

Abstract
The purpose of this article is to review the status and limitations of anatomic tumor response metrics including the World Health Organization (WHO) criteria, the Response Evaluation Criteria in Solid Tumors (RECIST), and RECIST 1.1. This article also reviews qualitative and quantitative approaches to metabolic tumor response assessment with 18F-FDG PET and proposes a draft framework for PET Response Criteria in Solid Tumors (PERCIST), version 1.0.
Methods
PubMed searches, including searches for the terms RECIST, positron, WHO, FDG, cancer (including specific types), treatment response, region of interest, and derivative references, were performed. Abstracts and articles judged most relevant to the goals of this report were reviewed with emphasis on limitations and strengths of the anatomic and PET approaches to treatment response assessment. On the basis of these data and the authors' experience, draft criteria were formulated for PET tumor response to treatment.
Results
Approximately 3,000 potentially relevant references were screened. Anatomic imaging alone using standard WHO, RECIST, and RECIST 1.1 criteria is widely applied but still has limitations in response assessments. For example, despite effective treatment, changes in tumor size can be minimal in tumors such as lymphomas, sarcoma, hepatomas, mesothelioma, and gastrointestinal stromal tumor. CT tumor density, contrast enhancement, or MRI characteristics appear more informative than size but are not yet routinely applied. RECIST criteria may show progression of tumor more slowly than WHO criteria. RECIST 1.1 criteria (assessing a maximum of 5 tumor foci, vs. 10 in RECIST) result in a higher complete response rate than the original RECIST criteria, at least in lymph nodes. Variability appears greater in assessing progression than in assessing response. Qualitative and quantitative approaches to 18F-FDG PET response assessment have been applied and require a consistent PET methodology to allow quantitative assessments. Statistically significant changes in tumor standardized uptake value (SUV) occur in careful test–retest studies of high-SUV tumors, with a change of 20% in SUV of a region 1 cm or larger in diameter; however, medically relevant beneficial changes are often associated with a 30% or greater decline. The more extensive the therapy, the greater the decline in SUV with most effective treatments. Important components of the proposed PERCIST criteria include assessing normal reference tissue values in a 3-cm-diameter region of interest in the liver, using a consistent PET protocol, using a fixed small region of interest about 1 cm3 in volume (1.2-cm diameter) in the most active region of metabolically active tumors to minimize statistical variability, assessing tumor size, treating SUV lean measurements in the 1 (up to 5 optional) most metabolically active tumor focus as a continuous variable, requiring a 30% decline in SUV for “response,” and deferring to RECIST 1.1 in cases that do not have 18F-FDG avidity or are technically unsuitable. Criteria to define progression of tumor-absent new lesions are uncertain but are proposed.
Conclusion
Anatomic imaging alone using standard WHO, RECIST, and RECIST 1.1 criteria have limitations, particularly in assessing the activity of newer cancer therapies that stabilize disease, whereas 18F-FDG PET appears particularly valuable in such cases. The proposed PERCIST 1.0 criteria should serve as a starting point for use in clinical trials and in structured quantitative clinical reporting. Undoubtedly, subsequent revisions and enhancements will be required as validation studies are undertaken in varying diseases and treatments.
Keywords: molecular imaging, oncology, PET/CT, anatomic imaging, RECIST, response criteria, SUV, treatment monitoring
Cancer will soon become the most common cause of death worldwide. For many common cancers, treatment of disseminated disease is often noncurative, toxic, and costly. Treatments prolonging survival by a few weeks and causing tumor shrinkage in only about 10%–15% of patients are in widespread use. Clearly, we need more effective therapies. With relatively low response rates in individual cancer patients, imaging plays a daily clinical role in determining whether to continue, change, or abandon treatment. Imaging is expected to have a major role not only in the individual patient but in clinical trials designed to help select which new therapies should be advanced to progressively larger and more expensive clinical trials.
The ultimate goal of new cancer therapies is cure. This goal, although sometimes achieved in hematologic malignancies, has rarely been achieved in disseminated solid cancers. A good cancer treatment should ideally prolong survival while preserving a high quality of life cost-effectively. To demonstrate prolonged survival in a clinical trial in some more slowly progressing cancers can take 5–10 y or longer. Such trials are expensive, not only in cost but in time.
The typical development pathway for cancer therapeutic drugs includes an evolution from phase I to phase II and to phase III clinical trials. In phase I trials, toxicity of the agent is typically assessed to determine what dose is appropriate for subsequent trials. Typically, the statistical power of phase I drug trials is inadequate to assess antitumor efficacy. In phase II trials, evidence of antitumor activity is obtained. Phase II trials can be done in several ways. One approach is to examine tumor response rate versus a historical control population treated with an established drug. New drugs with a low response rate are typically not moved forward to advanced clinical testing under such a paradigm. In such trials, tumor response has nearly always been determined anatomically. An alternative approach is to use a typically larger sample size and have a randomized phase II trial, in which the new treatment is given in one treatment arm and compared with a standard treatment (1–4). Once drug activity is shown—or suggested—in phase II, phase III trials are typically performed. Phase III trials are larger and typically have a control arm treated with a standard therapy. Not all phase III trials are successful, but all are costly.
Determining which innovative cancer therapeutics should be advanced to pivotal large phase III trials can be unacceptably delayed if survival is the sole endpoint for efficacy. Survival trials can also be complicated by deaths due to nonmalignant causes, especially in older patients in whom comorbidities are common. Additional complexities can include patients who progress on a clinical trial but who go on to have one of several nonrandomly distributed follow-up therapies—which can confound survival outcomes.
There is great interest in surrogate metrics for survival after investigational cancer treatments, such as response rate, time to tumor progression, or progression-free survival (5). Changes in tumor size after treatment are often, but not invariably, related to duration of survival. A variety of approaches to measuring response rate have been developed, beginning with the original reports by Moertel on physical examination in 1976 and continuing to the subsequent World Health Organization (WHO) criteria (1979), Response Evaluation Criteria in Solid Tumors (RECIST) (2000), and RECIST 1.1 (2009) (6–8). Response rate typically refers to how often a tumor shrinks anatomically and has been defined in several ways. Not uncommonly, complete response, partial response, stable disease, and progressive disease are defined as in the WHO and RECIST criteria (Tables 1–3) (8). This type of classification divides intrinsically continuous data (tumor size) into 4 bins, losing statistical power for ease of nomenclature and convenience (9).
TABLE 1.
| Target lesions | Nontarget lesions | New lesions | Overall response |
|---|---|---|---|
| CR | CR | No | CR |
| CR | Non-CR/non-PD | No | PR |
| CR | Not evaluated | No | PR |
| PR | Non-PD or not all evaluated | No | PR |
| SD | Non-PD or not all evaluated | No | SD |
| Not all evaluated | Non-PD | No | NE |
| PD | Any | Yes or no | PD |
| Any | PD | Yes or no | PD |
| Any | Any | Yes | PD |
CR = complete response; PR = partial response; SD = stable disease; NE = not evaluable; PD = progressive disease.
TABLE 3.
