

Abstract
Spongiform neurodegeneration is characterized by the appearance of vacuoles throughout the central nervous system. It has many potential causes, but the underlying cellular mechanisms are not well understood. Mice lacking the E3 ubiquitin ligase Mahogunin Ring Finger-1 (MGRN1) develop age-dependent spongiform encephalopathy. We identified an interaction between a “PSAP” motif in MGRN1 and the ubiquitin E2 variant (UEV) domain of TSG101, a component of the endosomal sorting complex required for transport I (ESCRT-I), and demonstrate that MGRN1 multimonoubiquitinates TSG101. We examined the in vivo consequences of loss of MGRN1 on TSG101 expression and function in the mouse brain. The pattern of TSG101 ubiquitination differed in the brains of wild-type mice and Mgrn1 null mutant mice: at 1 month of age, null mutant mice had less ubiquitinated TSG101, while in adults, mutant mice had more ubiquitinated, insoluble TSG101 than wild-type mice. There was an associated increase in epidermal growth factor receptor (EGFR) levels in mutant brains. These results suggest that loss of MGRN1 promotes ubiquitination of TSG101 by other E3s and may prevent its disassociation from endosomal membranes or cause it to form insoluble aggregates. Our data implicate loss of normal TSG101 function in endo-lysosomal trafficking in the pathogenesis of spongiform neurodegeneration in Mgrn1 null mutant mice.
Keywords: TSG101, Spongiform neurodegeneration, Mahogunin Ring Finger-1, MGRN1, Ubiquitination, Endocytic trafficking
1. Introduction
Spongiform encephalopathy, where vacuoles develop throughout the central nervous system (CNS), can be caused by infection with the conformationally altered form of the cellular prion protein (PrPSc), retroviruses including human immunodeficiency virus (HIV), murine leukemia virus (MuLV) or maedi-visna virus (MVV), and coronavirus [1], [2], [3], [4]. In humans, spongiform changes are also observed in Alzheimer's disease, diffuse Lewy body disease, Niemann–Pick disease (caused by lysosomal storage defects), Leigh disease (caused by mitochondrial dysfunction) and Canavan disease (caused by aspartoacylase deficiency) [5], [6], [7]. Inherited spongiform encephalopathies develop in mutant mice homozygous for null alleles of Mahogunin Ring Finger-1 (Mgrn1), Attractin (Atrn), superoxide dismutase 2 (Sod2), Fig4, Vac14 and the Kir4.1 potassium channel subunit [8], [9], [10], [11], [12], [13], [14], [15]. These genetic models provide an opportunity to gain insight into the mechanisms that underlie vacuolar changes in the CNS through the identification of the cellular role(s) of the mutated proteins. While the function of the proteins encoded by all of these genes is not known, several are known to affect mitochondrial function or endo-lysosomal trafficking.
The endo-lysosomal pathway plays a critical role in a variety of cellular activities including nutrient intake, signal transduction, and receptor down-regulation, recycling and degradation. Defects in endocytic trafficking are associated with numerous diseases including pigmentation, bleeding and neurodegenerative disorders [9], [16], [17], [18], [19], [20], [21], [22], [23], [24]. Transmembrane proteins are primarily internalized into cells via clathrin-mediated endocytosis, then delivered into early endosomes (EEs) where they are clustered and sorted into intraluminal vesicles to form multivesicular bodies (MVBs). When MVBs fuse with lysosomes, these vesicles and their contents are degraded by lysosomal enzymes. Sorting into MVBs is a critical regulatory step for many proteins and requires ubiquitination at several steps along the way [25]. Ubiquitination is a multistep process in which the small polypeptide ubiquitin is activated in an ATP-dependent manner and then covalently attached to target proteins through the concerted actions of ubiquitin activating (E1), conjugating (E2) and ligase (E3) proteins [26]. Ubiquitination is a sorting signal for many processes, such as trafficking toward the lysosome from many cellular compartments including the plasma membrane (where it promotes internalization), Golgi (where it can divert proteins to endosomes) and MVBs [reviewed by [27]].
Protein sorting into MVBs is regulated by endosomal sorting complexes required for transport (ESCRT)-I, II and III. The sorting process is initiated by the recruitment of hepatocyte growth factor (HGF)-regulated tyrosine kinase substrate (HRS, also commonly referred to as ESCRT-0) to the endosomal membrane through its FYVE domain, which binds to phosphatidylinositol-3-phosphate (PtdIns(3)P). HRS also interacts with ubiquitinated cargos through its ubiquitin-interacting motif (UIM), while a C-terminal “LIEL” sequence binds to clathrin. HRS recruits ESCRT-I to the sorting site via a PSAP motif, which binds to the ubiquitin E2 variant (UEV) domain of the ESCRT-I component TSG101. Ubiquitinated cargo are recognized by the UEV domain of TSG101 and the Npl4 zinc finger (N2F) domain of VPS36 in ESCRT-II. Cargo proteins are clustered by the ESCRT-III complex and their ubiquitin moieties removed by the deubiquitinating enzyme DOA4. Cargo are then sorted to the inner vesicles of MVBs and the ESCRT complexes are disassembled by the AAA-type ATPase VPS4 [28], [29], [30]. Ubiquitination and the ESCRT machinery are also involved in retroviral budding [31], [32]. Ubiquitinated GAG proteins of many viruses, including HIV, associate with TSG101 to recruit it and the ESCRT machinery to the plasma membrane, where the same processes that give rise to MVBs are used to bud virus particles out of the cell [33], [34], [35], [36].
Mice lacking the E3 ubiquitin ligase Mahogunin Ring Finger-1 (MGRN1) develop progressive, widespread spongiform neurodegeneration of the CNS that first becomes apparent between 9 and 12 months of age [11]. This phenotype is preceded by mitochondrial dysfunction and elevated oxidative stress [37]. Mgrn1 null mutant mice also have a defect in pigment-type switching that prevents them from producing yellow pigment [38], and aberrant patterning of the left–right body axis during development that causes lethal congenital heart defects in ∼ 50% of animals [39]. As an E3 ligase, MGRN1 may target a wide variety of proteins for ubiquitination to account for the broad spectrum of phenotypes observed in null mutants, or it may target a small number of proteins that have essential roles in multiple cell types to exert its pleiotropic effects. Here, we confirm a previous report that MGRN1 associates with and ubiquitinates TSG101, a component of ESCRT-I [40], and extend the analysis by examining the in vivo consequences of loss of MGRN1. We demonstrate that ubiquitination of TSG101 in the mouse brain is regulated in part by MGRN1 and that its solubility and pattern of ubiquitination change with age in normal mice and differs between wild-type and Mgrn1 null mutant mice. We propose a model in which endocytic trafficking in general, and TSG101 in particular, plays a central role in the pathogenesis of spongiform encephalopathy in Mgrn1 null mutant mice.
2. Materials and methods
2.1. Mice
Mgrn1 md-nc (null) mutant mice were originally obtained as frozen heterozygous F1 embryos (C3H × 101 strains) from Harwell Mammalian Genetics Unit (Harwell, UK) and have been maintained by brother-sister inbreeding of homozygotes to heterozygotes for over 21 generations. A line of coisogenic wild-type control mice was generated by intercrossing inbred (> 21 generations) Mgrn1 md-nc/+ mice to obtain Mgrn1 +/+ animals; this line has also been maintained by brother-sister inbreeding, with 3–4 generations separating them from the Mgrn1 md-nc allele-carrying line. C3H/HeJ mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and maintained at Cornell University by brother-sister inbreeding for up to 6 generations. Mice were maintained under standard conditions in the AALAC certified Transgenic Mouse Core Facility at Cornell University. All experiments involving mice adhered to the Public Health Service Policy on Humane Care and Use of Laboratory Animals and were approved by Cornell University IACUC.
