

Abstract
Perifosine is an orally bioavailable alkylphospholipid currently being tested in Phase II clinical trials as a potential anticancer drug. In this study, we have revealed a novel mechanism underlying perifosine's anticancer activity involving induction of cyclooxygenase 2 (COX-2) in human cancer cells. Perifosine induced apoptosis and/or cell cycle arrest in several lung and head and neck cancer cell lines. However, the combination of perifosine with low concentrations of celecoxib rendered cells less sensitive to perifosine both in cell culture systems and in lung cancer xenograft models. Subsequently, we examined the effects of perifosine on COX-2 expression and activity in a set of lung and head and neck cancer cell lines and found that perifosine rapidly and potently increased COX-2 levels and activity, the degrees of which correlated to perifosine's abilities to inhibit the growth of cancer cells. We also detected increased COX-2 levels in lung cancer xenografts treated with perifosine. Moreover, blockage of COX-2 induction by both antisense and siRNA approaches decreased cell sensitivity to perifosine. Collectively, these data indicate that the activation of COX-2 contributes to perifosine's anticancer activity including apoptosis induction and growth arrest. These data are clinically relevant as they suggest that the combination of perifosine and COX-2 inhibitors such as celecoxib, may produce a potential drug contradiction.
Keywords: Perifosine, cyclooxygenase-2, celecoxib, apoptosis
Introduction
Alkylphospholipids represent a class of antitumor agents that can induce apoptosis in tumors cells while sparing normal cells (1). These agents act at the cell membrane and disrupt membrane phospholipid metabolism (1). Miltefosine, the first alkalphospholipid to be tested in clinical trials, has been approved for topical treatment of cutaneous lymphomas and cutaneous metastases from breast cancer (2). Perifosine, in which the choline moiety of miltefosine is replaced by a cyclic aliphatic piperidy residue, has greater oral bioavailability with limited gastrointestinal side effects and better activity in preclinical models when compared with miltefosine. Perifosine effectively inhibited the growth of a variety of human tumor cell lines including melanoma, lung, prostate, colon and breast cancer cells and suppressed the growth of mammary carcinomas induced by DMBA in Sprague-Dawley rats (3). In addition, perifosine when combined with other cancer therapeutic agents exhibited enhanced apoptosis-inducing and anticancer activity (4-7). Perifosine is currently being tested in Phase II clinical trials and has shown single agent partial responses in certain types of cancer such as renal cell carcinoma (8).
How perifosine exerts its antitumor effect is not completely understood. Perifosine has been shown to inhibit the plasma membrane localization of Akt and Akt phosphorylation leading to antiproliferative effects in tumor cells (9). As well, perifosine causes cell cycle arrest in tumor cells by inducing p21WAF (10). Our recent study has shown that perifosine induces apoptosis in human lung cancer cells involving activation of the extrinsic death receptor-mediated apoptotic pathway (11).
The cyclooxygenase-2 (COX-2) inhibitor celecoxib is a marketed drug for use in humans for management of arthritis and acute pain as well as in patients with familial adenomatous polyposis for reduction of adenomatous colorectal polyps (12). In addition, celecoxib is currently being tested pre-clinically and clinically for its chemopreventive and therapeutic efficacy against a broad spectrum of epithelial malignancies, including lung cancers, either as a single agent or in combination with other agents (13, 14). In an effort to identify perifosine-based combination regimens, we unexpectedly found that celecoxib diminished perifosine's growth inhibitory effects in human non-small cell lung cancer (NSCLC) and head and neck cancer cell lines.
COX-2 catalyzes the conversion of arachadonic acid to prostaglandins (PGs). COX-2 expression is generally low or absent in most tissues but its expression is inducible upon stimulation by such molecules as cytokines and growth factors (15). COX-2 is usually undetectable in most normal tissues, but overexpressed in a variety of premalignant and malignant tissues (16). In general, COX-2 overexpression is associated with resistance to apoptosis, angiogenesis, tumorigenesis, and poor prognosis (15, 16), all of which are the scientific rationale for COX-2 as a preventive or therapeutic target of cancer. However, COX-2 has also been demonstrated to suppress tumorigenesis in one model of skin cancer (17), to be associated with good prognosis in some studies (18-20), and to induce cell cycle arrest (21) and apoptosis (22-27).
Interestingly, another member of the alkylphospholipid family, edelfosine, upregulates the expression of COX-2 which contributes to apoptotic death induced by edelfosine (28). Therefore, we investigated why celecoxib antagonized perifosine's anticancer effects by understanding the role of COX-2 in perifosine-mediated anticancer activity including cell cycle arrest and induction of apoptosis. Our results demonstrate that perifosine induces COX-2 expression, which contributes to perifosine-induced apoptosis and cell cycle arrest. Therefore, our study reveals a novel mechanism underlying perifosine's anticancer activity involving COX-2 activation.
Materials and Methods
Reagents
Perifosine (Fig. 1A) was supplied by Keryx Biopharmaceuticals, Inc (New York, NY) with a chemical purity of >99%. This agent was dissolved in PBS and stored at -20°C. Celecoxib was purchased from LKT Laboratories (St. Paul, MN). The other COX-2 inhibitors SC-58125 and DuP697 were purchased from Cayman Chemical (Ann Arbor, MI). The celecoxib derivative 2,5-dimethyl-celecoxib (DMC), which lacks COX-2-inhibitory activity (29), was provided by Dr. Axel H. Schönthal (University of Southern California, Los Angeles, CA). These agents were dissolved in DMSO and stored at -20°C. Stock solutions were diluted to the appropriate concentrations with growth medium immediately before use.
Fig. 1. Perifosine (A) inhibits the growth of human NSCLC cells (B) through induction of apoptosis (C) and G2/M arrest (D).

A, The chemical structure of perifosine. B, The indicated cell lines were treated with increased concentrations of perifosine (1-20 μM) for 3 days. Cell numbers were then estimated using the SRB assay. IC50 values were calculated from a dose-response curve. C and D, The indicated cell lines were treated with 10 μM perifosine for 48 h. The cells were trypsinized, harvested, and subjected to flow cytometric analysis for measuring apoptosis using Annexin V staining (C) and for analyzing cell cycle using propidium iodide staining (D). Columns, means of triplicate determinations; Bars, ± SDs.
Cell Lines and Cell Culture
The human NSCLC cell lines used in this study and their culture conditions were described previously (30). The H157 stable cell line that expresses antisense COX-2 (H157-AS) and its matched vector control line (H157-V) were described previously (31) and provided by Dr. S. M. Dubinett (University of California, Los Angeles, CA). The head and neck cancer cell line 686Ln and its derived cell lines M4c, M4d, M4e, which have high metastatic potential, were provided by Dr. Z. G. Chen in our institute (32).
Cell Viability Assay
Cells were cultured in 96-well cell culture plates and treated the next day with the agents indicated. Viable cell number was estimated using the sulforhodamine B (SRB) assay, as previously described (30).