| Characteristic | WHO | RECIST | RECIST 1.1 |
|---|---|---|---|
| Measurability of lesion at baseline | 1. Measurable, bidimensional* (product of LD and greatest perpendicular diameter) | 1. Measurable, unidimensional (LD only: size with conventional techniques ≥ 20 mm, with spiral CT ≥ 10 mm) | 1. Measurable, unidimensional (LD only: size with conventional techniques ≥ 20 mm, with spiral CT ≥ 10 mm; nodes: target short axis ± 15 mm, nontarget 10- to 15-mm nodes, normal < 10 mm) |
| 2. Nonmeasurable/evaluable (e.g., lymphangitic pulmonary metastases, abdominal masses) | 2. Nonmeasurable: all other lesions, including small lesions; evaluable is not recommended | 2. Nonmeasurable: all other lesions, including small lesions; evaluable is not recommended | |
| Objective response | 1. Measurable disease (change in sum of products of the LD and greatest perpendicular diameters, no maximal number of lesions specified): CR, disappearance of all known disease, confirmed at ≥4 wk; PR, ≥50% decrease from baseline, confirmed at ≥4 wk; PD, ≥25% increase of one or more lesions or appearance of new lesions; NC, neither PR nor PD criteria met | 1. Target lesions (change in sum of LD, maximum of 5 per organ up to 10 total [more than 1 organ]): CR, disappearance of all target lesions, confirmed at ≥4 wk; PR, ≥30% decrease from baseline, confirmed at 4 wk; PD, ≥20% increase over smallest sum observed or appearance of new lesions; SD, neither PR nor PD criteria met | 1. Target lesions (change in sum of LDs, maximum of 2 per organ up to 5 total [more than 1 organ]): CR, disappearance of all target lesions, confirmed at ≥4 wk; PR, ≥30% decrease from baseline, confirmed at 4 wk; PD, ≥20% increase over smallest sum observed and overall 5-mm net increase or appearance of new lesions; SD, neither PR nor PD criteria met |
| 2. Nonmeasurable disease: CR, disappearance of all known disease, confirmed at ≥4 wk; PR, estimated decrease of ≥50%, confirmed at 4 wk; PD, estimated increase of ≥25% in existent lesions or new lesions; NC, neither PR nor PD criteria met | 2. Nontarget lesions: CR, disappearance of all nontarget lesions and normalization of tumor markers, confirmed at ≥4 wk; PD, unequivocal progression of nontarget lesions or appearance of new lesions; non-PD, persistence of one or more nontarget lesions or tumor markers above normal limits | 2. Nontarget lesions: CR, disappearance of all nontarget lesions and normalization of tumor markers, confirmed at ≥4 wk; PD, unequivocal progression of nontarget lesions or appearance of new lesions; non-PD: persistence of one or more nontarget lesions or tumor markers above normal limits; PD must be “unequivocal” in nontarget lesions (e.g., 75% increase in volume); PD can also be new “positive PET” scan with confirmed anatomic progression. Stably positive PET is not PD if it corresponds to anatomic non-PD | |
| Overall response | 1. Best response is recorded in measurable disease | 1. Best response is recorded in measurable disease from treatment start to disease progression or recurrence | 1. Best response is recorded in measurable disease from treatment start to disease progression or recurrence |
| 2. NC in nonmeasurable lesions will reduce CR in measurable lesions to overall PR | 2. Non-PD in nontarget lesions will reduce CR in target lesions to overall PR | 2. Non-PD in nontarget lesions will reduce CR in target lesions to overall PR | |
| 3. NC in nonmeasurable lesions will not reduce PR in measurable lesions | 3. Non-PD in nontarget lesions will not reduce PR in target lesions | 3. Non-PD in nontarget lesions will not reduce PR in target lesions | |
| 4. Unequivocal new lesions are PD regardless of response in target and nontarget lesions | 4. Unequivocal new lesions are PD regardless of response in target and nontarget lesions | ||
| Duration of response | 1. CR: from date CR criteria are first met to date PD is first noted | 1. Overall CR: from date CR criteria are first met to date recurrent disease is first noted | 1. Overall CR: from date CR criteria are first met to date recurrent disease is first noted |
| 2. Overall response: from date of treatment start to date PD is first noted | 2. Overall response: from date CR or PR criteria are first met (whichever status came first) to date recurrent disease is first noted | 2. Overall response: from date CR or PR criteria are first met (whichever status came first) to date recurrent disease is first noted | |
| 3. In patients who achieve only PR, only period of overall response should be recorded | 3. SD: from date of treatment start to date PD is first noted | 3. SD: from date of treatment start to date PD is first noted | |
Lesions that can be measured only unidimensionally are considered measurable (e.g., mediastinal adenopathy or malignant hepatomegaly).
LD = longest diameter; CR = complete response; PR = partial response; PD = progressive disease; SD = stable disease; NC = no change.
The time to tumor progression and progression-free survival examine when the disease recurs or progresses (including death for progression-free survival). Because cancers typically grow before they cause death, these markers provide readouts of tumor growth often considerably before the patients die of tumor. These metrics have been shown in some, but not all, cancers to be predictive of survival. Notable exceptions have been identified in several metaanalyses (6–9).
Response rates must be viewed with some caution when one is trying to predict outcomes in newer cancer therapies that may be more cytostatic than cytocidal. With such newer treatments, lack of progression may be associated with a good improvement in outcome, even in the absence of major shrinkage of tumors as evidenced by partial response or complete response (2,3). To determine lack of progression by changes in tumor size requires regular and systematic assessments of tumor burden. Newer metrics such as PET may be more informative (10).
Surrogate endpoints for survival should provide earlier, hopefully correct, answers about the efficacy of treatment and should allow better decisions on whether a drug should be advanced from early phase I to phase II or III trials. Until now, for drug development and regulatory approval purposes, indices of efficacy of treatment of solid tumors have been based solely on systematic assessments of tumor size, including the WHO, RECIST, and International Workshop Criteria (IWC) for lymphoma. However, for many years, there has been evidence that nuclear medicine imaging techniques could provide unique, biologically relevant, and prognostically important information unavailable through anatomic imaging.
For example, using planar γ-camera imaging, Kaplan et al. showed that a positive 67Ga scan midway through or at the end of treatment of patients with diffuse large cell lymphoma predicted a poor outcome in comparison to patients whose scans had normalized, even if residual masses were over 10 cm in size (11). Using planar γ-camera imaging and SPECT of 67Ga citrate, Israel, Front, et al. from Haifa showed the utility of 67Ga scanning for monitoring response and showed that CTanatomic imaging was insufficient to reliably predict disease-free survival or survival in patients with Hodgkin disease or non-Hodgkin lymphoma after completing therapy (12–14). The poor predictive ability of CT was because residual masses on CT commonly were found to represent not viable tumor but rather scarring in both Hodgkin disease and non-Hodgkin lymphoma. 67Ga results, qualitatively reported as positive or negative, were significantly predictive of outcome, with a negative 67Ga scan predicting a favorable outcome (12,14,15). A positive or negative 67Ga scan after 1 cycle of treatment was also shown to be predictive of eventual response to therapy in both Hodgkin disease and non-Hodgkin lymphoma (12–14). Although the prognostic value of 67Ga in these settings is stronger than that of CT, 67Ga imaging has now been substantially supplanted by PET using 18F-FDG.
Di Chiro et al. demonstrated that a negative 18F-FDG PET scan could help distinguish brain tumor necrosis from viable tumor at the end of therapy, despite the overlapping anatomic appearance of brain tumor and necrosis on CT (16,17). Planar imaging and SPECT with 18F-FDG showed that breast cancers and lymphomas had qualitative declines in tracer uptake with effective treatment (18,19).
Quantitative 18F-FDG PET was introduced for the early sequential monitoring of tumor response of breast cancer in 1993 (20). Since then, there has been growing interest in using 18F-FDG PET to quickly assess whether a tumor is—or is not—responding to therapy (20). In the initial report, women with newly diagnosed breast cancer had a rapid and significant decline in standardized uptake value (SUV), influx rate for 18F-FDG determined by Patlak analysis (influx constant Ki), and estimated phosphorylation rate of 18F-FDG to FDG-6 phosphate (k3) within 8 d of the start of effective treatment. These parameters continued to decline with each progressive treatment in the responding patients, antedating changes in tumor size. By contrast, the nonresponding patients did not have a significant decline in their SUV. Since that report, there have been many others in a wide range of tumors (21,22). Abundant data now exist that PET is a useful tool for response assessment in a variety of diseases, at the end of treatment, at mid treatment, and when performed soon after treatment is initiated.
Quantitative nonanatomic imaging approaches can be used as a biomarker of cancer response to predict or assess the efficacy of treatments (23–25). PET with 18F-FDG appears to be one of the most powerful biomarkers introduced to date for clinical trials and for individual patients.
An evolving personalized cancer management paradigm is one in which a tumor biopsy is used to produce a genetic or epigenetic profile to help select the initial treatment and enrich for response. A baseline PET scan and a PET scan after 1 or 2 cycles of treatment could then be performed to determine whether the treatment was indeed effective in that specific tumor and patient (26,27). Rapid readouts of treatment effect and prompt shifting of patients from ineffective to effective therapies, as well as quick abandonment of ineffective therapies, is an extremely attractive possibility for personalized health care. Use of these so-called response-adaptive or risk-adaptive treatment approaches is expected to grow (28). Indeed, it is probable that the integration of imaging in which the exact effects of the therapeutic agent on a specific tumor in a specific patient are imaged will be much more potent than are predictions of response based on more traditional established prognostic information (29).
In the past 20 years, there has been remarkable growth in the use of 18F-FDG PET in cancer imaging, with PET now being used increasingly routinely in the diagnosis, staging, restaging, and treatment monitoring of many cancers. Despite the rapid integration of PET with 18F-FDG into clinical practice in individual patients, there has been relatively little systematic integration of PET into clinical trials of new cancer treatments. Such clinical trials and the regulatory agencies evaluating them rely mainly on anatomic approaches to assess response and progression. Part of the delay in integrating PET into phase I–III clinical trials as a response metric is due to the variability in study performance across centers and the lack of uniformly accepted, or practiced, treatment response metrics for PET. Recently, standardized approaches to the performance of PET and to machine calibrations have been articulated (30,31). Further, qualitative dichotomous (positive/negative) 18F-FDG PET readings at the end of treatment have recently been integrated into lymphoma response assessment in the IWC + PET criteria (32,33). Given the clinical importance and quantitative nature of PET, it is important to have methods to allow inclusion of PET response criteria into clinical trials, as well.