2.2. Protein interaction and ubiquitination studies
Yeast two-hybrid assays were performed using the ProQuest Two-Hybrid System with Gateway technology (Invitrogen, Carlsbad CA). The cDNA encoding the C-terminus of MGRN1 isoform I (encoding the region downstream of the RING domain, amino acids 316–532; Fig. 1 ) was fused to the GAL4 DNA binding domain (DB) in the pDEST32 bait vector and used to screen a mouse brain cDNA library (Invitrogen #11298-015). Plasmids encoding proteins that interacted with MGRN1 were isolated using a yeast plasmid isolation kit (Clontech, Mountain View, CA) and sequenced at the Cornell University Sequencing Core Facility. Tsg101 expression constructs were provided by Dr. Kay-Uwe Wagner, University of Nebraska Medical Center. HA-tagged wild-type ubiquitin was provided by Pengbo Zhou, Weill Medical College. GFP-tagged ubiquitin K0 (all lysine residues mutated to arginines, rendering it incapable of forming polyubiquitin chains) was obtained from Nico Dantuma via Addgene (plasmid 11934) [41].
Fig. 1.
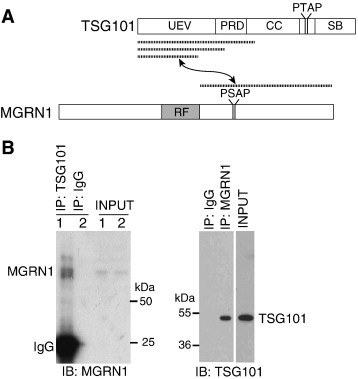
MGRN1 associates with TSG101. (A) Schematic of TSG101 and MGRN1 proteins. Regions of each protein found to interact with the other in yeast two-hybrid assays are indicated by dashed lines. UEV: ubiquitin E2 variant domain; PRD: proline rich domain; CC: coiled-coil domain; SB: steadiness box domain; RF: C3HC4 ring finger domain. The position of late-viral P[S/T]AP domains in TSG101 and MGRN1 are also indicated. (B) MGRN1 associates with TSG101. IP of lysates from untransfected HEK293T cells for TSG101 (left panel) or MGRN1 (right panel) followed by IB for MGRN1 (left panel) or TSG101 (right panel) confirmed association of the endogenous proteins. Clean-blot detection reagent was used in lieu of secondary antibody for TSG101 IB of MGRN1 IP products to eliminate the heavy chain IgG band that otherwise obscures the TSG101 signal.
Full-length MGRN1 isoform I was amplified by RT-PCR from C3H/HeJ mouse brain RNA using primers: GGGGACAAGTTTGTACAAAAAAGCAGGCTCCGGCTGGCCGGTAGAAAC and GGGGACCACTTTGTACAAGAAAGCTGGGTACTATACCAACAGAGCACGAC. PCR products were cloned using the Gateway system into the pEGFP-N1 vector (to express C-terminal GFP-tagged MGRN1) (Clontech). Isoform I was also amplified using primers CACCATGGGCTCCATCATGA and TTACTCCTCTATACCAACAG and cloned into pKH3 vector (to express N-terminal triple hemagglutinin (HA)-tagged MGRN1). The vectors were first converted to be Gateway compatible using the Gateway Vector Conversion System (Invitrogen). A SalI/BamHI fragment of IMAGE clone 6401088, which encodes full-length MGRN2 (RNF157), was purified and ligated into Gateway modified pEGFP-N1 to create a mammalian expression construct for MGRN2 where its final 3 amino acids are replaced by GFP.
MGRN1 isoform I in pEGFP-N1 was used as template for site-directed mutagenesis using QuickChange Site Directed Mutagenesis kit (Stratagene). Cysteine residues at positions 278 and 281 were mutated to alanine residues to generate MGRN1(AVVA)-GFP using forward primer GACAACAGCAGTGAGGCTGTGGTGGCCCTGTCAGACCTGCG and reverse primer CTGTTGTCGTCACTCCGACACCACCGGGACAGTCTGGACGC. This CVVC to AVVA mutation has previously been shown to inactivate the catalytic activity of MGRN1 [11]. The PSAP motif at amino acid position 384–387 (isoform I) was mutated to ASAA (subsequently referred to as MGRN1(ASAA)) using forward primer CCCCGGCTA-TCGCATCAGCTGCCCTCTATGAG and reverse primer GATGGGCTCATAGCCAGCTGCGATACTGTCAGAACTAG.
Plasmid DNA or siRNA against Mgrn1 (sense: GAACUCGGCCUAUCGCUACUU, antisense: PGUAGCGAUAGGCCGAGUUCUU, corresponding to amino acids 57–75 of human MGRN1) or a scrambled control siRNA (sense: GUACCGCACCGCUAGUCAU, antisense: AUGACUAGCGGUGCGGUAC) were transfected into Human Embryonic Kidney (HEK293T) cells for transient overexpression. Fig. S1 in the supplementary material demonstrates the efficiency of MGRN1 knockdown. For ubiquitination assays, some cells were treated with 50 μM of the proteasome inhibitor MG132 for 2–4 h before lysis. Cells were harvested in lysis buffer containing Complete protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN) 24–48 h after transfection, frozen and thawed 3 times, then used for TA purification or coimmunoprecipitation (coIP) using Protein G IP Kit (Sigma-Aldrich Corp., St. Louis, MO) or GrabIt IP kit (Calbiochem, Gibbstown, NJ) according to manufacturer's instructions. Mouse brains were homogenized in Cytobuster buffer (Calbiochem) containing Complete protease inhibitor cocktail prior to IP for TSG101. Proteins were separated by SDS-PAGE and transferred to Immobilon P membrane (Millipore, Billerica, MA) for immunoblotting. All data shown is representative of at least two independent experiments. Antibodies used: MGRN1 (#11285-1-AP, ProteinTech Group, Inc., Chicago, IL), TSG101 (#612696, BD Biosciences, San Jose, CA or #C-2, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), HA (#MMS-101P, Covance, Trenton, NJ), GFP (#JL-8, Clontech or #MAB3580, Millipore), Myc (#Ab32, Abcam, Cambridge, MA) and GAPDH (#Ab9482, Abcam). For IB of TSG101 on MGRN1 IP products, Clean-Blot Detection Reagent (Thermo Scientific, Rockford, IL) was used in place of a secondary antibody to avoid the heavy chain IgG band that is very close in size to TSG101.