Western Blot Analysis
Preparation of whole-cell protein lysates and Western blot analysis were described previously (33, 34). Mouse anti-caspase-3 monoclonal antibody was purchased from Imgenex (San Diego, CA). Rabbit anti-caspase-8, anti-caspase-9, and anti-poly(ADP-ribose)polymerase (PARP) polyclonal antibodies were purchased from Cell Signaling Technology (Beverly, MA). Rabbit Anti-COX-2 polyclonal antibody was purchased from Oxford Biomedical (Oxford, MI). Rabbit anti-G3PDH polyclonal antibody was purchased from Trevigen (Gaithersburg, MD). Rabbit anti-β-actin polyclonal antibody was purchased from Sigma. Secondary antibodies goat anti-mouse and goat anti-rabbit-horseradish peroxidase conjugates were purchased from Bio-Rad (Hercules, CA).
Apoptosis Assays
Apoptosis was detected either by analysis of caspase activation using Western blot analysis as described above or by Annexin V staining using Annexin V-PE apoptosis detection kit (BD Bioscience, San Jose, CA) following the manufacturer's instructions and analyzed by flow cytometry using the FACScan (Becton Dickinson, San Jose, CA).
Cell Cycle Assay
Control cells or cells treated with the indicated agents for 24 or 48 hours were harvested, washed with cold PBS and fixed in 70% ethanol. DNA was stained by incubating cells with PBS containing 50 μg/ml propidium iodide and 15 μg/ml RNase A for 20 minutes at room temperature. Fluorescence was measured using the FACScan and analyzed using CellQuest software (BD Biosciences, San Jose, CA).
Enzyme-Linked Immunosorbent Assay (ELISA) for Detection of PGE2 and 15d-PGJ2
Cells were cultured in 60 mm dishes in culture medium supplemented with 5% fetal bovine serum and treated with perifosine alone, celecoxib plus perifosine or celecoxib alone. After a 16-hour treatment, medium was collected and measured for the levels of PGE2 using the ELISA kit from Cayman Chemical (Ann Arbor, MI) and for the levels of 15d-PGJ2 using the ELISA kit from Assay Designs (Ann Arbor, MI) following the manufacturer's instructions. Because treatment with perifosine induces significant cell death, prostaglandin levels were normalized based on protein concentration for each sample after treatment.
Silencing of COX-2 Expression using siRNA
Silencing of COX-2 was achieved by transfecting siRNA using RNAifect transfection reagent (Qiagen, Valencia, CA) following the manufacturer's instructions. Control Stealth™ siRNA oligonucleotides targeting the sequence 5′-GAATTCCTCAATTCCCTCTCGCATT-3′ and COX-2 Stealth™ siRNA oligonucleotides targeting the sequence 5′-AAAGACTGGTATTTCATCTGCCTGC-3′ were purchased from Invitrogen (Carlsbad, CA). Cells were plated in 24-well plates and the next day transfected with siRNAs. Twenty-four hours later cells were re-plated and the next day treated with perifosine as indicated. Gene silencing effects were evaluated by Western blot analysis as described above.
Lung Cancer Xenografts and Treatments
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University. Four- to 6-week old (about 20 g of body weight) female athymic (nu/nu) mice (Taconic Hudson, NY) were housed under pathogen-free conditions in microisolator cages with laboratory chow and water ad libitum. 5 × 106 H358 cells or H460 cells in PBS were injected s.c. into the flank region of nude mice. When tumors reached certain size ranges (50-100 mm3), the mice were randomized into four groups (n = 6/group) according to tumor volumes and body weights for the following treatments: vehicle control, perifosine in PBS (20 or 30 mg/kg/day, og), celecoxib in DMSO (50 or 100 mg/kg/day; og), and the combination of perifosine and celecoxib. Tumor volumes were measured using caliper measurements once every two days and calculated with the formula V = π(length × width2)/6. After a 5-, 6-, 10- or 11-day treatment, the mice were sacrificed with CO2. The tumors were then removed, weighed and frozen in liquid nitrogen or fixed with 4% fomaldehyde. Certain portions of tumor tissues from each tumor were homogenized in protein lysis buffer for preparation of whole-cell protein lysates as described previously (30). Western blotting results were quantitated using NIH Image J (NIH, Bethesda, DC).
Immunohistochemistry (IHC) for Detection of COX-2 in Tumor Tissues
Five-micrometer formaldehyde fixed, paraffin-embedded sections from H358 tumor xenografts were dewaxed, hydrated though graded alcohols and microwaved in 100 mM sodium citrate for 5 min on high power and 10 min on low power for antigen retrieval. IHC analysis was performed following the protocol for the DakoCytomation EnVision + Dual Link System (Carpinteria, CA). Sections were incubated overnight at 4°C with rabbit polyclonal anti-COX-2 (1:500 dilution; Oxford Biomedical Research). COX-2 positive cells were counted using MetaMorph Imaging Software (Universal Imaging Corporation; Downington, PA).
Statistical Analysis
The statistical significance of differences between two groups was analyzed with two-sided unpaired Student's t tests when the variances were equal or with Welch's corrected t test when the variances were not equal, by use of Graphpad InStat 3 software (GraphPad Software, San Diego, CA). Data were examined as suggested by the same software to verify that the assumptions for use of the t tests held. Results were considered to be statistically significant at P < 0.05. All statistical tests were two-sided.
Results
Perifosine Inhibits the Growth of Human NSCLC Cells through Induction of Apoptosis and Cell Cycle Arrest
Human NSCLC cell lines exhibited varied sensitivities to perifosine (Fig. 1B). Among these cell lines, H460 and H358 were the most sensitive to perifosine, whereas H226 was resistant to perifosine. Both A549 and H157 exhibited intermediate sensitivities to perifosine (Fig. 1B). Detection of apoptosis and cell cycle alteration revealed that H460 cells primarily underwent apoptotic cell death (84.8 ± 1.1% in perifosine-treated cells vs. 7.7 ± 1.8% in PBS-treated cells), whereas H358 cells were very sensitive to G2/M arrest by perifosine (44.8 ± 2.9 vs. 21.6 ± 1.5 in PBS-treated cells) with limited sensitivity to undergo apoptotic cell death (18.4 ± 2.4% in perifosine treated cells vs. 10.8 ± 3.9% in PBS-treated cells). No apoptosis, but very weak G2/M arrest (25.8 ± 1.9% in perifosine-treated cells vs. 22.9 ± 0.9% in control cells), was detected in H226 cells exposed to 10 μM perifosine. A549 and H157 cells underwent both G2/M arrest and apoptosis upon perifosine treatment (P < 0.05 or 0.01) (Figs. 1C and 1D). Thus, it appears that perifosine induces apoptosis and/or G2/M arrest leading to inhibition of the growth of human NSCLC cells.
Celecoxib Reduces Perifosine's Anticancer Activity in Cell Culture and in vivo
We were interested in enhancing perifosine's anticancer activity. To this end, we tested the effects of perifosine in combination with other cancer therapeutic agents including celecoxib. Unexpectedly, the presence of celecoxib at the tested concentrations ranging from 5 to 20 μM, significantly attenuated perifosine-mediated growth-inhibitory effects in both H460 and H358 cells (P < 0.01 or 0.001) (Fig. 2A). Similarly, other COX-2 inhibitors, including SC-58125 and DUP697, also significantly protected cells from perifosine-induced growth inhibition. However, the celecoxib derivative, DMC, which lacks COX-2-inhibitory activity, failed to protect cells from perifosine-induced cell death (Supplemental Fig. S1).