This article attempts to address the status and limitations of currently applied anatomic tumor response metrics, including WHO, RECIST, and the new RECIST 1.1 criteria. It then reviews the qualitative and quantitative approaches used to date in PET treatment response assessment, including the IWC + PET criteria for lymphoma and the European Organization for Research and Treatment of Cancer (EORTC) criteria for PET. Finally, it proposes, on the basis of the literature reviewed and the authors' experience, a draft framework for PET Response Criteria in Solid Tumors (PERCIST, version 1.0). These criteria may be useful in future multicenter trials and may serve as a starting point for further refinements of quantitative PET response. They may also provide some guidance for clinical quantitative structured reporting on individual patients.
Methods
Selected articles obtained using Internet search tools, including PubMed and syllabi from meetings (e.g., Clinical PET and PET/CT syllabus, Radiological Society of North America, 2007), were identified. Publications resulting from database searches and including the main search terms RECIST, positron, FDG, ROI (region of interest), cancer, lymphoma, PET, WHO, and treatment response were included. The search strategy for relevant 18F-FDG PET studies articulated by Mijnhout et al. was also applied (34,35). These were augmented by key references from those studies, as well as the authors' own experience with PET assessments of treatment response, informal discussions with experts on PET treatment response assessment, and pilot evaluations of clinical data from the authors' clinical practice. Limitations and strengths of the anatomic and functional methods to assess treatment response were evaluated with special attention to studies that had applied qualitative or quantitative imaging metrics, had determined the precision of the method, and had histologic correlate or outcome data available. On the basis of these data, proposed treatment response criteria including PET were formulated, drawing from both prior anatomic models (notably WHO, RECIST, and RECIST 1.1) and the EORTC PET response draft criteria (36). These conclusions were based on a consensus approach among the 4 authors. Thus, a systematic review and a limited Delphilike approach augmented by key data were undertaken to reach consensus in a small group. For demonstration purposes, 18F-FDG PET scans obtained at our institution on 1 of 2 GE Healthcare PET/CT scanners were analyzed with several tools, including a tool for response assessment.
Results
Searches for the word RECIST on PubMed produced 406 references. Searching for WHO & treatment & response & cancer produced 404 references in December 2008. Searching for IWC & lymphoma & PET produced 6 references. Searching for PET or positron & treatment & response produced 3,336 references. Searching for FDG & treatment & response produced 1,024 references. Limitation of the latter search to humans resulted in 934 potential references. Searching for FDG and SUV produced 1,012 references on January 7, 2009. The abstracts of many were reviewed by the authors, and the seemingly most relevant full articles were examined in detail. Additional references were identified from the reference lists of these articles. Given the large extent of the available literature and the limited time and personnel available to produce this initial review, some major references may not have been identified.
The results of this review are presented in 3 main areas: anatomic response criteria, PET metabolic response criteria, and rationale for the proposed PERCIST criteria.
Anatomic Response Criteria
A scientific approach to assessing cancer treatment response was notably applied by Moertel and Hanley (6). They evaluated the consistency of assessment of tumor size by palpation among 16 experienced oncologists using 12 simulated masses and routine clinical examination skills. Two pairs of the 12 masses were identical in size. When a 50% reduction in tumor dimensions (perpendicular diameters) was taken as a significant reduction in size, the frequency of detecting a tumor response was about 7%–8% because of chance differences in measurement values. If a 25% reduction in the product of the perpendicular diameters of the tumors was considered a response, an unacceptably high false tumor reduction occurred 19%–25% of the time because of variability in the measurement technique. This study quantified for the first time the variability in determinations of tumor size by experts due to measurement error using metrics available at that time. Moertel and Hanley thus recommended that a true tumor response would need to be greater than 50% so as to avoid these random responses due to measurement variance.
As measurement tools are developed, a key question is their intrinsic variability from study to study. Lower variability (i.e., higher precision) means that smaller treatment-induced effects in tumor characteristics can be identified. This does not necessarily mean, however, that the treatment-induced changes identified are medically relevant.
WHO Criteria
Moertel and Hanley's work and the development of a variety of promising anticancer therapies, mainly cytotoxics, in the 1960s and 1970s brought about a clear need for standardization of response criteria. Because CT of the body was not in widespread use until the early 1980s, most tumor measurements were obtained by palpation or chest radiographs. In 1979, WHO attempted to standardize treatment response assessment by publishing a handbook of criteria for solid tumor response (7). The proposed WHO methods included determining the product of the bidimensional measurement of tumors (i.e., greatest perpendicular dimensions), summing these dimensions over all tumors, and then categorizing changes in these summed products as follows: complete response—tumor has disappeared for at least 4 wk; partial response—50% or greater reduction in sum of tumor size products from baseline confirmed at 4 wk; no change—neither partial response nor complete response nor progressive disease; and progressive disease—at least a 25% increase in tumor size in one or more lesions, with no complete response, partial response, or stable disease documented before increase in size, or development of new tumor sites.
Reviewing the data of Moertel and Hanley, one would be concerned that the progressive disease category in WHO might be easy to achieve by chance changes in measurement (i.e., a 25% increase in the product of 2 measurements could occur with an approximately 11% increase in each dimension). In addition, the WHO criteria were not explicit on such factors as how many tumor foci should be measured, how small a lesion could be measured, and how progression should be defined. Thus, despite efforts at standardization, the WHO criteria did not fully standardize response assessment. The WHO criteria are still in use in some trials and are the criteria used to define clinical response rates in many trials from the past 2 decades—which are important reference studies. Although not as commonly used at present, familiarity with the WHO response criteria is essential for comparison with more recent studies using RECIST, especially as relates to the issue of when tumors progress. The WHO criteria are summarized in Table 3.
RECIST
The RESIST criteria were published in 2000 and resulted from the recognition of some limitations of the WHO criteria (8). The criteria were developed as a primary endpoint for trials assessing tumor response. In addition, between the time of development of the WHO criteria and development of RECIST, cross-sectional imaging with CT and MRI entered the practice of oncology. RECIST specified the number of target lesions to assess (up to 10), though it did not give substantial guidance on how they were to be selected, except that there should not be more than 5 per organ. RECIST assumed that transaxial imaging would be performed, most commonly with CT, and specified that only the single longest dimension of the tumor should be mentioned. Thus, RECIST implemented a unidimensional measurement of the long axis of tumors. RECIST also clearly stated that the sum of these unidimensional measurements was to be used as the metric for determining response. RECIST also specified the minimum size of the lesions to be assessed, typically 1 cm using modern CT with 5-mm or thinner slices. Lesions of adequate size for measurement are described as “measurable.” There are also designations of “target” and “nontarget” lesions (Tables 1–3). All target lesions are measurable. Some nontarget lesions are measurable. Both can contribute to disease progression and to complete response (Tables 1–3).
The RECIST categories for response include complete response—disappearance of all tumor foci for at least 4 wk; partial response—a decline of at least 30% in tumor diameters for at least 4 wk; stable disease—neither partial response nor progressive disease; and progressive disease—at least a 20% increase in the sum of all tumor diameters from the lowest tumor size. A 20% increase in tumor dimensions results in a 44% increase in the bidimensional product, substantially greater than the WHO progression criterion of 25%. One would predict progression to be later, and possibly less frequent, using RECIST than using WHO. This has been the case, and earlier progression is seen in about 7% of patients using WHO versus RECIST (8). Thus, time to disease progression can be shorter with WHO than with RECIST (for the identical patient data). When progression is due to new tumor foci (which occurs about half the time in some reports), the 2 methods would be expected to be concordant in indicating progression of disease (8). Overall, quite good concordance was seen with the 2 methods. The RECIST and WHO criteria are contrasted in Table 3.
Another consideration for anatomic and functional imaging is that many of the changes in response, from partial response to complete response, or from stable disease to partial response, are at the border zones between response groups (i.e., 48% vs. 52% change in tumor size in WHO, or 28%–32% change in RECIST (nonresponse vs. partial response, for example). These border zones are frankly quite artificial, as changes in tumor size occur on a continuum. This is why continuous, so-called waterfall, plots of fractional shrinkage or growth of tumors are becoming increasingly popular as a means of graphically displaying tumor response data (1,2,10). It is to avoid such problems that PERCIST includes providing a specific percentage reduction in the SUV (SUV lean, or SUL) from baseline, as well as noting when the information is available—the number of weeks from the start of treatment.
Therasse, Verweij, et al. recently reviewed the use of RECIST in about 60 papers and American Society of Clinical Oncology meeting abstracts (37,38). The expected delay in progression detection versus WHO was observed. In addition, recognition of challenges in certain pediatric tumors, unusually shaped tumors such as mesotheliomas, and tumors with a great deal of central necrosis or cystic changes, such as gastrointestinal stromal tumor (GIST), were noted. Overall, however, the authors believed that RECIST had been highly successful but that some improvements were needed.