2.3. Western analysis of mouse tissues
For analysis of TSG101 expression, brains were rapidly dissected from at least 2 animals of each genotype, then homogenized in a Dounce homogenizer on ice in RIPA buffer (50 mM Tris, 150 mM NaCl, 1% w/v Triton X-100) containing Complete protease inhibitor cocktail. For solubility assay, brains were homogenized in Triton X-100 buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100) with Complete protease inhibitor cocktail using a TissueLyser (Qiagen, Valencia, CA). The homogenate was centrifuged at 15,000g for 20 min at 4 °C, and the supernatant collected as the detergent-soluble fraction. The resulting pellet was washed three times in Triton X-100 buffer then homogenized in Triton X-100 buffer supplemented with 2% SDS to collect the detergent-insoluble fraction. Protein concentrations were determined using BCA Protein Assay (Thermo Scientific) and 50 μg of protein separated by SDS-PAGE (10% w/v acrylamide). Proteins were transferred to Immobilon P membrane and immunoblotted using anti-TSG101 antibody (#C-2, Santa Cruz). Blots were stripped and reblotted for GAPDH to verify equal protein loading. RIPA-extracted brain proteins from wild-type and Mgrn1 md-nc/md-nc mice were also subjected to IP for TSG101 (as described above) and blotted for TSG101 and ubiquitin (FK2 (#PW0150), Biomol International Inc., Plymouth Meeting, PA) to confirm that the high-molecular weight bands observed with the TSG101 antibody represent multiubiquitinated TSG101.
2.4. Viral budding assay
HEK293T cells were plated in 12-well tissue culture dishes at a density of 5 × 105 cells/well in 1 mL DMEM (Cellgro) supplemented with 10% FBS and l-glutamine. The following day, each well was transfected with 0.5 μg of HIV-1 GAG [42] and 1.0 μg of either GFP, GFP-VPS4(EQ) [43], wild-type MGRN1-GFP or MGRN1(AVVA)-GFP plasmids (“treatments”), using 3 μl Lipofectamine 2000 (Invitrogen) per well. HIV-1 GAG and VPS4(EQ) plasmids were generously provided by Dr. Volker Vogt. Transfection efficiency was monitored by GFP fluorescence and estimated to be equivalent (∼ 60%) for all samples. Twenty-four hours post-transfection, media was collected and spun at 2000 rpm for 1 min to pellet debris. Cleared media was spun at 13,000 rpm in a microcentrifuge for 100 min to pellet virus-like particles (VLPs), which were resuspended in 1× sample loading buffer (60 mM Tris–HCl, pH 6.8, 5% glycerol, 3% SDS, 1 M BME, 0.025% bromophenol blue) and boiled (95 °C for 5 min) for western analysis. Cells were collected into lysis buffer (50 mM Tris–HCl pH 8.0, 1% Igepal CA-630, 1 mM EDTA, 10 mM iodoacetamide with Complete protease inhibitor cocktail), incubated on ice for 15 min, and centrifuged at 13000 rpm for 10 min. Supernatants in sample loading buffer were boiled as for VLP preps. Samples (∼ 20% of total for VLP preparations, ∼ 3% of total for cell lysates) were separated on 10% polyacrylamide gels under standard Laemmli conditions and electrophoretically transferred to Immobilon P membrane (Millipore). Immunoblotting was performed under standard conditions, using rabbit anti-HIV CA polyclonal antibody (HIV-1SF2 p24 Antiserum, NIH AIDS Research and Reference Reagent Program #4350). Blots were visualized using a VersaDoc 5000 (BioRad). Each gel contained one set of samples for each treatment. Band intensities were quantified using Quantity One software (BioRad), correcting for background using the signal from a lane for a sample prepared from cells transfected only with GFP vector (no GAG DNA). Samples on the same gel/blot were compared to one another to control for inter-blot variability. The relative budding index was calculated as the ratio of GAG signal in media to GAG signal in cell lysates for each transfection relative to that of the GAG + GFP (control) samples run on the same blot. Differences between the relative budding index value of each treatment and the GFP control were assessed using a pairwise 2-tailed Student's T-test.
3. Results
3.1. MGRN1 associates with TSG101
Yeast two-hybrid screens using the region of MGRN1 C-terminal to the RING domain yielded several partial Tsg101 cDNA clones as strong interactors (Fig. 1A). We confirmed that MGRN1 associated with TSG101 in mammalian cells by several means, including coIP of endogenous MGRN1 and TSG101 from untransfected HEK293T cells (Fig. 1B). All of the Tsg101 cDNA clones isolated from yeast-two hybrid screens contained sequences encoding the N-terminal ubiquitin E2 variant (UEV) domain (Fig. 1A). Some clones extended further C-terminal into the adjacent proline-rich domain (PRD) and/or coiled coil (CC) region. Analysis of the MGRN1 amino acid sequence revealed a PSAP motif that is highly conserved among vertebrate MGRN1 homologs (Fig. 2A). This motif is found in all vertebrate MGRN1 isoforms. Interestingly, bovine MGRN1 contains a short replacement of sequence that disrupts the PSAP motif but replaces it with a functionally interchangeable PTAP sequence (Fig. 2A). P[S/T]AP motifs are considered “late viral domains” and the UEV domain of TSG101 has previously been shown to interact with this motif in a variety of proteins including HRS, several viral GAG proteins and the TAL ubiquitin ligase [31], [44], [45], [46]. To test whether the PSAP motif of mouse MGRN1 mediates its association with TSG101, site-directed mutagenesis was performed to convert it to ASAA (MGRN1(ASAA)-GFP). CoIP and immunoblotting assays confirmed that MGRN1(ASAA)-GFP did not associate with TSG101 (Fig. 2B).
Fig. 2.
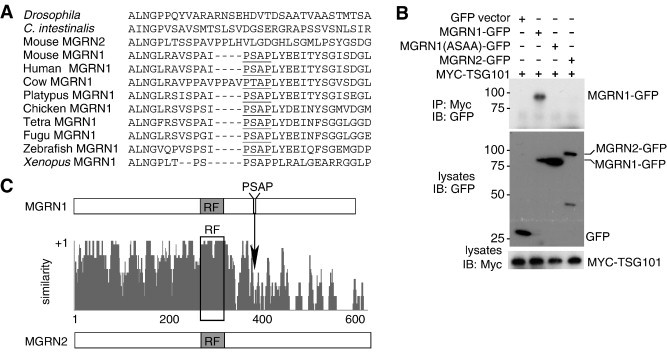
A conserved PSAP motif in MGRN1 mediates its association with TSG101. (A) Vertebrate MGRN1 homologs contain a PSAP “late-viral” motif that is not found in the related protein, MGRN2 (RNF157) or invertebrate MGRN homologs. (B) HEK293T cells were transfected with Myc-tagged TSG101 and control GFP, MGRN2-GFP, wild-type MGRN1-GFP, or mutant MGRN1(ASAA)-GFP (PSAP motif mutated to ASAA). Lysates were subjected to IP using an antibody against Myc (to IP TSG101) and blotted for GFP. Only wild-type MGRN1-GFP coimmunoprecipitated with TSG101, indicating that the PSAP motif is required for their association. (C) Schematic showing protein structures of and amino acid identity between MGRN1 and MGRN2. MGRN2 does not contain a PSAP motif.
Neither Drosophila, sea squirt nor worm MGRN proteins contain a P[S/T]AP motif (Fig. 2A), suggesting that association between MGRN1 and TSG101 is a relatively recent evolutionary event. Vertebrates contain a homologous gene, Mgrn2 (Rnf157), that encodes a C3HC4 RING-containing protein 69% identical to MGRN1 (Fig. 2C). MGRN2, like invertebrate MGRN homologs, lacks a P[S/T]AP motif (Fig. 2A,C). MGRN1 and MGRN2 are most similar across the region N-terminal to the location of the PSAP motif in MGRN1 (Fig. 2C). We transiently transfected HEK293T cells with GFP-tagged MGRN2 and Myc-tagged TSG101 expression constructs and used IP and IB to confirm that MGRN2 did not associate with TSG101 (Fig. 2B).
