak syndrome subtype includes a valuable listing of all the HPS gene mutations reported to date. [DOI] [PubMed] [Google Scholar]
- 125.Weiss HJ, Witte LD, Kaplan KL, Lages BA, Chernoff A, et al. Heterogeneity in storage pool deficiency: studies on granule-bound substances in 18 patients including variants deficient in alpha-granules, platelet factor 4, beta-thromboglobulin, and platelet-derived growth factor. Blood. 1979;54:1296–319. [PubMed] [Google Scholar]
- 126.Westbroek W, Adams D, Huizing M, Koshoffer A, Dorward H, et al. Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol. 2007;127:2674–7. doi: 10.1038/sj.jid.5700899. [DOI] [PubMed] [Google Scholar]
- 127.Westbroek W, Lambert J, Bahadoran P, Busca R, Herteleer MC, et al. Interactions of human Myosin Va isoforms, endogenously expressed in human melanocytes, are tightly regulated by the tail domain. J Invest Dermatol. 2003;120:465–75. doi: 10.1046/j.1523-1747.2003.12068.x. This study shows alternative splicing of MYO5A, involving six exons. In melanocytes, the presence of the alternative exon F is required for actin-dependent melanosome transport by indirect interaction with Rab27a and direct interaction with Melanophilin. Skin hypopigmentation in Griscelli syndrome is not caused by defects in melanosome biogenesis, but by ineffective capturing of melanosomes by the peripheral actin network. [DOI] [PubMed] [Google Scholar]
- 128.Winder SJ. Structural insights into actin-binding, branching and bundling proteins. Curr Opin Cell Biol. 2003;15:14–22. doi: 10.1016/s0955-0674(02)00002-9. [DOI] [PubMed] [Google Scholar]
- 129.Wu X, Jung G, Hammer JA., 3rd Functions of unconventional myosins. Curr Opin Cell Biol. 2000;12:42–51. doi: 10.1016/s0955-0674(99)00055-1. [DOI] [PubMed] [Google Scholar]
- 130.Wu X, Rao K, Zhang H, Wang F, Sellers J, et al. Identification of an organelle receptor for myosin-Va. Nat Cell Biol. 2002;4:271–8. doi: 10.1038/ncb760. [DOI] [PubMed] [Google Scholar]
- 131.Youssefian T, Cramer EM. Megakaryocyte dense granule components are sorted in multivesicular bodies. Blood. 2000;95:4004–7. [PubMed] [Google Scholar]
- 132.zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14:827–34. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]