RECIST 1.1
The RECIST group, which included representatives from, among others, the EORTC, the National Cancer Institute (NCI), the National Cancer Research Network, and industry, recently reported new response criteria for solid tumors, RECIST 1.1 (39). This version of RECIST, reported in January 2009, includes several updates and modifications to refine the prior RECIST criteria. Notably, RECIST 1.1 made use of a data warehouse of images and outcomes provided from a variety of clinical trials, allowing assessment of changes in tumor size based on several formulae. Although the original RECIST included size measurements of up to 10 lesions, with a maximum of 5 for any single organ; simulations in RECIST 1.1 assessed the use of 1, 2, 3, or 5 target lesions, versus the original 10. They found strong agreement in response classifications using fewer than 10 lesions, even using just 1 lesion, but even better concordance when 5 lesions were used. In randomized studies in which tumor progression is the major concern, RECIST 1.1 suggests that just 3 lesions may be used, not 5. Thus, there are potentially 50%–70% fewer tumor measurements with RECIST 1.1 than with RECIST. RECIST 1.1 also suggests that the largest lesions be used for response, as long as they are distinctly capable of being measured.
RECIST 1.1 also dealt with lymph nodes differently than did the original RECIST criteria. In the original RECIST, the longest axis of lymph nodes was to be measured and the lymph nodes had to disappear completely to secure a complete response. In RECIST 1.1, nonnodal lesions had to be 1 cm in size or larger (long axis) to be considered measurable. By contrast, in RECIST 1.1, the short axis of lymph nodes is measured; short-axis lengths greater than 1.5 cm are considered suitable for measurement, and nodes with short axes under 1 cm are considered normal. If a node disappears nearly completely and cannot be precisely measured, it is assigned a value of 5 mm. If totally absent, it becomes 0 mm. The difference between RECIST and RECIST 1.1 in lymph nodes is that the lymph node size can decline to greater than 0 and still be considered a complete response. Thus, with RECIST 1.1, especially in diseases in which lymph nodes represent a significant fraction of the total tumor burden, criteria for a complete response are less stringent than with the original RECIST. In the simulation data used in the RECIST 1.1 study, if nodal disease predominated, 23% of cases would move from partial response to complete response, whereas about 10% would move from partial response to stable disease. It should be noted that short-axis nodal diameter is added to long axis of other tumors to result in an overall tumor burden assessment in measurable lesions. This reclassification to an increased complete response rate for node-dominant disease is a major change and may be controversial as regards comparing RECIST with RECIST 1.1.
The overall definition of progressive disease also changed in RECIST 1.1 by requiring an absolute increase in the sum of the tumor dimensions of at least 5 mm. This requirement prevents a minimal (<5-mm sum of tumor long axes) 20% increase from being categorized as progressive disease. The new RECIST 1.1 criteria offer guidance on what constitutes unequivocal progression of nonmeasurable or nontarget disease. There is also a brief discussion in RECIST 1.1 of the implications of a newly positive PET scan with 18F-FDG in disease otherwise not considered to be progressing—the PET scan must be taken seriously as recurrence (39–41). Methods for classifying anatomic response in RECIST and RECIST 1.1 are detailed in Tables 1–3.
Although these anatomic criteria may appear to be arcane, the RECIST criteria and now, quite likely, the RECIST 1.1 criteria are or will be used in virtually every clinical trial of new solid tumor therapeutics, as response is essentially always measured. Further, regulatory agencies have accepted RECIST as the de facto standard in response assessment for clinical trials in many countries. Familiarity with the implications of trials in which response is measured using the WHO, RECIST, and RECIST 1.1 criteria is essential, as they are not identical and do not produce identical results.
Limitations of Anatomic Response Criteria
Although RECIST has been used quite extensively for the past 8 y, some concerns about the method have not been fully addressed, even in RECIST 1.1. One issue is the fundamental statistical issue of reducing intrinsically continuous data on tumor size and tumor response to a series of 4 bins of response (i.e., complete response, partial response, stable disease, and progressive disease). With such reductionism, potential valuable information that may be important is lost (1,2,4,10). For example, with some newer cancer treatments that are mainly cytostatic, longstanding stable disease is a highly beneficial outcome. Indeed, examples of such effects include the behavior of GIST tumors, in which tumor size shrinks slowly but patients live for long periods with stable disease (42,43). Similar findings of prolonged life, with limited antitumor size response by RECIST, have been seen in hepatomas treated by sorafenib (44,45). Thus, there have been attempts to use tumor characteristics other than size to assess response. For example, the Choi criteria that have been developed for GIST include assessments of the size and CT Hounsfield units of tumors before and after treatment. With the Choi criteria, a 10% decrease in size or a 15% decrease in CT Hounsfield units is associated with a good response. Although these are potentially difficult measures to make precisely, it has been generally agreed that RECIST is not adequate for GIST (42,46,47). Additional anatomic characteristics of GIST, such as the development of mural nodules, but not necessarily with tumor growth because of the predominantly cystic nature of the tumors, are indicative of progression and of a poor outcome (48,49).
Limitations of RECIST in predicting response are noted clearly in the SHARP trial, in which sorafenib, an inhibitor of vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and Raf, was used in a randomized placebo-controlled trial in patients with hepatoma. In this trial of over 602 hepatoma patients who had not received previous therapy, only about 2% of the treated group and 1% of the control group had a partial response by RECIST, a figure that might lead one to conclude the drug to be inactive. However, the main endpoints of the trial were not tumor response but rather survival and progression-free survival. Because hepatomas have a bad prognosis and there is a high death rate, survival studies are feasible. At the time the study was ended, median overall survival was 10.7 mo in the sorafenib group and 7.9 mo in the placebo group (P < 0.001). The median time to radiologic progression was 5.5 mo in the sorafenib group and 2.8 mo in the placebo group (P < 0.001). Thus, clearly prolonged survival of about 3 mo was seen in this group of patients with advanced hepatocellular carcinoma treated with sorafenib, in comparison to patients treated with placebo. This substantial improvement in survival was associated with stable (not shrinking) anatomic disease (45).
In hepatomas, alternative criteria to RECIST have been developed, referred to as the EASL (European Association for the Study of the Liver) criteria (44,50). These criteria rely on contrast enhancement patterns after vascular interventional therapies and appear superior to RECIST in this limited setting. Similarly, in mesotheliomas and pediatric tumors, modifications of RECIST dealing with the peculiarities of these tumors are in place (51–53,53A).
An additional consideration for RECIST is that the most precise estimates are achieved when the same reader assesses the baseline and follow-up studies. More misclassifications and variance in response are seen when a different reader assesses the baseline and follow-up studies (54).
Tumor size is a clearly important parameter, and there is some evidence that the more rapidly a tumor shrinks, the more likely it is that the response will be durable. For example, in lymphomas, patients whose tumors shrink the most rapidly are most likely to do well, and they may need less treatment (55). Estimates of tumor volume may prove more useful than 1-dimensional methods of tumor assessment in evaluating tumor response. Caution, however, is needed even with volumes; in neoadjuvant therapy of lung cancer, early changes in lung cancer volume were shown not to be predictive of histologic response (56). Tumor histologic status was well associated with changes in tumor volumes in neoadjuvant therapy of colorectal cancer, however (57). The use of continuous as opposed to discrete sets of response has been suggested. Such continuous assessments may then lend themselves well to randomized phase II trials in which the response metrics can be compared using more standard statistical testing than concordance or κ-statistics (4).
Lymphoma
Lymphomas have had a somewhat different approach to response assessment than solid tumors. Briefly, residual or even bulky masses after therapy completion are frequent in both Hodgkin disease and non-Hodgkin lymphoma but correlate poorly with survival (58). Masses often do not regress completely after adequate (curative) treatment because of residual fibrosis and necrotic debris. The anatomic response categories of “complete remission unconfirmed” or “clinical complete remission” were created in recognition of the problem that, particularly in patients with lymphoma, anatomic response criteria often underestimate the chemotherapeutic effect (59). Patients with stable disease by conventional anatomic criteria may be cured. It has been demonstrated that adding PET to the posttherapy CT is especially useful in identifying which of these patients have achieved a satisfactory functional remission (60,61). The reader should be aware that there are well-established anatomic metrics of response in lymphoma (59). These metrics have recently been updated and modified to include PET at the end of therapy because of the limitations of anatomic imaging (Tables 4 and 5) (32,33).
TABLE 4.