3.2. MGRN1 multimonoubiquitinates TSG101
Association of MGRN1 with TSG101 suggested that TSG101 may be a target for MGRN1-mediated ubiquitination. To test this hypothesis, HEK293T cells were transfected with HA-ubiquitin and either empty GFP vector, MGRN1-GFP, catalytically inactive MGRN1(AVVA)-GFP, or siRNA against Mgrn1. In the presence or absence of the proteasome inhibitor MG132, multiubiquitination of TSG101 was increased in MGRN1-GFP transfected cells relative to vector-transfected cells (which contain endogenous MGRN1) and greatly reduced in cells expressing MGRN1(AVVA)-GFP or Mgrn1 siRNA (Fig. 3A, B). A short exposure of Fig. 3A shows that more multiubiquitinated TSG101 is present in cells expressing MGRN1-GFP than GFP alone, even in the presence of the proteasome inhibitor MG132 (Fig. S2 in the supplementary material). Experiments using GFP-ubiquitin-K0 (in which all lysines have been mutated to alanines, rendering it incapable of forming polyubiquitin chains) in place of wild-type ubiquitin showed a similar pattern of MGRN1-dependent ubiquitination of TSG101, indicating that TSG101 is multimonoubiquitinated by MGRN1 (Fig. 3C). We confirmed that MGRN1(AVVA)-GFP was still able to associate with TSG101; in fact, TSG101 appeared to associate more strongly with MGRN1(AVVA)-GFP than with wild-type MGRN1-GFP (Fig. 3D). Catalytically inactive mutants of other E3s have also been shown to bind their target proteins more strongly [i.e., 47].
Fig. 3.
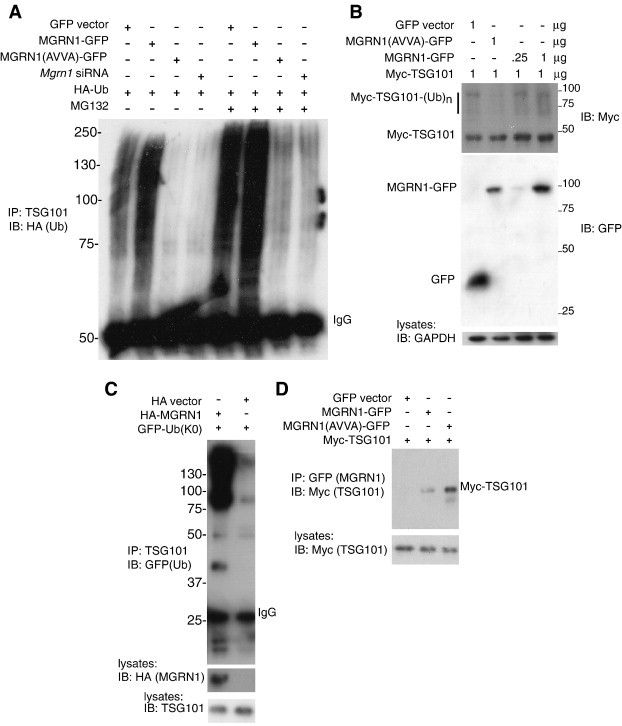
MGRN1 multimonoubiquitinates TSG101. (A) MGRN1 ubiquitinates TSG101. Duplicate sets of HEK293T cells were transfected with indicated plasmids. One set of plates was treated with proteasome inhibitor MG132 prior to collection of lysates. Lysates were subjected to IP for endogenous TSG101 and blotted for HA (ubiquitin). Regardless of MG132 treatment, TSG101 was multiubiquitinated in cells expressing empty GFP vector or wild-type MGRN1-GFP, with a stronger signal in cells overexpressing MGRN1. Very little ubiquitinated TSG101 was detected in cells expressing catalytically inactive MGRN1 (MGRN1(AVVA)-GFP) or siRNA against Mgrn1. (B) Dose-dependent effect of MGRN1 on TSG101 ubiquitination. HEK293T cells were transfected with indicated amounts (μg) of each plasmid. Lysates were blotted for Myc (TSG101), GFP (GFP or tagged MGRN1) and GAPDH (control for protein loading). A smear of ubiquitinated TSG101 was detected in cells expressing MGRN1, with a stronger signal in cells expressing higher amounts of MGRN1. The intensity of the signal for multiubiquitinated TSG101 (Myc-TSG101-(Ub)n) was much reduced in cells expressing catalytically inactive MGRN1 (AVVA mutant). (C) MGRN1 multimonoubiquitinates TSG101. HEK293T cells were transfected with indicated plasmids and lysates subjected to IP for endogenous TSG101. IP products were blotted for GFP to detect ubiquitin-K0 (contains no lysine residues to allow for the formation of polyubiquitin chains). A significant increase in multimonoubiquitinated TSG101 was observed in cells overexpressing MGRN1. Lysates were blotted for HA (to detect HA-MGRN1; HA produced from empty vector ran off the bottom of the gel) and TSG101 to confirm expression. (D) HEK293T cells were transfected with indicated plasmids. Lysates were subjected to IP for GFP (MGRN1) and blotted for Myc (TSG101) to demonstrate that catalytically inactive MGRN1 (AVVA mutant) not only still binds TSG101, but binds it more tightly than wild-type MGRN1: Myc-TSG101 expression was equal in the lysates prior to IP but much stronger in the IP lane from cells expressing MGRN1(AVVA)-GFP.
3.3. MGRN1 regulates TSG101 in the mouse brain
As our primary interest was whether loss of MGRN1-mediated ubiquitination of TSG101 contributes to the development of spongiform neurodegeneration in Mgrn1 null mutant mice, we used western analysis to examine TSG101 expression in brain protein extracts from wild-type, Mgrn1 heterozygotes and Mgrn1 null mutant mice (Figs. 4A and S3 in the supplementary material). Several TSG101 bands were observed, consistent with unmodified (∼ 44 kDa) and post-translationally modified forms, with the latter being consistent in size with bands described by others as ubiquitinated TSG101. We confirmed that the high molecular weight bands represented ubiquitinated forms of TSG101 by immunoprecipitating TSG101 from brain lysates and showing that these bands were detected with antibodies against TSG101 and ubiquitin (Fig. 4B). As we did not distinguish whether they represent multimono- or poly-ubiqutinated TSG101, or both, these bands will be referred to as multiubiquitinated TSG101.
Fig. 4.