Fig. 2. Celecoxib protects NSCLC cells from perifosine-induced decrease in cell survival (A) and apoptosis (B and C) and antagonizes perifosine's anticancer activity in mouse xenograft models (D).
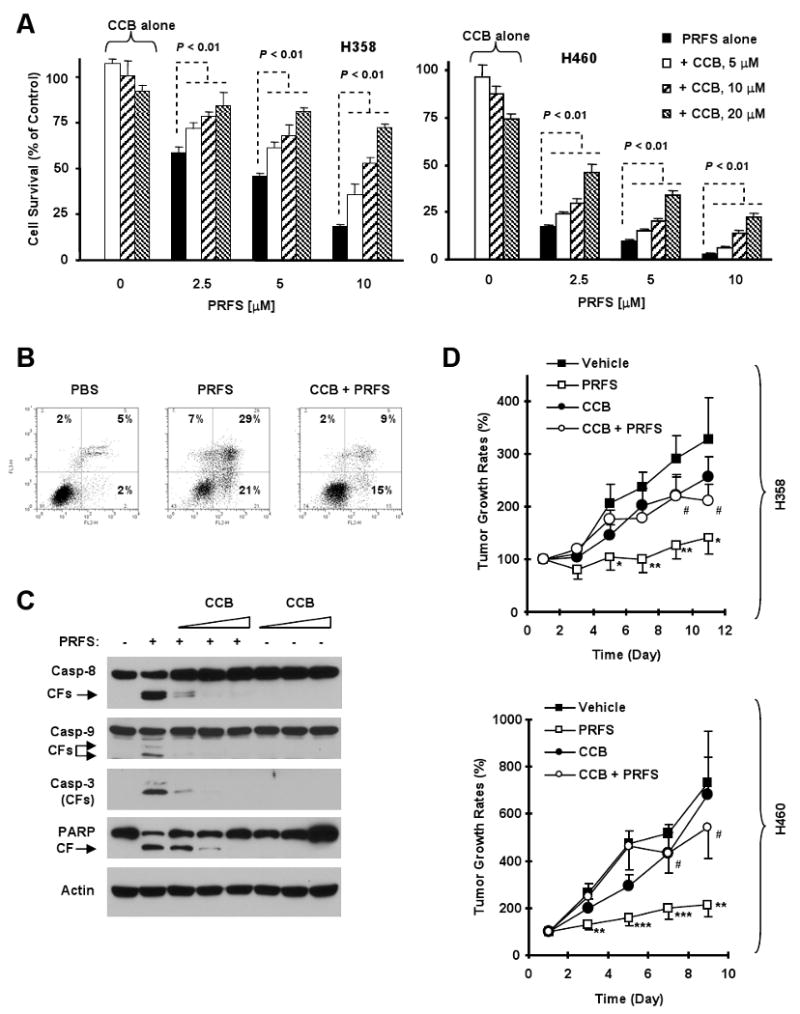
A, Both H358 and H460 cells were treated with perifosine alone, celecoxib alone or perifosine plus celecoxib at the indicated concentrations for 48 hours. Cell survival was estimated using the SRB assay. Columns, means of four replicate determinations; Bars, ± SD. P values at all combination treatments are at least < 0.01 when compared with perifosine alone. B, H460 cells were treated with 10 μM perifosine in the absence and presence of 10 μM celecoxib for 24 h and analyzed for apoptosis by Annexin V staining. C, H460 cells were treated with 5 μM perifosine in the absence or presence of increasing doses of celecoxib (5, 10, 20 μM) for 12 h. The cells were harvested for preparation of whole-cell protein lysates and detection of caspase and PARP cleavage by Western blot analysis. CF, cleaved from. D, The indicated lung cancer xenografts were treated with vehicle, 20 mg/kg perifosine, 50 mg/kg celecoxib or the combination of celecoxib and perifosine for 9 days (H460) or 30 mg/kg perifosine, 100 mg/kg celecoxib or the combination for 11 days (H358). Tumor sizes were measured once every two days. Tumor growth rates were calculated by comparing with the initial size of each individual tumor. Each measurement is a mean ± SE (n = 6). * P < 0.05, ** P < 0.01, and *** P < 0.001 compared to vehicle control. # P< 0.05 compared with perifosine treatment. PRFS, perifosine; CCB, celecoxib.
Furthermore, we analyzed the effects of perifosine on induction of apoptosis and G2/M arrest in the presence of celecoxib. Perifosine induced 50% of H460 cells to undergo apoptosis in the absence of celecoxib, but only 24% apoptosis in the presence of celecoxib (Fig. 2B). Perifosine alone caused cleavage of caspase-8, caspase-9, caspase-3 and PARP. However, the presence of celecoxib abrogated perifosine's ability to cause cleavage of caspases and PARP (Fig. 2C). Thus, these results clearly show that celecoxib protects cells from perifosine-induced apoptosis. Since H358 cells are more sensitive to G2/M arrest when treated with perifosine, we also examined the impact of celecoxib on perifosine's ability to induce G2/M arrest in this cell line. We found that perifosine alone induced 70% of cells in G2/M phase, but only arrested 39% of cells in G2/M phase in the presence of celecoxib (supplemental Fig. S2). Therefore, we conclude that celecoxib also protects cells from perifosine-induced cell cycle arrest.
We also determined whether celecoxib antagonized perifosine's anticancer efficacy in vivo using lung cancer xenografts in mice. As presented in Fig. 2D, perifosine alone significantly inhibited the growth of both H460 and H358 xenografts (p < 0.05). Celecoxib alone at the tested doses (50 or 100 mg/kg) had minimal effects on the growth of either tumors. When perifosine was combined with celecoxib, perifosine lost its activity to inhibit the growth of lung tumors in both xenograft models (p < 0.05). Thus, it is apparent that celecoxib also antagonizes perifosine's anticancer activity in vivo.
Perifosine Increases COX-2 Expression and Activity in NSCLC Cells and in Lung Cancer Xenografts
Since the addition of celecoxib could abrogate perifosine-induced cell death and growth arrest, we next determined whether perifosine modulated COX-2 expression and activity in NSCLC cells. By Western blot analysis, we detected dose-dependent increases in COX-2 expression in H460, H358, A549 and H157 cells, but not in H226 cells (Fig. 3A, top panel). Of these NSCLC cell lines, H460 and H358 cells exhibited the highest degree of COX-2 induction (Figs. 3A) and are the most sensitive to perifosine (Fig. 1B), whereas the H226 cells that did not show COX-2 induction after perifosine treatment were the least sensitive to perifosine (Fig. 1B), suggesting that COX-2 induction by perifosine correlates with sensitivity to perifosine. Importantly, the induction of COX-2 in the H460 and the H358 cells lines occurred as early as 6 hours after perifosine treatment (Fig. 3A, bottom panel), suggesting that COX-2 induction is an early event in response to perifosine treatment. Perifosine had no effect on the expression of COX-1 in H460 or H358 cell lines (data not shown). In addition, significantly increased levels of the prostaglandins, PGE2 and 15d-PGJ2, were detected in cells exposed to perifosine (P < 0.05 or 0.01) (Fig. 3B), indicating that perifosine increases COX-2 activity as well. Collectively, these results demonstrate that perifosine increases COX-2 expression and activity in human NSCLC cells.