Response Definitions for Clinical Trials: Lymphoma Response (33)
| Response | Definition | Nodal masses | Spleen, liver | Bone marrow |
|---|---|---|---|---|
| CR | Disappearance of all evidence of disease | (a) 18F-FDG–avid or PET-positive before therapy must be PET-negative after therapy; mass of any size is permitted if PET is negative; (b) variably 18F-FDG–avid or PET-negative; regression to normal size on CT | Not palpable, nodules disappeared | Infiltrate has cleared on repeated biopsy; if indeterminate by morphology, immunohistochemistry should be negative for CR |
| PR | Regression of measurable disease and no new sites | ≥50% decrease in SPD of up to 6 largest dominant masses; no increase in size of other nodes; (a) 18F-FDG–avid or PET-positive before therapy; one or more PET-positive at previously involved site; (b) variably 18F-FDG–avid or PET-negative; regression on CT |
≥50% decrease in SPD of nodules (for single nodule in greatest transverse diameter); no increase in size of liver or spleen | Irrelevant if positive before therapy; cell type should be specified |
| SD | Failure to attain CR/PR or PD | (a) 18F-FDG–avid or PET-positive before therapy; PET positive at prior sites of disease and no new sites on CT or PET; (b) variably 18F-FDG–avid or PET-negative; no change in size of previous lesions on CT | ||
| Relapsed disease or PD | Any new lesion or increase of previously involved sites by ≥50% from nadir | Appearance of new lesions > 1.5 cm in any axis, ≥50% increase in SPD of more than one node, or ≥50% increase in longest diameter of previously identified node > 1 cm in short axis; lesions PET-positive if 18F-FDG–avid lymphoma or PET-positive before therapy | >50% increase from nadir in SPD of any previous lesions | New or recurrent involvement |
CR = complete remission; PR = partial remission; SPD = sum of product of diameters; SD = stable disease; PD = progressive disease.
TABLE 5.
| Characteristic | Hicks criteria | IWC + PET (lymphoma) |
|---|---|---|
| Measurability of lesion at baseline | 1. 18F-FDG–avid | 1. 18F-FDG–avid tumor; baseline PET scan is desirable |
| 2. Standardized display with normalization to liver | 2. Variably 18F-FDG–avid tumor; 18F-FDG baseline PET scan is required | |
| 3. Follow-up PET at least 3 wk after last chemotherapy session or at least 8–12 wk after last radiation therapy session | ||
| Objective response | Complete metabolic response: 18F-FDG–avid lesions revert to background of normal tissues in which they are located | Complete response in 18F-FDG–avid tumors: no focal or diffuse increased 18F-FDG uptake over background in location consistent with tumor, regardless of CT abnormality; new lung nodules in lymphoma patient without history of lung involvement (regardless of 18F-FDG avidity) are not considered lymphoma; increased focal or multifocal marrow uptake is not considered tumor unless biopsy is done |
| Partial metabolic response: “significant reduction in SUV in tumors” | Noncomplete response: diffuse or focal uptake exceeding mediastinal blood pool if >2 cm in size; in nodes < 2 cm diameter, uptake of 18F-FDG greater than background is positive; lesions > 1.5 cm in size in liver or spleen with uptake equal to or greater than spleen are considered tumor | |
| SMD: “no visible change in metabolic activity of tumors” | Partial remission: see Table 3 | |
| Progressive metabolic disease: “increase in intensity or extent of tumor metabolic activity or new sites” | Progressive disease: see Table 3 | |
Although limited in their early assessment of treatment response, and somewhat variable in terms of outcome prediction, WHO, RECIST, and RECIST 1.1 are the standard anatomic response assessments currently accepted by most regulatory agencies, and RECIST, in particular, is in widespread use in clinical trials. By contrast, it is infrequent for these response criteria to be used in routine clinical practice. Although the criteria are quite detailed, variance in response occurs because of measurement errors and the inability of anatomic processes to quickly detect functional changes in tumors resulting from early effective treatment. The delayed readouts from anatomic imaging mean that it is difficult to quickly use anatomic imaging to modify treatments in individual patients. Functional imaging with PET offers major advantages.
Metabolic Response Criteria
This entire supplement to The Journal of Nuclear Medicine is devoted to treatment response assessment using PET, mainly with 18F-FDG, though other tracers have shown promise. The general principles for assessing treatment response with 18F-FDG PET have been articulated elsewhere for several different disease types. Although a range of factors has been associated with 18F-FDG uptake, there appears to be a rather strong relationship between 18F-FDG uptake and cancer cell number in a substantial number of studies (62,63). Consequently, it is reasonable to expect that declines in tumor 18F-FDG uptake would be seen with a loss of viable cancer cells and that increases in tumor glucose use and volume of tumor cells would be expected in progressive tumor. Clear in such studies is the inability of 18F-FDG to detect minimal tumor burden versus no tumor burden (64–66).
The conceptual framework for PET tumor response is shown in Figure 1. PET is capable of detecting cancers that are smaller than depicted on CT. In addition, as a quantitative technique, the binary readings typically applied in clinical diagnosis do not need to be applied. As we have previously discussed in The Journal of Nuclear Medicine, cancers are usually not diagnosed until they reach a size of 10–100 g, or 1010–1011 cells. In the idealized setting, standard cancer therapies kill cancer cells by first-order kinetics; a given dose will kill the same fraction, not the same number, of cancer cells regardless of the size of the tumor. Thus, a dose of therapy that produces a 90% (1 log) reduction in tumor mass will have to be repeated 11 times to eliminate a newly diagnosed cancer comprising 1011 cells (26,27).
FIGURE 1.
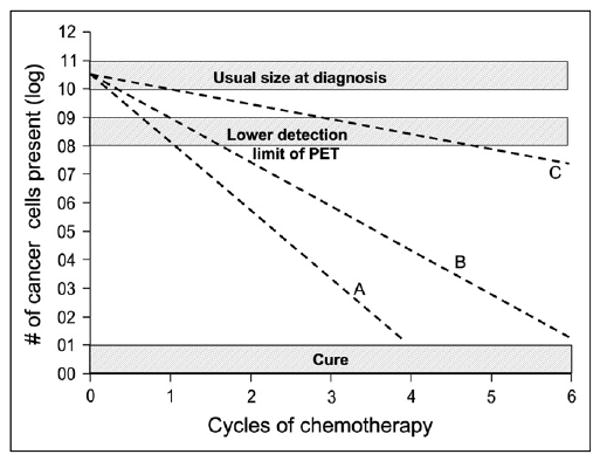
Kinetics of tumor cell kill and relation to PET. Line A represents brisk tumor response that would produce cure after only 4 cycles of chemotherapy. Line B represents minimum rate of tumor cell kill that will lead to cure in 6 cycles of treatment. Both lines would be associated with negative PET scan after 2 cycles of chemotherapy. In contrast, line C represents rate of tumor cell kill that would be associated with negative PET scan after 4–6 cycles but would not produce cure. Importantly, PET scan for line C would likely be positive after 3 cycles (27).
With current PET systems, the limit of resolution for detecting typical cancers by 18F-FDG PET generally ranges between a 0.4- and 1.0-cm diameter (67,68), which translates into a tumor size roughly of 0.1–0.5 to 1.0 g or 108–109 cells. It follows that PET likely can measure only the first 2 logs of tumor cell kill, depending on the initial size of the tumor. Thus, a negative PET scan at the end of therapy can mean there are no cancer cells present or that there are as many as 107 cells. Although a completely negative PET scan at the end of therapy typically suggests a good prognosis, it does not necessarily correspond to an absence of cancer cells. Several studies have demonstrated the inability of 18F-FDG PET to detect minimal tumor burden versus no tumor burden (64–66). On the contrary, in the absence of inflammation, a positive 18F-FDG PET scan after several cycles of treatment is usually a harbinger of residual tumor. Because it is not possible for PET in its current form to detect microscopic burden, efforts to read to a high sensitivity, although well-intentioned, may yield excessive false-positive rates. Thus, it would probably be important to maintain the specificity of the technique in readings and in response assessments, in order to maximize the utility of the method.
As is apparent in Figure 1, the time to normalization of the PET scan is also important, as this time should reflect the rate of cell kill and, therefore, predict the likelihood of cure, per our simple model. Because a true-positive PET scan at the end of 2 cycles suggests that fewer than 1 or 2 logs of tumor cells have been eliminated, it is unlikely that the 10 or 11 logs needed for cure will be eradicated by standard-duration 8-cycle treatments. A true-negative scan after 1 or 2 cycles implies the opposite; that is, the rate of tumor cell kill for this tumor is sufficient to produce cure—or at least a valuable remission (Fig. 1).
In the earliest studies of cancer treatment response with PET, sequentially evaluating 18F-FDG uptake in breast cancers before and at varying times after treatment, declines in 18F-FDG uptake were seen with each successive treatment cycle in patients who were responding well (20). By contrast, lesser or no decline in 18F-FDG uptake was seen in the nonresponders. Those patients with a continuing decline in 18F-FDG uptake over time were the most likely to have complete pathologic responses by histology at the end of therapy. Tumor 18F-FDG uptake also declined more rapidly than did tumor size with effective treatment.
A large body of evidence supports these general principles in a wide range of human cancers evaluated with PET, including esophageal, lung, head and neck, and breast cancers and lymphoma (21,69–71). Patients whose PET scans convert from positive to negative after treatment more commonly have complete pathologic responses and typically better disease-free survival and overall survival than patients whose scans remain positive. Quite striking is that prognostic stratification between high and low 18F-FDG uptake after (or during) treatment is typically preserved across disease types regardless of whether the changes in 18F-FDG uptake are assessed qualitatively (often visually) or quantitatively, using a variety of cut-point thresholds for percentage decline in SUV or a cutoff value in absolute SUV. Readers are referred to several references for further examples of risk stratification with PET (63,72–85).