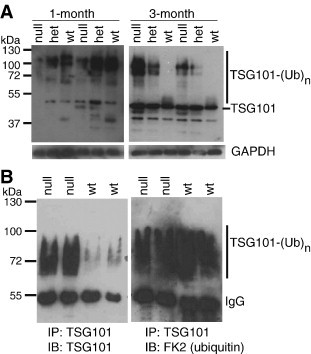
Age- and MGRN1-dependent changes in TSG101 ubiquitination in the mouse brain. (A) Immunoblotting (IB) of whole brain protein lysates from wild-type (wt), Mgrn1 heterozygous (het) and Mgrn1 null mutant mice for TSG101. In wild-type mice, high levels of multiubiquitinated TSG101 (TSG101-(Ub)n) were observed in lysates from 1-month-old animals, but most TSG101 expression in older (3-month-old) animals was unmodified or monoubiquitinated. At 1-month of age, the levels of multiubiquitinated TSG101 were lower in Mgrn1 null mutant brains than in wild-type brains, but at 3-months of age, there was more multiubiquitinated TSG101 in the brains of mutant mice. At both ages, heterozygotes had levels intermediate to those of null mutants and wild-type mice. A 3-min exposure is shown for 1 month samples, a 1-min exposure for 3-month samples (3-min exposure of both shown in Supplemental Fig. S3B). Multiubiquitinated TSG101 was observed in samples from 3-month-old mice when blots were imaged longer (not shown). (B) Brain lysates from 6-month-old wild-type (wt) and Mgrn1 null mutant mice were subjected to IP for TSG101. Duplicate blots were immunoblotted for TSG101 and ubiquitin (FK2 antibody, which recognizes mono- and poly-ubiquitinated proteins), respectively. Both antibodies detected the same high molecular weight bands, indicating that they represent multiubiquitinated TSG101.
More multiubiquitinated TSG101 was observed in brain lysates from 1-month-old mice than from 3- and 6-month-old wild-type mice (Figs. 4A and S3 in the supplementary material), although high molecular weight forms of TSG101 were detected in samples from older animals when blots were imaged longer (or following IP for TSG101, as shown in Fig. 4B). Multiubiquitinated TSG101 was present at reduced levels in the brains of 1-month-old Mgrn1 heterozygotes and Mgrn1 null mutant mice relative to controls. In 3- or 6-month-old animals, however, the bands representing multiubiquitinated TSG101 were stronger in the brains of heterozygotes and Mgrn1 null mutant mice than in wild-type animals and than in 1-month old heterozygotes and Mgrn1 null mutants (Figs. 4A and S3B). A band consistent in size with monoubiquitinated TSG101 was present in brain lysates from 3- and 6-month-old wild-type mice but not in 1-month-old wild-type mice nor in Mgrn1 md-nc/+ or Mgrn1 md-nc/md-nc mice at any of the ages examined. At all ages, differences in the levels of TSG101 and its modified forms were associated with the number of functional Mgrn1 alleles, where samples from heterozygotes had levels intermediate to those from wild-type and Mgrn1 null mutant mice.
TSG101 plays a crucial role in sorting monoubiquitinated proteins, such as the epidermal growth factor receptor (EGFR), into MVBs to traffic them to the lysosome for degradation. If TSG101 function is impaired in the brains of Mgrn1 null mutant mice, EGFR trafficking would be expected to be disrupted. Western blotting demonstrated a significant increase in EGFR levels in the brains of 1-month-old Mgrn1 null mutant mice relative to wild-type mice (Fig. 5 ). There was also a slight increase in EGFR levels in the brains of 6-month-old Mgrn1 null mutant mice (Fig. 5).
Fig. 5.
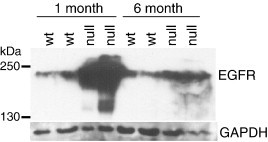
EGFR, which requires functional TSG101 for its endosomal transport to the lysosome to be degraded, accumulates in the brains of Mgrn1 null mutant mice. Brain protein lysates from wild-type (wt) and Mgrn1 null mutant animals of the indicated ages were subjected to IB for EGFR. Protein loading is indicated by IB for GAPDH.
TAL-dependent ubiquitination of TSG101 has previously been reported to increase TSG101 solubility [44]. Since loss of MGRN1 led to the accumulation of ubiquitinated TSG101 in the brains of 3-month-old (and older) Mgrn1 null mutant mice, we examined whether the solubility of TSG101 also changed with age or was affected by loss of MGRN1. Brain proteins from 1- and 6-month-old Mgrn1 null mutant and wild-type mice were extracted in Triton X-100 buffer and the resulting pellet extracted with SDS-containing buffer. Immunoblotting for TSG101 demonstrated that very little of the multiubiquitinated TSG101 in 1-month-old wild-type brains was soluble in Triton X-100 while a greater proportion was soluble in 6-month-old wild-type brains (Fig. 6 ). Most TSG101 in the brains of 1-month-old Mgrn1 null mutants was not ubiquitinated and was soluble in Triton X-100. There was a significant increase in soluble and insoluble TSG101 in the brains of 6-month-old Mgrn1 null mutants, with the majority of insoluble TSG101 being ubiquitinated in those samples (Fig. 6). Thus, upon prolonged absence of MGRN1 from the mouse brain, most of the multiubiquitinated TSG101 that accumulates is insoluble.
Fig. 6.
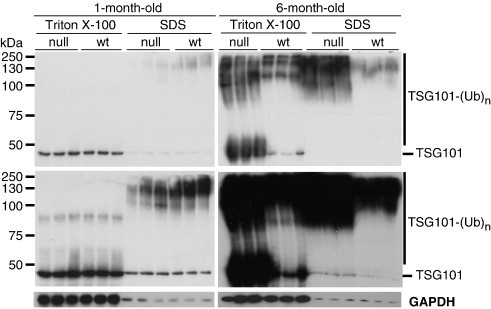
Loss of MGRN1 results in the accumulation of insoluble, high molecular weight (multiubiquitinated) forms of TSG101. Sequential extraction of brain proteins from 1- and 6-month-old wild-type (wt) and Mgrn1 null mutant mice was performed to isolate proteins based on their solubility (proteins in Triton X-100 fractions are more soluble than those in SDS fractions). Fractions were subjected to IB to detect unmodified and ubiquitinated TSG101 (TSG101-(Ub)n). The same blots were imaged for 1 min (top panels) and 10 min (bottom panels), and blotted for GAPDH as a loading control. In the brains of young animals, multiubiquitinated TSG101 was predominantly observed in the SDS fraction. In older animals, multiubiquitinated TSG101 was observed in both fractions, although more was present in the SDS fraction in the brains of Mgrn1 null mutants than in wild-type mice.
3.4. MGRN1 is not required for viral budding
We examined the release of HIV-1 GAG from cells to test whether MGRN1-dependent ubiquitination of TSG101 was also required for VLP budding. HEK293T cells were mock-transfected or transfected with HIV-1 GAG and either a mutant VPS4 that disrupts viral budding (GFP-VPS4(EQ)), empty GFP vector, wild-type MGRN1-GFP or catalytically inactive MGRN1 (MGRN1(AVVA)-GFP). GAG release from cells as VLPs was detected by IB for GAG on proteins extracted from the cell media, while GAG retained in the cells was detected by IB for GAG on cell lysates. The budding index was determined as the ratio of secreted GAG/intracellular GAG in treated samples relative to control (GAG + GFP), averaged over 3 experiments. While the VPS4(EQ) mutant greatly diminished GAG release, as expected, the addition of excess wild-type or catalytically inactive MGRN1 had no significant effect (Fig. 7A, B). This suggests that the effect of MGRN1-dependent ubiquitination on TSG101 function may be restricted to endo-lysosomal trafficking.
Fig. 7.