Fig. 3. Perifosine increases COX-2 expression (A) and prostaglandin production (B) in NSCLC cells and induces COX-2 expression in lung cancer xenografts (C and D).
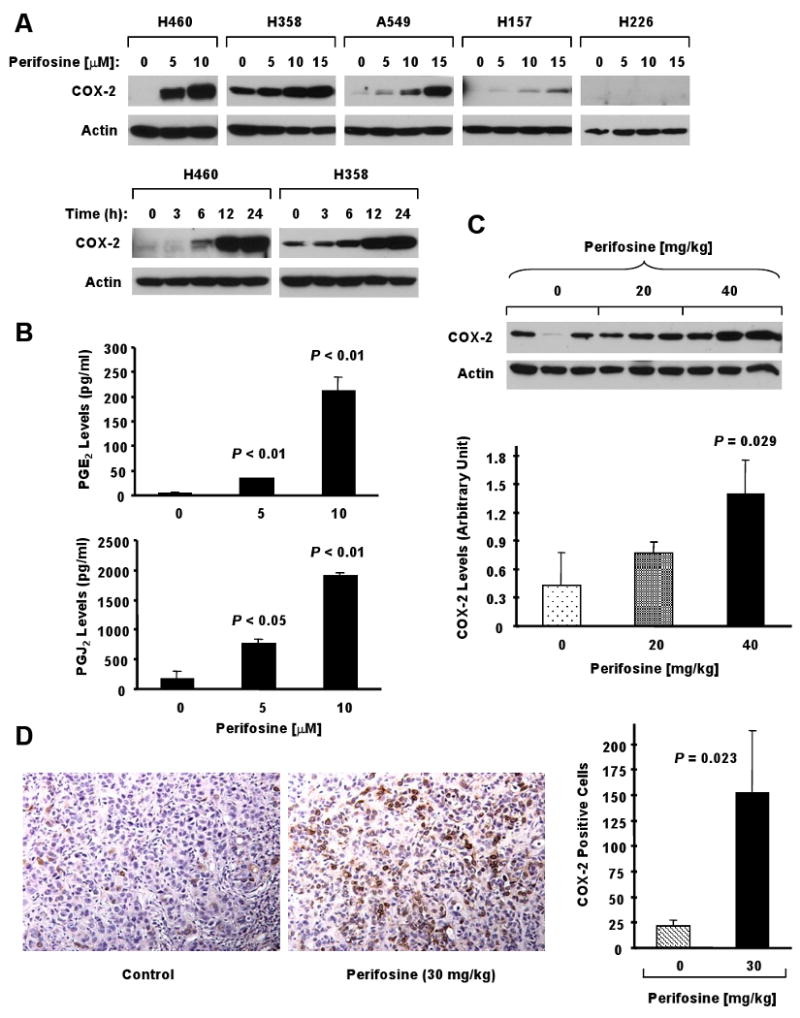
A, The indicated cell lines were treated with the given concentrations of perifosine for 8 h (top panel) or with 10 μM perifosine for the indicated times (bottom panel). The cells were subjected to preparation of whole-cell protein lysates and subsequent Western Blot analysis. Actin served as a loading control. B, H460 cells were treated with the indicated concentrations of perifosine for 16 h and then subjected to estimation of PGE2 levels and 15d-PGJ2 levels in media using ELISA kits according to the manufacturers' instructions. Columns, means of duplicate determinations; Bars, ± SD. C, Western blot analysis of COX-2 expression in H358 tumor lysates after 6 days of treatment with vehicle control, 20 mg/kg perifosine, or 40 mg/kg perifosine by oral gavage in nude mice. These data were also quantitated using the NIH ImageJ software (bottom panel). D, IHC staining of COX-2 expression in H358 tumors after 5 days of treatment with vehicle control or 30 mg/kg perifosine by oral gavage in nude mice. The COX-2 positive cells were scored using the MetaMorph Imaging Software and graphed (right panel). The representative IHC results of COX-2 were shown in the left panel. The results in C and D are the means of triplicate tumors (n = 3). Bars, ± SDs.
To determine whether perifosine increased COX-2 expression in vivo, we treated H358 lung cancer xenografts with perifosine or vehicle control and detected COX-2 levels in these xenografts by both Western blotting (6-day treatment) and immunohistochemistry (5-day treatment). As we observed in cell cultures, we detected significant increased levels of COX-2 by Western blot analysis (Fig. 3C) and immunohistochemistry (Fig. 3D) in perifosine-treated xenografts compared to vehicle-treated tumors (P < 0.05). Thus, perifosine also increases COX-2 levels in tumor tissues in vivo.
Celecoxib Inhibits Perifosine-induced COX-2 Expression and Activity
To determine whether celecoxib at the tested concentrations indeed inhibits perifosine-induced COX-2 activation, we analyzed the effects of perifosine on the production of PGE2 and 15d-PGJ2 in the presence and absence of celecoxib. As presented in Fig. 4A, perifosine alone increased the levels of PGE2 and 15d-PGJ2; however, addition of celecoxib significantly inhibited the perifosine-induced increase in PGE2 and 15d-PGJ2 (P < 0.05). Moreover, we found that the presence of celecoxib also abrogated perifosine's ability to increase COX-2 expression (Fig. 4B). Collectively, these results clearly indicate that celecoxib at the tested concentration ranges inhibits perifosine-induced COX-2 activation.
Fig. 4. Celecoxib inhibits perifosine-induced prostaglandin production (A) and COX-2 expression (B) in human NSCLC cells.
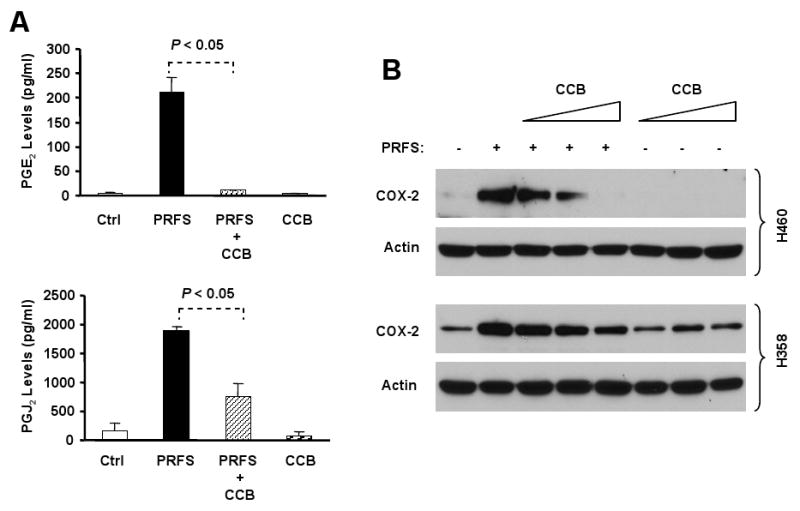
A, H460 cells were exposed to 10 μM perifosine in the absence or presence of 10 μM celecoxib or celecoxib alone for 16 hours. The media were then collected for measurement of PGE2 (top panel) or 15dPGJ2 (bottom panel) levels by ELISA kits. Columns, means of duplicate determinations; Bars, ± SDs. PRFS, perifosine. B, The indicated cell lines were treated with 5 μM perifosine in the absence and presence of increasing concentrations of celecoxib (5, 10, 20 μM) or celecoxib alone (5, 10, 20 μM) for 12 hours. The cells were subjected to preparation of whole-cell protein lysates and subsequent Western Blot analysis for detection of the given proteins. PRFS, perifosine; CCB, celecoxib.