Because a growing body of data suggests that patients whose scans rapidly normalize are those most likely to have a favorable outcome, a disease-assessment scan performed soon after the beginning of treatment provides much information predictive of subsequent outcomes (85). Often, early changes in 18F-FDG uptake are not complete and may be difficult to visualize. In this setting, quantitation of 18F-FDG uptake may provide a better assessment than does qualitative analysis (57,86). It is also clear that for certain noncytotoxic agents, such as imatinib mesylate (Gleevec; Novartis), PET scans normalize much more quickly than anatomic changes, thus providing a better early prediction of outcome (43,87).
How Is Response Determined on PET?
Two basic approaches can be considered for assessing the metabolic changes of treatment: qualitative and quantitative. Another issue is whether a response scale should be binary (yes/no for response) or continuous (giving varying degrees of response). An additional and not fully resolved issue is whether the most metabolically active region of the tumor should be assessed or whether the entire tumor burden glycolysis and volume should be assessed. Not fully resolved, as well, is what constitutes a negative scan, a problem not unique to 18F-FDG PET (88).
Qualitative
PET scans for diagnosis and cancer staging in clinical practice are typically interpreted using qualitative methods in which the distribution and intensity of 18F-FDG uptake in potential tumor foci are compared with tracer uptake in normal structures such as the blood pool, muscle, brain, and liver. Qualitative interpretations include a great deal of information, such as clinical experience, expectations of disease patterns for specific diseases, and knowledge of normal variants and artifacts. It might be expected that conversion of a markedly positive PET scan to a totally negative scan at the end of therapy could be done quite well with qualitative methods. Indeed, this has commonly been the method used in PET studies performed at the conclusion of therapy.
The IWC + PET criteria developed through the efforts of Juweid and Cheson dichotomize PET results into positive and negative relative to the intensity of tracer uptake, as compared with the blood pool or nearby normal structures (Table 4). Such an approach is attractive, and this dichotomous reporting has been used by many investigators in lymphoma, as reviewed by Kasamon et al. (27). However, there are pitfalls to this approach, because intermediate patterns of tracer uptake with intermediate prognostic significance have been described. One of these patterns was described by Mikhaeel et al. and termed minimal residual uptake. In a retrospective study of 102 patients evaluated with 18F-FDG PET at mid treatment for aggressive lymphoma, 19 patients had scans with minimal residual uptake and had an estimated 5-y progression-free survival of 59.3%, closer to the 88.8% for the PET-negative group (n = 50) than to the 16.2% for the PET-positive group (n = 52), but seemingly different (89). Kaplan–Meier analyses showed strong associations between the mid-therapy 18F-FDG PET results and progression-free survival (P < 0.0001) and overall survival (P < 0.01). In clinical practice, classification of minimal residual uptake seems to be the most challenging. Other approaches to lymphoma PET scoring using a 5-point visual scale have also been implemented in risk-adaptive clinical trials (90).
Investigators in Melbourne have used the visual qualitative analysis criteria noted in Table 5 to predict outcomes at the end of therapy for non–small cell lung, colon, esophageal, and metastatic breast cancers (82,84,91–94), with excellent risk stratification capability between positive and negative scans. Hicks has argued for qualitative assessments and has emphasized the considerable value of the reader's perception in excluding treatment-induced alterations from actual disease progression. Other investigators have found qualitative imaging to be more accurate than quantitative imaging, such as in lung cancer nodal assessment (72). In studies of neoadjuvant therapy of colorectal cancer, we have found that multipoint qualitative assessments of treatment response on 18F-FDG PET perform somewhat less well than quantitative assessments such as maximal SUV (SUVmax) or total lesion glycolysis (57). Given these results and those reviewed for lymphoma and by Weber and others, it is clear that qualitative assessments of tumor response carry with them considerable prognostic information.
There are, however, surprisingly few data on the reproducibility of qualitative readings of PET for diagnosis or for treatment response. Reproducibility is important for clinical practice and clinical trials. In addition, there are not nearly as many data qualitatively evaluating PET response to treatment soon after treatment has been started as there are at the conclusion of treatment. The likely reason is that the changes in PET findings at the conclusion of treatment are far more substantial than those observed early after treatment has begun, and that early clinical trials with PET (and reimbursement for PET) focused, at least in the United States, on the restaging scenario at the conclusion of a course of treatment.
The performance of PET diagnostic readers has been compared, to a limited extent. Moderate concordance in diagnostic accuracy was found for interpretations of PET scans of the axilla in women with untreated breast cancer. Three experienced readers had a comparable accuracy of 0.7–0.76 (area under the curve) (95) in over 300 patients evaluated independently by each reader. In lung cancer, moderate agreement in mediastinal staging by PET, especially of trained readers, has been reported, with κ-values of 0.65 (96). After radiotherapy of head and neck cancer, variability in reporting has been seen by qualitative methods, with an intraclass κ of 0.55. In 17% of cases, indeterminate readings were rendered (i.e., neither positive nor negative), indicating the difficulty of dichotomizing the inherently continuously variable PET uptake patterns (97). This is possibly similar to the “minimal residual uptake” category reported in treated lymphomas by Mikhaeel's group (89,98).
In lymphoma, in which a dichotomous, positive/negative PET scoring system has been applied (Table 4), some variability in reporting has been observed among readers. In one report, false-positive PET readings were not uncommon, occurring in about 50% of PET-negative cases of non-Hodgkin lymphoma when read by less experienced readers. Indeed, only a 56% concurrence rate was seen between less experienced readers and experts (99) in assessments of non-Hodgkin lymphoma disease activity. These figures may be reflective of inexperienced readers without benefit of PET/CT but suggest that some level of discordance qualitatively is to be expected. Although mainly qualitative readings have been used at the end of therapy in lymphoma treatment response, in mid-treatment monitoring both qualitative and quantitative readings have been used.
We have used a 5-point visual assessment scale in our patients with non-Hodgkin lymphoma during therapy, and a 4-point scale in colorectal cancer after treatment, recognizing that response does likely represent a continuum of intensities of uptake (57,90). These approaches have not been fully studied for reproducibility among readers but likely have been made more consistent by limiting the number of readers of the study. For earlier subtle changes in tumor uptake before treatment effect is complete, quantitation may be more desirable and perhaps essential for consistent reporting among readers. Certainly, more information is needed on the reproducibility of qualitative reporting of treatment response in the therapy-monitoring setting.
Quantitative
Because PET is intrinsically a quantitative imaging method, quantitative measurement of early treatment-induced changes is an attractive potential tool for measuring subclinical response and more complete changes. The feasibility of detecting small changes in tumor glucose metabolism quantitatively was demonstrated over 15 years ago in studies of neoadjuvant treatment of primary breast cancer, for which declines in SUV of 20%–50% were seen, depending on the time from the start of treatment. These declines were evident using Ki, SUV, and the k3 rate constant (20). More than 30 different ways to monitor tumor response have been discussed, but the SUV appears to be the most widely applied, generally correlating well with more complex analytic approaches (100,101).
The SUV is a widely used metric for assessing tissue accumulation of tracers. SUV can be normalized to body mass, lean body mass (SUL), or body surface area. Body surface area and SUL are less dependent on body habitus across populations than is SUV based on total body mass. In a single patient of stable weight, all 3 SUV normalization approaches will give comparable percentage changes with treatment, as the normalization terms cancel out mathematically. However, the absolute change in SUV with effective treatment and the absolute amount of change in SUV to be significantly different from a prior scan will differ on the basis of the metric used.
The determination of SUV is dependent on identical patient preparation and adequate scan quality that is similar between the baseline and follow-up studies. Ideally, the scans should be performed on the same scanner with comparable injected doses of 18F-FDG and comparable uptake times before scanning. Absolute and rigorous standardization of the protocol for PET is required to achieve reproducible SUVs. Standardization has been well summarized in a consensus document from the National Institutes of Health and a recent report from The Netherlands (30,31). SUL is preferred by many over SUV normalized by body surface area, as the SUL values are relatively close to (though usually somewhat less than) SUVs normalized on the basis of total body mass (30,102,103). SUL is typically more consistent from patient to patient than is total-body-mass SUV, as patients with high body mass indices have high normal organ SUVs because 18F-FDG does not significantly accumulate in white fat in the fasting state (102,103).
ROI selection is a key aspect of determining tumor SUV, tumor Ki, or any quantitative PET parameter. A wide variety of SUV ROI selection metrics has been used: manually defined ROIs; irregular isocontour ROIs based on a fixed percentage of the maximal pixel in the tumor (e.g., 41%, 50%, 70%, 75%, or 90% of the maximum); irregular isocontour ROIs based on a fixed SUV threshold (e.g., SUV = 2.5); irregular isocontour ROIs based on a background-level threshold (e.g., relevant background + 2–3 SDs); and small fixed-dimension ROIs centered over the highest-uptake part of the tumor (e.g., 15-mm-diameter circles or spheres or 12 × 12 mm squares, giving rise to a parameter sometimes called SUV peak). In addition, SUV is frequently obtained from the pixel with the SUVmax and, although not usually determined in this way, it could be considered to be a single-pixel ROI.