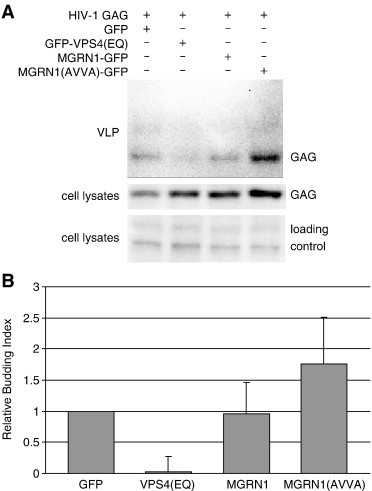
Overexpression of neither wild-type nor catalytically inactive MGRN1 significantly affects HIV-1 GAG release from cells. HEK293T cells were cotransfected with an HIV-1 GAG p55 expression construct and either a GFP control, a positive control for budding inhibition (GFP-VPS4(EQ)), or GFP fusion constructs expressing wild-type MGRN1 or catalytically inactive MGRN1 (AVVA mutant). (A) Representative immunoblots from 1 of 3 replicate experiments. Top panel: GAG IB of virus-like particle (VLP) preps from media of transfected cells. Middle panel: GAG IB of cell lysates of transfected cells. Bottom panel: non-specific bands from upper region of cell lysate blot demonstrate even loading. (B) Graphical representation of the effect of each treatment on VLP budding, averaged from 3 replicate experiments. Relative budding index (y axis) represents the ratio of GAG signal in VLPs to GAG signal in cell lysates of each transfection relative to that of GAG + GFP control samples. VPS4(EQ) reduced GAG release (p < 0.02) while neither wild-type nor catalytically inactive MGRN1 had a significant effect (p > 0.3). The standard error of the GAG + GFP transfected samples is 0 because the budding index of these samples is defined as 1 within each experiment.
4. Discussion
TSG101 is a highly conserved and essential protein required for sorting ubiquitinated cargo to the inner vesicles of MVBs. Here, we confirm and extend the work of Kim et al. [40], showing that the vertebrate-specific E3 ubiquitin ligase MGRN1 binds and multimonoubiquitinates TSG101 to regulate its function in endo-lysosomal trafficking in vivo. We focused on the effect of loss of MGRN1 in the mouse brain on the regulation of TSG101, where the relationship between Mgrn1 genotype and levels of multiubiquitinated TSG101 (which was lower in homozygous null mutants than in heterozygotes) indicates that MGRN1 is responsible for ubiquitinating a significant portion of TSG101 in the brains of young (1-month-old) wild-type mice. The PSAP motif in MGRN1 that mediates its association with TSG101 is not present in invertebrate MGRN homologs, nor in the vertebrate family member MGRN2 (RNF157). This implies that the ability of MGRN1 to associate with TSG101 is a relatively recent evolutionary event and either has an important vertebrate-specific function or plays a role in vertebrate cells that is performed by another E3 ligase in non-vertebrates. We also observed age-dependent changes in the pattern of TSG101 expression in the brains of wild-type mice, with more TSG101 multiubiquitinated in the brains of young (1-month-old) animals than in adult mice. This suggests that MGRN1-dependent TSG101-mediated endocytic trafficking may be particularly important during development and that only low levels of activity are required for normal adult CNS function.
The fact that ubiquitinated TSG101 is reduced but not absent in the brains of young Mgrn1 null mutant mice indicates that MGRN1 is not the only E3 ligase that targets TSG101 at this age. The accumulation of multiubiquitinated TSG101 in the brains of older animals that lack MGRN1 suggests that, upon prolonged absence or reduction of MGRN1, TSG101 is recognized and ubiquitinated by another E3. At least two other E3s, MDM2 and TAL (also known as LRSAM1), have been reported to ubiquitinate TSG101 [44], [45], [48], [49]. TAL has been reported to multimonoubiquitinate TSG101 to regulate its cargo-sorting and viral budding functions [44], and to polyubiquitinate TSG101 and target it for proteasomal degradation when it is not part of the ESCRT complex [45]. TAL, like MGRN1, is not found in invertebrates. This suggests multiple changes in the regulation of TSG101 during vertebrate evolution.
Interestingly, TAL and MGRN1 have different effects on the viral budding and EGFR trafficking functions of TSG101: knock-down of TAL expression by siRNA increased VLP release; overexpression of wild-type TAL increased EGFR levels and inhibited VLP (GAG) release; and overexpression of catalytically inactive TAL reduced EGFR levels [44]. Overexpression of MGRN1 had no effect on GAG budding and loss of MGRN1 resulted in elevated levels of EGFR. TAL-mediated ubiquitination of TSG101 increased its solubility [44]. Since the solubility of the ubiquitinated TSG101 that accumulates in the brains of Mgrn1 null mutant mice was reduced, TAL is probably not the E3 that ubiquitinates TSG101 in the brains of older animals lacking MGRN1. The fact that multiubiquitinated TSG101 accumulates in the brains of adult Mgrn1 null mutant mice indicates that it is not targeted for proteasomal degradation, while its reduced solubility suggests that it may form insoluble cytoplasmic aggregates or be membrane-bound. If the latter, multimonoubiquitination by MGRN1 may be necessary for TSG101 to complete its ESCRT-I function and subsequently be released from endosomal membranes. The increase in EGFR levels in the brains of Mgrn1 null mutant mice is consistent with MGRN1-dependent ubiquitination of TSG101 being required for its normal function in MVB sorting and receptor down-regulation.
While further studies will be needed to determine whether disrupted TSG101 function in fact causes the spongiform neurodegeneration observed in Mgrn1 null mutant mice, there are at least two possible mechanisms by which it could do so. The first is related to the fact that endo-lysosomal trafficking of EGFR is required for its down-regulation and degradation in the lysosome [50], [51], [52]. We observed increased levels of EGFR in the brains of Mgrn1 null mutant mice, particularly in 1-month-old animals. Lysosomal trafficking of EGFR has also been reported to be disrupted in cells in which MGRN1 was depleted by siRNA, with prolonged activation of mitogen-activated protein (MAP) kinase signaling [40]. Exposure of mixed cortical cultures (containing neurons and astrocytes) to 100 ng/ml of EGF caused neuronal death associated with features of necrosis and increased free radical generation [53]. The EGFR-specific tyrosine kinase inhibitor C56 blocked the effect of excess EGF, indicating that the neurotoxicity was mediated through EGFR signaling. Thus, accumulation of activated EGFR could be responsible for (or at least contribute to) elevated levels of oxidatively damaged proteins observed in the brains of Mgrn1 null mutant mice [37] and subsequent vacuolation.
Another possibility is that the vacuoles in the brains of Mgrn1 null mutant mice represent accumulated endocytic compartments. Mammalian cells lacking TSG101 showed disrupted bulk transport to the lysosome as well as structural rearrangement of the early endosome to form enlarged vacuoles that were often folded into multicisternal structures reminiscent of the abnormal membranous compartments that accumulate in yeast “Class E” vacuolar protein sorting (vps) mutants [54], [55]. We propose that any mutation that severely disrupts endo-lysosomal trafficking would cause the accumulation of endocytic compartments that could, over time, enlarge to fill the cell. Loss-of-function mutations in Fab1 (endosomal phosphatidylinositol PtdIns(3)P 5-kinase) in Drosophila or Vac14 (a regulator of phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2) synthesis) or Fig4 (a PtdIns(3,5)P2 5-phosphatase) in mice have also been shown to disrupt endo-lysosomal trafficking and cause cellular vacuolation and spongiform neurodegeneration [9], [10], [17], [56]. Thus, defects in intracellular trafficking represent a common cellular mechanism for causing spongiform neurodegeneration. It remains to be determined whether vacuoles are the result of aberrant trafficking of and signaling by specific receptors, the accumulation of endosomal compartments, or a combination of the two.