Blockade of COX-2 Induction via Genetic Manipulations Attenuates Perifosine's Anticancer Activity
To robustly demonstrate the role of COX-2 induction on perifosine's anticancer activity, we further evaluated perifosine's ability to inhibit the growth of NSCLC cells when COX-2 induction was blocked using either antisense or gene silencing approaches. Perifosine exerted a dose-dependent effect on COX-2 induction in vector control H157-V cells; however, this effect was substantially abrogated in H157-AS cells, which express antisense COX-2 gene (Fig. 5A). Accordingly, the H157-AS cells were significantly less sensitive to perifosine compared to parental (H157-P) or H157-V cells (P < 0.01 or less) (Fig. 5B). Since H157 cells are more susceptible to undergo G2/M arrest upon perifosine treatment (Fig. 1), we also compared perifosine's effects on G2/M arrest in these cell lines. As expected, the effect of perifosine on G2/M arrest was abrogated in H157-AS cells compared to H157-P and H157-V cells (supplemental Fig. S3). In addition, we silenced COX-2 expression in H460 cells through transfection of COX-2 siRNA and then examined its impact on perifosine's apoptosis-inducing effect. Transfection of COX-2 siRNA successfully inhibited perifosine-mediated COX-2 induction (Fig. 5C). Correspondingly, we detected much less cleaved PARP (Fig. 5C) and annexin V-positive cells (Fig. 5D) in COX-2 siRNA-transfected cells than in control siRNA-transfected cells. Specifically, we detected 29.6 ± 0.8% of apoptotic cells in control siRNA-transfected cells, but only 15.8 ± 0.2% of apoptotic cells in COX-2-transfected cells (Fig. 5D), which is significantly less than that in control siRNA-transfected cells (P < 0.01). Collectively, these results provide a robust support for a critical role of COX-2 activation in mediating perifosine's anticancer activity.
Fig. 5. Inhibition of COX-2 induction by expression of antisense COX-2 (A and B) or transfection of COX-2 siRNA (C and D) protects against perifosine-induced growth inhibition.
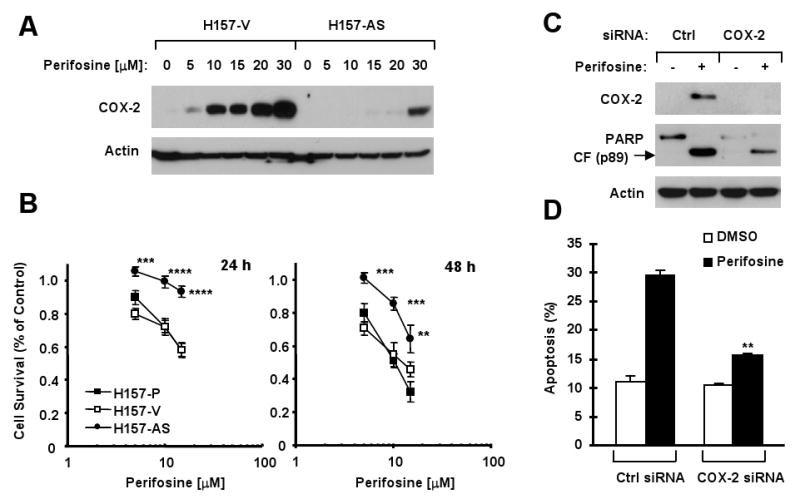
A, Western blot analysis of COX-2 expression in H157 vector control (H157-V) and H157 antisense COX-2 (H157-AS) cells after treatment with the indicated concentrations of perifosine for 8 h. B. The indicated cell lines were treated with the given concentrations of perifosine for 24 h or 48 h. The cells were subjected to estimation of cell survival using the SRB assay. Points, means of four replicate determinations; Bars, ± SD. **, P < 0.01, ***, P < 0.001 and ****, P < 0.0001 compared with H157-P or H157-V. C and D, H460 cells were transfected with control siRNA (Ctrl) or COX-2 siRNA using RNAifect transfection reagent for 48 h and then treated with 10 μM perifosine. After 24 h, the cells were subjected to preparation of whole-cell protein lysates and subsequent Western blot analysis for the given proteins (C) or to annexin V staining and flow cytometry for detection of apoptosis (D). Columns, means of duplicate determinations; Bars, ± SDs. **, P < 0.01 compared with perifosine in control siRNA-transfected cells.
Perifosine Also Induces COX-2-dependent Apoptosis in Head and Neck Cancer Cells
To determine whether perifosine exerts COX-2-dependent apoptosis and shows antagonism with celecoxib in other types of cancer cells, we further examined the effects of perifosine on COX-2 expression in 686Ln and its derivative cell lines M4c, M4d and M4e, which exhibited differential responses to perifosine (Fig. 6A). As we found in the NSCLC cell lines, the M4c, M4d and M4e cell lines which expressed much higher levels of COX-2 after perifosine treatment compared to the 686Ln cell line (Fig. 6B) were much more sensitive to perifosine than the 686Ln cells (Fig. 6A). As well, perifosine was able to induce COX-2 expression in the M4e cells as early as 3 hours showing that COX-2 induction is an early event. The induction of COX-2 reached peak levels at 6 h and was maintained up to 24 h, indicating that COX-2 induction is also a sustained event (supplementary Fig. S4). We next examined the effects of perifosine combined with celecoxib on the growth of these head and neck cancer cells including examination of apoptosis. As presented in Fig. 6C, in the presence of celecoxib at the tested concentrations ranging from 5 to 20 μM, perifosine-mediated growth-inhibitory effects were significantly attenuated in both M4c and M4e cells (P < 0.01 or 0.001). The addition of celecoxib also substantially reduced apoptosis induced by perifosine as was examined by annexin V analysis and cleavage of caspase-3 and PARP (Fig. 6D.) Thus, the protection from perifosine-induced cell death by the addition of celecoxib is also clearly demonstrated in human head and neck cancer cell lines. Moreover, the presence celecoxib abrogated perifosine-induced COX-2 expression (Fig. 6D, right panel), further supporting an important role of COX-2 in perifosine-induced apoptosis.