As part of this special contribution, we have ascertained the methods for ROI selection in determining SUV in cancer studies in over 1,000 reports. The use of varying regions of interest to determine SUV over the past decade is shown in Figure 2. It is apparent that SUVmax is growing in use and is the de facto standard, given its widespread use. A close examination of the graph shows a growing use of SUV peak, as well. The isocontour and manual ROIs have also been applied in some studies. Given that the use of SUVmax is so commonly reported, it might seem to be the “best” method. However, the wide use of SUVmax may also be due its being easily measured using current commercial workstations. To simply recommend SUVmax as the preferred treatment response parameter would be easy, as it should also be most resistant to partial-volume issues in small tumors. However, this recommendation must be taken with some trepidation as SUVmax is highly dependent on the statistical quality of the images and the size of the maximal pixel (104). For SUVmax to be used routinely, its performance characteristics should be well understood, including its reproducibility versus other approaches.
FIGURE 2.
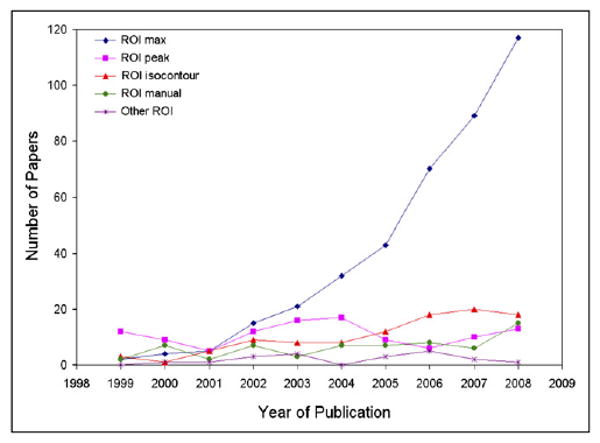
Number of papers that included use of tumor ROIs, as function of year of publication. Papers were identified by Medline search that queried for FDG AND SUV OR “standard uptake value” OR “standardized uptake value” OR “standardised uptake value”). Only human 18F-FDG oncology studies were included. ROI max refers to maximal pixel in tumor. ROI peak refers to small (typically 15 × 15 mm) fixed-size ROI centered on most metabolically active part of tumor. ROI isocontour refers to irregular ROI defined by isocontour set at, for example, some percentage of maximal pixel. ROI manual refers to manually drawn ROI. Only a subset of these papers describes response assessment studies.
A fundamental biologic question underlying choices of regions of interest is whether the total tumor volume or the maximally metabolically active portion of the tumor is most important. Intuitively, both would seem important and desirable to determine. However, concepts of stem cell biology suggest that the most critically important parts of tumors are the most aggressive portions, which may not be the entire tumor. This controversial concept is under study for many cancers (105–108). In practice, much of the early development of PET for treatment response was in the setting of a single tumor, as neoadjuvant therapy or as palliative treatment. Most papers focus on a single or a few tumor foci in ROI selection. However, the total lesion volume and its metabolic activity, known as the total lesion glycolysis, effective glycolytic volume, or total glycolytic volume (calculated in similar manners—mean SUV of the total tumor times × total tumor volume, in mL), are potentially important parameters for studying the behavior of the total tumor (109–112). For the purposes of this article, although the terms represent similar indices, we will refer to total lesion glycolysis in discussions of response based on total lesion volume and its metabolic activity.
To use quantitative metrics to assess treatment response, one must know their performance characteristics. We are aware of 5 reports on the test–retest reproducibility of PET with 18F-FDG in cancer, and the major methods and protocols of these studies are summarized in Table 6 (100,113–115). Overall, the reproducibility of quantitative PET parameters in the test–retest setting has varied depending on lesion size and the methods for image acquisition, reconstruction, and analysis. The lowest variability in PET quantitative parameters is in the 6%–10% range, but up to 42% variability has been reported. In the test–retest setting, ROI and lesion size seem to be important for SUV reproducibility whereas reproducibility appears less dependent on glucose correction factors (113,114) and the reconstruction method used (filtered backprojection vs. ordered-subset expectation maximization) (100).
TABLE 6.
Summary of Studies on Test–Retest Reproducibility of Untreated Tumors Without Interval Therapy
| Study | Pts/lesions | No. and time between PET scans | Imaging and reconstruction parameters | Variables and ROIs | Major findings |
|---|---|---|---|---|---|
| Minn 1995 | 10 pts; 10 lesions; primary lung cancer ≥ 2 cm | 2 scans; mean 1.8 ± 1.8 d | PET alone/68Ge AC; dynamic acquisition × 60 min; 3.4-mm slice thickness (n = 8); 6.75-mm slice thickness (n = 2); 128 × 128 matrices; FBP 0.3 Hanning filter; ∼8 mm FWHM; axial resolution not given | Maximal SUL 1.2 × 1.2 cm; ROI 4 × 4 pixels (“peak”) | Test–retest mean percentage difference between scans/correlation (SUL: 10% ± 7%/0.987; Ki: 10% ± 8%/0.969; SUL glucose correction: 6% ± 6%/0.995; K1: 24% ± 15%/0.812; k2: 42% ± 31%/0.0.765; k3: 24% ± 13%/0.953) |
| Weber 1999 | 16 pts; 50 lesions; various cancers; tumor volume 0.8–111 mL | 2 scans; mean 3 ± 3 d | PET alone/68Ge AC; dynamic acquisition × 70 min; 3.4-mm slice thickness; 128 × 128 matrices (4 × 4 mm); FBP 0.4 Hanning filter; ∼8 mm FWHM; axial resolution ∼5 mm | SUV bw in 50% threshold around maximal 18F-FDG ROI (mean diameter 32 ± 36 mm, range 12–60 mm) | Mean percentage difference in SUV for test–retest is ∼10%; 0.9 SUV unit required for significant change; greater variability in smaller lesions; glucose correction, no significant differences |
| Nakamoto 2002 | 10 pts; lung cancer | 2 scans; within 1 wk | Reassessment of Minn data; same parameters for image acquisition and reconstruction | Maximal SUL in 1 × 1 pixel anywhere in tumor; highest average SUL in 4 × 4 pixels in tumor; effective glycolytic volume (SUL × volume) | Mean percentage difference between scans (maximal SUL: 11.3% ± 8%; mean SUL: 10.1% ± 8.2%; effective glycolytic volume: 10.1% ± 8%; mean percentage differences slightly reduced with glucose correction) |
| Krak 2005 | 11 pts; 29 lesions; NSCLC; median volume ∼9 cm3* | 2 scans; 2 consecutive days | PET alone/68Ge AC; dynamic acquisition × 60 min; 2.5-mm slice thickness; 128 × 128 matrices; FBP 0.5 Hanning filter; OSEM (2 iterations, 16 subsets); ∼7 mm FWHM; axial resolution not given | FBP vs. OSEM; SUL ROIs (manual; 15 mm fixed; 50%, 75% threshold; single pixel maximum) | Test–retest reproducibility similar for FBP vs. OSEM; mean percentage differences of SUV between 2 scans (8%–10% ± 7%–8% for manual and 15-mm fixed ROI; 12%–14% ± 11 %–13% for threshold methods; 13% ± 11%–12% for single-pixel SUVmax); mean percent differences of ROI volume (23% ± 20% for 50% threshold; 55% ± 35% for 75% threshold); ICC highest for 15-mm fixed ROI (0.95); ICC for threshold/single-pixel SUVmax 0.89–0.91 |
| Nahmias 2008 | 26 pts; 26 lesions; various cancers; tumor size not given | 2 scans; mean 3 ± 2 d | PET/CT (CT AC); static acquisition; 90 min after tracer injection; 2.5-mm slice thickness; 256 × 256 matrices; OSEM (4 iterations,16 subsets); ∼8 mm FWHM; axial resolution ∼8 mm | Manual ROI definition in axial slice with most 18F-FDG uptake; mean SUV 9- to 17-mm circular ROI (30% of maximum guide); single-pixel SUVmax in 2.5 × 2.5 × 2 mm ROI | Mean SUV (large manual ROI) test–retest: high correlation (r = 0.99, 95% confidence interval (CI) 0.99–1.00); mean difference 0.01 ± 0.27 SUV (95% CI ± 0.53 SUV); absolute difference mean SUV < 0.5 SUV; SUVmax test–retest mean difference −0.05 ± 1.14 SUV (95% CI ± 2.2 SUV) (larger absolute difference in SUVmax as SUVmax increased, frequently more than 1.5 SUV units with SUVmax over 7.5) |
PET metabolic tumor volume; CT size not given.