Acknowledgements
We thank Dr. Volker Vogt and members of his laboratory for HIV-1 GAG reagents and advice on the viral budding assay, and Drs. Kay-Uwe Wagner and Pengbo Zhou for plasmids. HIV-1 SF2 p24 antiserum was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. The project described was supported by Grant Number R01AG022058 from the National Institute On Aging to T.M.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute On Aging or the National Institutes of Health.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbadis.2009.08.009.
Appendix A. Supplementary data
References
- 1.Bilello J.A., Pitts O.M., Hoffman P.M. Characterization of a progressive neurodegenerative disease induced by a temperature-sensitive Moloney murine leukemia virus infection. J. Virol. 1986;59:234–241. doi: 10.1128/jvi.59.2.234-241.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Everall I., Luthert P., Lantos P. A review of neuronal damage in human immunodeficiency virus infection: its assessment, possible mechanism and relationship to dementia. J. Neuropathol. Exp. Neurol. 1993;52:561–566. doi: 10.1097/00005072-199311000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Jacomy H., Talbot P.J. Vacuolating encephalitis in mice infected by human coronavirus OC43. Virology. 2003;315:20–33. doi: 10.1016/S0042-6822(03)00323-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Georgsson G., Palsson P.A., Panitch H., Nathanson N., Petursson G. The ultrastructure of early visna lesions. Acta Neuropathol. (Berl). 1977;37:127–135. doi: 10.1007/BF00692058. [DOI] [PubMed] [Google Scholar]
- 5.Baslow M.H. Canavan's spongiform leukodystrophy: a clinical anatomy of a genetic metabolic CNS disease. J. Mol. Neurosci. 2000;15:61–69. doi: 10.1385/JMN:15:2:61. [DOI] [PubMed] [Google Scholar]
- 6.Smith T.W., Anwer U., DeGirolami U., Drachman D.A. Vacuolar change in Alzheimer's disease. Arch. Neurol. 1987;44:1225–1228. doi: 10.1001/archneur.1987.00520240007003. [DOI] [PubMed] [Google Scholar]
- 7.Iseki E., Li F., Kosaka K. Close relationship between spongiform change and ubiquitin-positive granular structures in diffuse Lewy body disease. J. Neurol. Sci. 1997;146:53–57. doi: 10.1016/s0022-510x(96)00282-1. [DOI] [PubMed] [Google Scholar]
- 8.Filosto M., Tomelleri G., Tonin P., Scarpelli M., Vattemi G., Rizzuto N., Padovani A., Simonati A. Neuropathology of mitochondrial diseases. Biosci. Rep. 2007;27:23–30. doi: 10.1007/s10540-007-9034-3. [DOI] [PubMed] [Google Scholar]
- 9.Chow C.Y., Zhang Y., Dowling J.J., Jin N., Adamska M., Shiga K., Szigeti K., Shy M.E., Li J., Zhang X., Lupski J.R., Weisman L.S., Meisler M.H. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., Zolov S.N., Chow C.Y., Slutsky S.G., Richardson S.C., Piper R.C., Yang B., Nau J.J., Westrick R.J., Morrison S.J., Meisler M.H., Weisman L.S. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc. Natl. Acad. Sci. U. S. A. 2007;104:17518–17523. doi: 10.1073/pnas.0702275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He L., Lu X.Y., Jolly A.F., Eldridge A.G., Watson S.J., Jackson P.K., Barsh G.S., Gunn T.M. Spongiform degeneration in mahoganoid mutant mice. Science. 2003;299:710–712. doi: 10.1126/science.1079694. [DOI] [PubMed] [Google Scholar]
- 12.He L., Gunn T.M., Bouley D.M., Lu X.Y., Watson S.J., Schlossman S.F., Duke-Cohan J.S., Barsh G.S. A biochemical function for attractin in agouti-induced pigmentation and obesity. Nat. Genet. 2001;27:40–47. doi: 10.1038/83741. [DOI] [PubMed] [Google Scholar]
- 13.Neusch C., Rozengurt N., Jacobs R.E., Lester H.A., Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melov S., Schneider J.A., Day B.J., Hinerfeld D., Coskun P., Mirra S.S., Crapo J.D., Wallace D.C. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat. Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 15.Lebovitz R.M., Zhang H., Vogel H., Cartwright J., Jr, Dionne L., Lu N., Huang S., Matzuk M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gissen P., Johnson C.A., Morgan N.V., Stapelbroek J.M., Forshew T., Cooper W.N., McKiernan P.J., Klomp L.W., Morris A.A., Wraith J.E., McClean P., Lynch S.A., Thompson R.J., Lo B., Quarrell O.W., Di Rocco M., Trembath R.C., Mandel H., Wali S., Karet F.E., Knisely A.S., Houwen R.H., Kelly D.A., Maher E.R. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat. Genet. 2004;36:400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 17.Rusten T.E., Vaccari T., Lindmo K., Rodahl L.M., Nezis I.P., Sem-Jacobsen C., Wendler F., Vincent J.P., Brech A., Bilder D., Stenmark H. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol. 2007;17:1817–1825. doi: 10.1016/j.cub.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 18.Metzler M., Legendre-Guillemin V., Gan L., Chopra V., Kwok A., McPherson P.S., Hayden M.R. HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2. J. Biol. Chem. 2001;276:39271–39276. doi: 10.1074/jbc.C100401200. [DOI] [PubMed] [Google Scholar]
- 19.Nixon R.A., Cataldo A.M. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J. Alzheimers Dis. 2006;9:277–289. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- 20.Parkinson N., Ince P.G., Smith M.O., Highley R., Skibinski G., Andersen P.M., Morrison K.E., Pall H.S., Hardiman O., Collinge J., Shaw P.J., Fisher E.M. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B) Neurology. 2006;67:1074–1077. doi: 10.1212/01.wnl.0000231510.89311.8b. [DOI] [PubMed] [Google Scholar]
- 21.Stein M.P., Dong J., Wandinger-Ness A. Rab proteins and endocytic trafficking: potential targets for therapeutic intervention. Adv. Drug Deliv. Rev. 2003;55:1421–1437. doi: 10.1016/j.addr.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Skibinski G., Parkinson N.J., Brown J.M., Chakrabarti L., Lloyd S.L., Hummerich H., Nielsen J.E., Hodges J.R., Spillantini M.G., Thusgaard T., Brandner S., Brun A., Rossor M.N., Gade A., Johannsen P., Sorensen S.A., Gydesen S., Fisher E.M., Collinge J. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005;37:806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 23.Lee J.A., Beigneux A., Ahmad S.T., Young S.G., Gao F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 24.Bronfman F.C., Escudero C.A., Weis J., Kruttgen A. Endosomal transport of neurotrophins: roles in signaling and neurodegenerative diseases. Dev. Neurobiol. 2007;67:1183–1203. doi: 10.1002/dneu.20513. [DOI] [PubMed] [Google Scholar]
- 25.Piper R.C., Katzmann D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007;23:519–547. doi: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 27.Urbe S. Ubiquitin and endocytic protein sorting. Essays Biochem. 2005;41:81–98. doi: 10.1042/EB0410081. [DOI] [PubMed] [Google Scholar]