Fig. 6. Effects of Perifosine on the growth of human head and neck cancer cells (A) and COX-2 expression (B) and protective effects of celecoxib on perifosine-induced decrease in cell survival (C) and apoptosis (D).
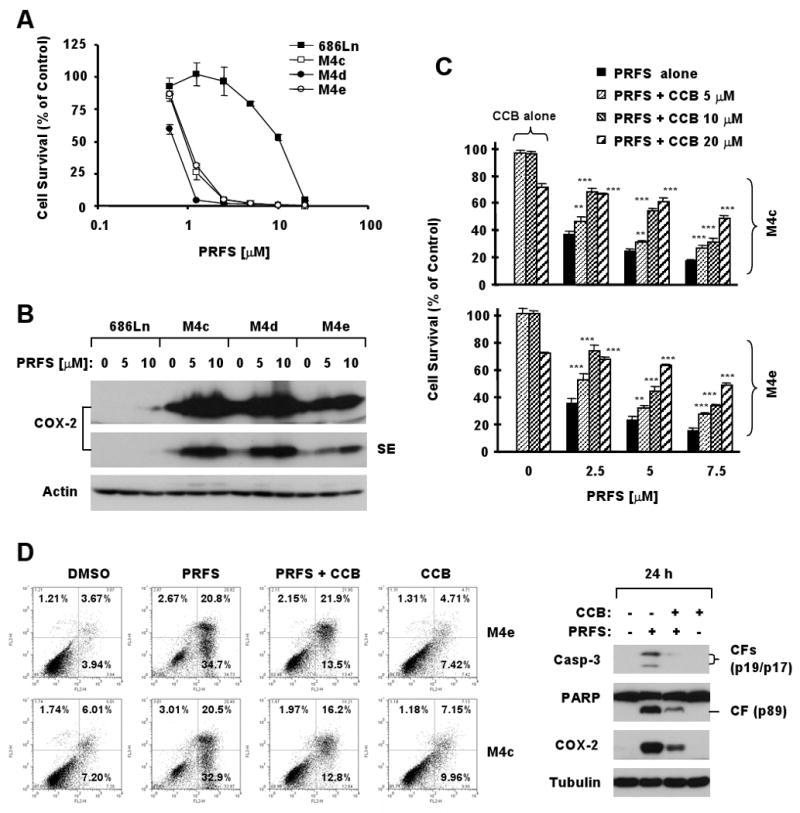
A, The indicated cell lines were treated with increased concentrations of perifosine (1-20 μM) for 3 days and cell numbers were estimated using the SRB assay. B, The indicated cell lines were treated with the given concentrations of perifosine for 8 h (upper panel). The cells were subjected to preparation of whole-cell protein lysates and subsequent Western blot analysis. Actin served as a loading control. C, The indicated cell lines were treated with the given concentrations of perifosine alone, celecoxib alone and their respective combinations for for 48 h and then subjected to the SRB assay for estimation of cell survival. Columns, means of four replicate determinations; Bars, ± SD. **, P < 0.01 and ***, P < 0.001 compared with perifosine alone. D, The indicated cell lines were treated with 5 μM perifosine alone, 10 μM celecoxib alone, or their combinaiton for 48 h (left panel). The cells were harvested and subjected to flow cytometric analysis for measuring apoptosis using annexin V staining (left panel). In addition, the same treatments were done in M4c cells for 24h to prepare whole-cell protein lysates for detection of the indicated proteins by Western Blot analysis (right panel). Tubulin served as a loading control. PRFS, perifosine; CCB, celecoxib;CF, cleavedform.
Discussion
Induction of apoptosis and cell cycle arrest by perifosine in other types of cancer cells was documented previously (10, 35, 36). Perifosine was reported to induce both G1 and G2/M arrest in glioma cells and head and neck squamous carcinoma cells (10, 35). However, we found that all of the tested human NSCLC cells which responded to perifosine underwent G2/M arrest albeit with various degrees, indicating that perifosine primarily induces G2/M arrest in human NSCLC cells. Given that perifosine also induces apoptosis in these cell lines, we conclude that perifosine induces both apoptosis and G2/M arrest, leading to inhibition of the growth of human NSCLC cells.
Importantly, the current study reveals a novel and unique mechanism underlying perifosine-mediated anticancer activity in human NSCLC and head and neck cancer cells which is COX-2 dependent. Specifically we demonstrate that perifosine induces apoptosis and/or cell cycle arrest in human cancer cells through induction and activation of COX-2 based on the following compelling evidence: First, perifosine increases COX-2 expression in both cell cultures and in mouse lung cancer xenografts. The degree of COX-2 induction is associated with perifosine's potency in inhibiting the growth of cancer cells; Second, the COX-2 specific inhibitor celecoxib as well as other COX-2 inhibitors suppress perifosine-induced COX-2 upregulation and activation and protect cells from perifosine-induced apoptosis or G2/M arrest. In addition, celecoxib's derivative, DMC, which lacks a COX-2-inhibitory effect, failed to abrogate perifosine's antitumor effect suggesting that induction of COX-2 activity is important in mediating apoptosis and cell cycle arrest induced by perifosine. Finally, blockade of COX-2 induction using either antisense or siRNA approaches attenuates perifosine's ability to induce growth arrest or apoptosis. To our knowledge, this is the first study to demonstrate a critical role of COX-2 activation in mediating perifosine's anticancer activity.
Given that previous studies have shown pro-apoptotic or tumor suppressive roles of COX-2 in various model systems (17, 21-25, 28), our current finding on the critical role of COX-2 activation in mediating perifosine's anticancer activity should not be surprising. The fundamental questions of how COX-2 mediates perifosine-induced apoptosis or cell cycle arrest and how perifosine increases COX-2 expression are of interest for further exploration in the future. 15d-PGJ2, one of the prostaglandins generated by COX-2, can act as a PPARγ ligand and is proapoptotic (37). A recent study suggests that edelfosine, another alkylphospholipid, induces COX-2-dependent apoptosis through production of 15d-PGJ2 (28). The authors showed that edelfosine could induce the expression and transcriptional activity of PPARγ in H-ras transformed human breast epithelial cells suggesting 15d-PGJ2 may be acting through PPARγ to induce apoptosis. In our study, we detected increased levels of 15d-PGJ2 in cells exposed to perifosine, however, perifosine had no effect on the transcriptional activity of PPARγ (data not shown.). Another recent study showed that the chemotherapeutic agents, paclitaxel, cisplatin and 5-fluorouracil, could induce COX-2 expression in cervical carcinoma cells and the inhibition of COX-2 by siRNA or the addition of the selective COX-2 inhibitor, NS-398, rendered the cells less sensitive to apoptosis-induced by these chemotherapeutic agents (26). The authors showed that addition of 15d-PGJ2 induced apoptosis and inhibition of lipocalin-type PGD synthase which contributes to prostaglandin synthesis could prevent the chemotherapeutic-induced apoptosis. Therefore, whether the production of prostagladins, specifically, 15d-PGJ2 directly contributes to perifosine-induced COX-2-dependent apoptosis in our system needs further investigation. Our unpublished data show that perifosine increased the activity of the COX-2 promoter, suggesting that perifosine likely induces COX-2 expression at the transcriptional level. Therefore, a detailed analysis of the COX-2 promoter may help us reveal the mechanism by which perifosine increases COX-2 expression.