- 28.Hurley J.H. ESCRT complexes and the biogenesis of multivesicular bodies. Curr. Opin. Cell Biol. 2008 doi: 10.1016/j.ceb.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katzmann D.J., Odorizzi G., Emr S.D. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 30.Williams R.L., Urbe S. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell. Biol. 2007;8:355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- 31.Garrus J.E., von Schwedler U.K., Pornillos O.W., Morham S.G., Zavitz K.H., Wang H.E., Wettstein D.A., Stray K.M., Cote M., Rich R.L., Myszka D.G., Sundquist W.I. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 32.Demirov D.G., Ono A., Orenstein J.M., Freed E.O. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. U. S. A. 2002;99:955–960. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blot V., Perugi F., Gay B., Prevost M.C., Briant L., Tangy F., Abriel H., Staub O., Dokhelar M.C., Pique C. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J. Cell Sci. 2004;117:2357–2367. doi: 10.1242/jcs.01095. [DOI] [PubMed] [Google Scholar]
- 34.Martin-Serrano J., Zang T., Bieniasz P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001;7:1313–1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- 35.Luttge B.G., Shehu-Xhilaga M., Demirov D.G., Adamson C.S., Soheilian F., Nagashima K., Stephen A.G., Fisher R.J., Freed E.O. Molecular characterization of feline immunodeficiency virus budding. J. Virol. 2007 doi: 10.1128/JVI.02337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patton G.S., Morris S.A., Chung W., Bieniasz P.D., McClure M.O. Identification of domains in gag important for prototypic foamy virus egress. J. Virol. 2005;79:6392–6399. doi: 10.1128/JVI.79.10.6392-6399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun K., Johnson B.S., Gunn T.M. Mitochondrial dysfunction precedes neurodegeneration in Mahogunin (Mgrn1) mutant mice. Neurobiol. Aging. 2007;28:1840–1852. doi: 10.1016/j.neurobiolaging.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller K.A., Gunn T.M., Carrasquillo M.M., Lamoreux M.L., Galbraith D.B., Barsh G.S. Genetic studies of the mouse mutations mahogany and mahoganoid. Genetics. 1997;146:1407–1415. doi: 10.1093/genetics/146.4.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cota C.D., Bagher P., Pelc P., Smith C.O., Bodner C.R., Gunn T.M. Mice with mutations in Mahogunin ring finger-1 (Mgrn1) exhibit abnormal patterning of the left–right axis. Dev. Dyn. 2006;235:3438–3447. doi: 10.1002/dvdy.20992. [DOI] [PubMed] [Google Scholar]
- 40.Kim B.Y., Olzmann J.A., Barsh G.S., Chin L.S., Li L. Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol. Biol. Cell. 2007;18:1129–1142. doi: 10.1091/mbc.E06-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergink S., Salomons F.A., Hoogstraten D., Groothuis T.A., de Waard H., Wu J., Yuan L., Citterio E., Houtsmuller A.B., Neefjes J., Hoeijmakers J.H., Vermeulen W., Dantuma N.P. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev. 2006;20:1343–1352. doi: 10.1101/gad.373706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ako-Adjei D., Johnson M.C., Vogt V.M. The retroviral capsid domain dictates virion size, morphology, and coassembly of gag into virus-like particles. J. Virol. 2005;79:13463–13472. doi: 10.1128/JVI.79.21.13463-13472.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bishop N., Woodman P. ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol. Biol. Cell. 2000;11:227–239. doi: 10.1091/mbc.11.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amit I., Yakir L., Katz M., Zwang Y., Marmor M.D., Citri A., Shtiegman K., Alroy I., Tuvia S., Reiss Y., Roubini E., Cohen M., Wides R., Bacharach E., Schubert U., Yarden Y. Tal, a Tsg101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004;18:1737–1752. doi: 10.1101/gad.294904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDonald B., Martin-Serrano J. Regulation of Tsg101 expression by the steadiness box: a role of Tsg101-associated ligase. Mol. Biol. Cell. 2008;19:754–763. doi: 10.1091/mbc.E07-09-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pornillos O., Higginson D.S., Stray K.M., Fisher R.D., Garrus J.E., Payne M., He G.P., Wang H.E., Morham S.G., Sundquist W.I. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 2003;162:425–434. doi: 10.1083/jcb.200302138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Itahana K., Mao H., Jin A., Itahana Y., Clegg H.V., Lindstrom M.S., Bhat K.P., Godfrey V.L., Evan G.I., Zhang Y. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12:355–366. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 48.Li L., Liao J., Ruland J., Mak T.W., Cohen S.N. A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc. Natl. Acad. Sci. U. S. A. 2001;98:1619–1624. doi: 10.1073/pnas.98.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng T.H., Cohen S.N. Human MDM2 isoforms translated differentially on constitutive versus p53-regulated transcripts have distinct functions in the p53/MDM2 and TSG101/MDM2 feedback control loops. Mol. Cell. Biol. 2007;27:111–119. doi: 10.1128/MCB.00235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Longva K.E., Blystad F.D., Stang E., Larsen A.M., Johannessen L.E., Madshus I.H. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol. 2002;156:843–854. doi: 10.1083/jcb.200106056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yokouchi M., Kondo T., Houghton A., Bartkiewicz M., Horne W.C., Zhang H., Yoshimura A., Baron R. Ligand-induced ubiquitination of the epidermal growth factor receptor involves the interaction of the c-Cbl RING finger and UbcH7. J. Biol. Chem. 1999;274:31707–31712. doi: 10.1074/jbc.274.44.31707. [DOI] [PubMed] [Google Scholar]
- 52.Ettenberg S.A., Magnifico A., Cuello M., Nau M.M., Rubinstein Y.R., Yarden Y., Weissman A.M., Lipkowitz S. Cbl-b-dependent coordinated degradation of the epidermal growth factor receptor signaling complex. J. Biol. Chem. 2001;276:27677–27684. doi: 10.1074/jbc.M102641200. [DOI] [PubMed] [Google Scholar]
- 53.Cha Y.K., Kim Y.H., Ahn Y.H., Koh J.Y. Epidermal growth factor induces oxidative neuronal injury in cortical culture. J. Neurochem. 2000;75:298–303. doi: 10.1046/j.1471-4159.2000.0750298.x. [DOI] [PubMed] [Google Scholar]
- 54.Doyotte A., Russell M.R., Hopkins C.R., Woodman P.G. Depletion of TSG101 forms a mammalian “Class E” compartment: a multicisternal early endosome with multiple sorting defects. J. Cell Sci. 2005;118:3003–3017. doi: 10.1242/jcs.02421. [DOI] [PubMed] [Google Scholar]
- 55.Katzmann D.J., Stefan C.J., Babst M., Emr S.D. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J. Cell Biol. 2003;162:413–423. doi: 10.1083/jcb.200302136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin N., Chow C.Y., Liu L., Zolov S.N., Bronson R., Davisson M., Petersen J.L., Zhang Y., Park S., Duex J.E., Goldowitz D., Meisler M.H., Weisman L.S. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27:3221–3234. doi: 10.1038/emboj.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.