In this study, perifosine-induced caspase-8 activation is clearly inhibited by celecoxib (Fig. 2C). We previously showed that perifosine induces DR5 expression which contributes to perifosine-induced apoptosis (11). Moreover, it has been shown that 15d-PGJ2 increases DR5 expression (38). Thus, it is plausible to speculate a link between perifosine-induced COX-2 and DR5 upregulation. Our preliminary data indeed show that celecoxib is able to abrogate perifosine-induced DR5 expression (Elrod and Sun, unpublished data). Our ongoing work in this direction may shed light on the mechanisms underlying COX-2-induced apoptosis.
We did not see complete protection from perifosine-induced cell killing or arrest with celecoxib, or blockade of COX-2 induction using either an antisense or gene knockdown strategy, suggesting that COX-2 is not the only mechanism mediating perifosine's anticancer activity in human NSCLC cells. It has been documented that perifosine inhibits Akt activity (9, 36). Our own study shows that perifosine inhibits Akt activity leading to perifosine-induced apoptosis in human NSCLC cells (11). Thus, further exploration of the relationship between COX-2 activation and Akt inhibition is also of interest in the future.
Celecoxib is a prescribed drug for treatment of inflammation and pain. In the field of oncology, celecoxib is an approved drug in the clinic for the adjuvant treatment of familial adenomatous polyposis, an inherited syndrome that predisposes individuals to colon cancer. In addition, celecoxib has been tested in various clinical trials for either cancer chemoprevention or therapy (13, 16). The clinically achievable peak plasma concentrations of celecoxib in humans after oral administration of a single dose of 800 mg are approximately 3.2-5.6 μM (39). At these concentration ranges, celecoxib significantly attenuated perifosine's growth-inhibitory effects. Consistently, celecoxib significantly diminished perifosine's activity against the growth of lung cancer xenografts in mouse models (Fig. 2). Collectively, these results clearly indicate that celecoxib antagonizes perifosine's anticancer activity both in vitro and in vivo. As mentioned above, the COX-2 inhibitor, NS 398 can render cervical carcinoma cells less sensitive to apoptosis induced by chemotherapeutic agents (26). These data and our current study add to the clinical relevance of COX-2 inhibition by NSAIDS in patients also being administered anticancer agents. The immediate clinical impact of our study is that caution should be taken when combining COX-2 inhibitors such as celecoxib with perifosine, so as not to inhibit the efficacy of perifosine. Given that COX-2 induction appears to be associated with cell sensitivity to perifosine-induced apoptosis or cell cycle arrest as demonstrated in our study, it may also be important in the clinical practice to consider COX-2 induction or activity as a predictive biomarker for perifosine-based cancer therapy.
Supplementary Material
Acknowledgments
We thank Dr. A. H. Schönthal (University of Southern California, Los Angeles, CA) for providing DMC, Dr. S. M. Dubinett (University of California, Los Angeles, CA) for providing antisense COX-2 stable cell lines and Dr. Z. G. Chen (Emory University, Atlanta, GA) for providing the head and neck cancer cell lines.
Grant support: The Georgia Cancer Coalition Distinguished Cancer Scholar award (S-Y. S), Department of Defense grants W81XWH-04-1-0142-VITAL (S-Y. Sun for Project 4), National Cancer Institute SPORE P50 grant CA128613-01 (S-Y. Sun for Project 2), and American Cancer Society Postdoctoral Fellowship PF-07-033-01-CCG (H.A. Elrod).
Footnotes
Conflict of interest: None
Notes: H.A. Elrod is a recipient of an American Cancer Society Fellowship. F.R. Khuri and S-Y. Sun are Georgia Cancer Coalition Distinguished Cancer Scholars.
References
- 1.Ruiter GA, Verheij M, Zerp SF, van Blitterswijk WJ. Alkyl-lysophospholipids as anticancer agents and enhancers of radiation-induced apoptosis. Int J Radiat Oncol Biol Phys. 2001;49:415–9. doi: 10.1016/s0360-3016(00)01476-0. [DOI] [PubMed] [Google Scholar]
- 2.Jendrossek V, Handrick R. Membrane targeted anticancer drugs: potent inducers of apoptosis and putative radiosensitisers. Curr Med Chem Anticancer Agents. 2003;3:343–53. doi: 10.2174/1568011033482341. [DOI] [PubMed] [Google Scholar]
- 3.Hilgard P, Klenner T, Stekar J, Nossner G, Kutscher B, Engel J. D-21266, a new heterocyclic alkylphospholipid with antitumour activity. Eur J Cancer. 1997;33:442–6. doi: 10.1016/s0959-8049(97)89020-x. [DOI] [PubMed] [Google Scholar]
- 4.Dasmahapatra GP, Didolkar P, Alley MC, Ghosh S, Sausville EA, Roy KK. In vitro combination treatment with perifosine and UCN-01 demonstrates synergism against prostate (PC-3) and lung (A549) epithelial adenocarcinoma cell lines. Clin Cancer Res. 2004;10:5242–52. doi: 10.1158/1078-0432.CCR-03-0534. [DOI] [PubMed] [Google Scholar]
- 5.Rahmani M, Reese E, Dai Y, et al. Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res. 2005;65:2422–32. doi: 10.1158/0008-5472.CAN-04-2440. [DOI] [PubMed] [Google Scholar]
- 6.Nyakern M, Cappellini A, Mantovani I, Martelli AM. Synergistic induction of apoptosis in human leukemia T cells by the Akt inhibitor perifosine and etoposide through activation of intrinsic and Fas-mediated extrinsic cell death pathways. Mol Cancer Ther. 2006;5:1559–70. doi: 10.1158/1535-7163.MCT-06-0076. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Luwor R, Lu Y, Liang K, Fan Z. Enhancement of antitumor activity of the anti-EGF receptor monoclonal antibody cetuximab/C225 by perifosine in PTEN-deficient cancer cells. Oncogene. 2006;25:525–35. doi: 10.1038/sj.onc.1209075. [DOI] [PubMed] [Google Scholar]
- 8.Stephenson J, Schreeder M, Waples J, et al. Perifosien (P), active as a single agent for renal cell carcinoma (RCC), now in phase I trials combined with tryosine kinase inhibitors (TKI) Journal of Clinical Oncology, 2007 ASCO Annual Meeting Proceddings Part I. 2007;25:15622. [Google Scholar]
- 9.Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2:1093–103. [PubMed] [Google Scholar]
- 10.Patel V, Lahusen T, Sy T, Sausville EA, Gutkind JS, Senderowicz AM. Perifosine, a novel alkylphospholipid, induces p21(WAF1) expression in squamous carcinoma cells through a p53-independent pathway, leading to loss in cyclin-dependent kinase activity and cell cycle arrest. Cancer Res. 2002;62:1401–9. [PubMed] [Google Scholar]
- 11.Elrod HA, Lin YD, Yue P, et al. The alkylphospholipid perifosine induces apoptosis of human lung cancer cells requiring inhibition of Akt and activation of the extrinsic apoptotic pathway. Mol Cancer Ther. 2007;6:2029–38. doi: 10.1158/1535-7163.MCT-07-0004. [DOI] [PubMed] [Google Scholar]
- 12.Frampton JE, Keating GM. Celecoxib: a review of its use in the management of arthritis and acute pain. Drugs. 2007;67:2433–72. doi: 10.2165/00003495-200767160-00008. [DOI] [PubMed] [Google Scholar]
- 13.Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Control. 2002;9:28–35. doi: 10.1177/107327480200902S04. [DOI] [PubMed] [Google Scholar]
- 14.Grosch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. 2006;98:736–47. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
- 15.Zha S, Yegnasubramanian V, Nelson WG, Isaacs WB, De Marzo AM. Cyclooxygenases in cancer: progress and perspective. Cancer Lett. 2004;215:1–20. doi: 10.1016/j.canlet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4:431–6. doi: 10.1016/s1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 17.Bol DK, Rowley RB, Ho CP, et al. Cyclooxygenase-2 overexpression in the skin of transgenic mice results in suppression of tumor development. Cancer Res. 2002;62:2516–21. [PubMed] [Google Scholar]
- 18.Yegnasubramanian S, Kowalski J, Gonzalgo ML, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975–86. doi: 10.1158/0008-5472.can-03-3972. [DOI] [PubMed] [Google Scholar]
- 19.Nakopoulou L, Mylona E, Papadaki I, et al. Overexpression of cyclooxygenase-2 is associated with a favorable prognostic phenotype in breast carcinoma. Pathobiology. 2005;72:241–9. doi: 10.1159/000089418. [DOI] [PubMed] [Google Scholar]
- 20.O'Kane SL, Cawkwell L, Campbell A, Lind MJ. Cyclooxygenase-2 expression predicts survival in malignant pleural mesothelioma. Eur J Cancer. 2005;41:1645–8. doi: 10.1016/j.ejca.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 21.Trifan OC, Smith RM, Thompson BD, Hla T. Overexpression of cyclooxygenase-2 induces cell cycle arrest. Evidence for a prostaglandin-independent mechanism. J Biol Chem. 1999;274:34141–7. doi: 10.1074/jbc.274.48.34141. [DOI] [PubMed] [Google Scholar]
- 22.Xu Z, Choudhary S, Voznesensky O, et al. Overexpression of COX-2 in human osteosarcoma cells decreases proliferation and increases apoptosis. Cancer Res. 2006;66:6657–64. doi: 10.1158/0008-5472.CAN-05-3624. [DOI] [PubMed] [Google Scholar]
- 23.Tang HY, Shih A, Cao HJ, Davis FB, Davis PJ, Lin HY. Resveratrol-induced cyclooxygenase-2 facilitates p53-dependent apoptosis in human breast cancer cells. Mol Cancer Ther. 2006;5:2034–42. doi: 10.1158/1535-7163.MCT-06-0216. [DOI] [PubMed] [Google Scholar]
- 24.Hinz B, Ramer R, Eichele K, Weinzierl U, Brune K. Up-regulation of cyclooxygenase-2 expression is involved in R(+)-methanandamide-induced apoptotic death of human neuroglioma cells. Mol Pharmacol. 2004;66:1643–51. doi: 10.1124/mol.104.002618. [DOI] [PubMed] [Google Scholar]
- 25.Davaille J, Gallois C, Habib A, et al. Antiproliferative properties of sphingosine 1-phosphate in human hepatic myofibroblasts. A cyclooxygenase-2 mediated pathway. J Biol Chem. 2000;275:34628–33. doi: 10.1074/jbc.M006393200. [DOI] [PubMed] [Google Scholar]
- 26.Eichele K, Ramer R, Hinz B. Decisive role of cyclooxygenase-2 and lipocalin-type prostaglandin D synthase in chemotherapeutics-induced apoptosis of human cervical carcinoma cells. Oncogene. 2007 doi: 10.1038/sj.onc.1210962. [DOI] [PubMed] [Google Scholar]
- 27.Lin HY, Tang HY, Keating T, et al. Resveratrol is pro-apoptotic and thyroid hormone is anti-apoptotic in glioma cells: both actions are integrin and ERK mediated. Carcinogenesis. 2008;29:62–9. doi: 10.1093/carcin/bgm239. [DOI] [PubMed] [Google Scholar]
- 28.Na HK, Inoue H, Surh YJ. ET-18-O-CH3-induced apoptosis is causally linked to COX-2 upregulation in H-ras transformed human breast epithelial cells. FEBS Lett. 2005;579:6279–87. doi: 10.1016/j.febslet.2005.09.094. [DOI] [PubMed] [Google Scholar]
- 29.Kardosh A, Soriano N, Liu YT, et al. Multitarget inhibition of drug-resistant multiple myeloma cell lines by dimethyl-celecoxib (DMC), a non-COX-2 inhibitory analog of celecoxib. Blood. 2005;106:4330–8. doi: 10.1182/blood-2005-07-2819. [DOI] [PubMed] [Google Scholar]
- 30.Sun SY, Yue P, Dawson MI, et al. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res. 1997;57:4931–9. [PubMed] [Google Scholar]
- 31.Krysan K, Merchant FH, Zhu L, et al. COX-2-dependent stabilization of survivin in non-small cell lung cancer. Faseb J. 2004;18:206–8. doi: 10.1096/fj.03-0369fje. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X, Su L, Pirani AA, et al. Understanding metastatic SCCHN cells from unique genotypes to phenotypes with the aid of an animal model and DNA microarray analysis. Clin Exp Metastasis. 2006;23:209–22. doi: 10.1007/s10585-006-9031-0. [DOI] [PubMed] [Google Scholar]
- 33.Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst. 2004;96:1769–80. doi: 10.1093/jnci/djh322. [DOI] [PubMed] [Google Scholar]
- 34.Sun SY, Yue P, Wu GS, et al. Mechanisms of apoptosis induced by the synthetic retinoid CD437 in human non-small cell lung carcinoma cells. Oncogene. 1999;18:2357–65. doi: 10.1038/sj.onc.1202543. [DOI] [PubMed] [Google Scholar]
- 35.Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65:7429–35. doi: 10.1158/0008-5472.CAN-05-1042. [DOI] [PubMed] [Google Scholar]
- 36.Hideshima T, Catley L, Yasui H, et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood. 2006;107:4053–62. doi: 10.1182/blood-2005-08-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Na HK, Surh YJ. Induction of cyclooxygenase-2 in Ras-transformed human mammary epithelial cells undergoing apoptosis. Ann N Y Acad Sci. 2002;973:153–60. doi: 10.1111/j.1749-6632.2002.tb04626.x. [DOI] [PubMed] [Google Scholar]
- 38.Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- 39.Niederberger E, Tegeder I, Vetter G, et al. Celecoxib loses its anti-inflammatory efficacy at high doses through activation of NF-kappaB. Faseb J. 2001;15:1622–4. doi: 10.1096/fj.00-0716fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.