Pts = patients; AC = attenuation correction; FBP = filtered backprojection; FWHM = full width at half maximum; bw = body weight; NSCLC = non–small cell lung cancer; OSEM = ordered-subset expectation maximization; ICC = intraclass correlation coefficient.
Minn et al. (116) first demonstrated that although kinetic modeling with nonlinear regression is conceptually more attractive than SUV, it is not as reproducible in the test–retest setting as is the simpler Patlak-derived Ki or the SUV. Because both Ki and SUV (or SUL or body-surface-area SUV) correlate well with kinetic modeling results, full kinetic modeling approaches are not typically undertaken in treatment response monitoring with 18F-FDG.
Ki is an attractive parameter and may be helpful when the SUV after treatment is low (117). However, Ki requires a period of dynamic scanning, a process typically more time consuming and restricted in the spatial location evaluated than whole-body PET. Further, only limited standard software is available for generation of Ki values.
The size of the ROI affects the reproducibility of SUV. SUVs obtained from larger, fixed ROIs are more reproducible than single-pixel SUVs (110,115, 118). Comparing the test–retest studies in Table 6, one can see that the ROI used by Minn in 1995 (113) was 39-fold larger in volume than that used by Nahmias and Wahl (115) in 2008 for single-voxel SUVmax (438 mm3 vs. 12.5 mm3). For equal sensitivity, there would be 39-fold fewer counts in the maximal pixel using modern PET scanners, versus the volume applied originally in determining the statistical precision of PET in the test–retest setting using older equipment with thicker slices and smaller matrices.
The assessment of Nakamoto et al. (110) of the data of Minn et al. (113) used a smaller maximal pixel volume, but it was still about 19 times larger than the volume of a single voxel used in many current scanners. Weber et al. (114) used regions of interest much larger than those of Minn et al., presumably increasing statistical reliability. Further, data from Nahmias and Wahl (115) were obtained at 90 min after injection and not the 50- to 60-min time used by Minn (113), meaning radioactive decay further reduced the total counts.
Reproducibility data from individual patients are likely of greatest practical interest in evaluating the degree of change required to determine that a change is significant between 2 studies. Weber et al. (114), using a larger ROI, reported that 0.9 SUV unit was needed for a significant change. Concordantly, Nahmias and Wahl (115) showed in test–retest studies that absolute differences in mean SUV obtained from a large ROI did not exceed 0.5 SUV unit and that the absolute differences in mean SUV decreased as mean SUV increased. In contrast, the absolute difference between SUVmax increased to over 1.5 SUV units in a substantial number of cases in which the SUVmax was over 7.5 (i.e., the hotter tumors). Thus, there are differences in the behaviors of SUVmax and mean SUV in terms of reproducibility that likely will have a direct impact on the fractional and absolute changes required to have a significant difference between a baseline and a follow-up scan.
The large ROI of Nahmias (115) showed superb test–retest performance; however, the size of their circular ROI was both manually determined and manually positioned, and thus it may be difficult to routinely achieve such low variability at other centers. Larger ROIs may be too big for small tumors such as nodes to be optimally assessed, as well.
These human data are augmented by phantom and modeling data. Boellaard et al. also showed that SUVmax variability increases as the lesion matrix size is increased from 128 × 128 to 256 × 256. They also showed that the variability increases with lower counts as the patient size increased (and the statistical quality decreased) (104).
The appeal of the single maximal pixel value is undeniable, but it is clear that with modern scanners and many small voxels, it is not as reproducible as larger ROIs and that larger changes in SUVmax between studies are needed for significance (104). This is mainly because of noise effects on SUV, which induce a positive bias in the recovery coefficient for SUVmax. As lesions get larger and hotter, there is also a statistical bias to higher single-pixel SUVmax simply because of the number of counts available. This raises concern, especially given the widespread and growing use of this parameter in clinical studies with PET, and caution must be applied in the use of single-pixel SUVmax for assessing small changes induced by treatment. For these reasons, it is probably important to have a minimum ROI for PET metrics of maximal tumor activity to ensure adequate statistical quality and intrastudy comparability.
Methods for determining total lesion glycolysis are still evolving. Choosing a threshold based on a single maximal pixel value in the tumor carries with it the variability inherent in determining a single-pixel value and is driven by that value (104,109,112,119,120). Investigators have also found poor reproducibility for tumor volume estimates (also applied to calculate total lesion glycolysis) using thresholding methods based on the maximal pixel value. After treatment, thresholding methods for tumor volume determination may extend to include too much normal tissue (118). The use of thresholds such as “anything 3 SDs or greater above background is tumor” is one approach that has been applied to defining lung cancer volumes on PET, avoiding the uncertainty of SUVmax (121). A background threshold approach has been developed as a tool for defining metabolic tumor volumes for mesotheliomas with good initial success, choosing 3 SDs above background levels for segmentation (111). Other approaches include determining the lesion volume not from PET but from the CT of the PET/CT (122). These methods hold great promise for providing the tumor burden, which may be quite important as a complement and addition to SUV.
One other approach, akin to total lesion glycolysis, is the multiplication of SUVmax × tumor width to provide a combined glycolysis × size parameter. Such approaches may be useful in response assessment but have not been extensively assessed. They could suffer from the variance intrinsic in the metabolic and anatomic methods, potentially reducing the precision of the methods, but initial results are encouraging in esophageal cancer treatment assessment (123).
Comparing tumor activity to background is an attractive way to minimize variability and to potentially ensure the quality of scans from test to retest. A variety of backgrounds has been used. Thighs, back muscle, liver, and mediastinum, for example, have been measured. Pacquet et al. showed that liver SUV is quite stable over time, when measured as a mean on a single slice in the right lobe of the liver centrally, as is mean mediastinal blood pool (124). Paquet et al. reported that mean SUL in the mediastinum was 1.33 ± 0.21 and 1.30 ± 0.21 (within-patient coefficient of variation, 12.3%) on test–retest. Mean SUL in the liver showed slightly less variance (within-patient coefficient of variation, 10.8%) and was 1.49 ± 0.25 and 1.45 ± 0.20. Glucose correction and use of the SUVmax in the liver or blood pool resulted in considerably higher variance and were not recommended for normalization. Similar results for normal organ uptakes were reported by Minn et al. in limited tissues, as well as by Wahl et al., among others (20,113). These values were slightly higher than mean blood-pool values. Krak et al. recommended the use of SUL for monitoring treatment response, as well, although they favored glucose correction (100).
A variety of methods has been used to determine the change in SUV with treatment. SUVmax in a single pixel, background-corrected values, larger or smaller ROIs, and total lesion glycolysis have been used, among others. The prospective data of Weber et al. are among the most compelling (125). Based on the differences seen in test–retest studies, they evaluated changes in SUV in cases that met the following characteristics: tumor clearly visible, large enough, and hot enough (2 × blood-pool background). Using a 1.5-cm ROI, they showed in lung, gastric, and esophageal cancers that declines in 18F-FDG uptake of 20%–35% after 1–2 doses of therapy are predictive of outcomes, with the larger the drop, the greater being the beneficial effect. In esophageal cancer, for example, Weber et al. found a drop of greater than 35% in SUV to be a good predictor of response (125). In neoadjuvant gastric cancer therapy, in which tumors with an SUV of more than 1.35 times the mean liver SUV + 2 SDs were assessed, the mean decline in SUV was about 50% in responders and 18% in nonresponders (126).
Weber has argued that any drop of more than 20% is significant and should be called a response on the basis of reproducibility considerations (Radiological Society of North America syllabus). However, in most studies, larger drops in SUV of more than 30%–35% are seen and associated with a good outcome. In lymphomas, at mid therapy, a drop in SUV of 65.7% was best at separating favorable from unfavorable responses and appeared superior to visual examination (accuracy visual, 65.2%; SUV reduction, 76%; tumor-to-background ratio, 74%; and SUV floor, 74%) in a study by Lin et al. (86). Although quantitative analysis appeared superior to visual analyses (though it must be cautioned that this was using a retrospective cutoff value and there was considerable overlap in the best responding and less well responding groups quantitatively—as well as a fine continuous scale for quantitation but a coarser approach for visual analyses), the several quantitative approaches appeared quite comparable. The authors favored the percentage decline in SUV. It appears that many methods of quantification can produce valuable prognostic information on treatment response using PET.
Another issue in PET treatment response is whether an absolute SUV floor or threshold (such as blood-pool background in the non-Hodgkin lymphoma PET criteria) or a percentage decline in SUV is most important. The advantages to a percentage drop in SUV versus a floor are that the percentage drop is likely easier to calculate than the absolute SUV; many measurement issues become less important when test–retest studies are done, because the technical issues are constant across studies. Modeling studies have shown that the ratios of SUV are less dependent on ROI choice than are absolute SUV determinations (104). An SUV floor carries the advantage of allowing a baseline PET scan to be obtained at another center to verify the 18F-FDG avidity of the tumor, but such a baseline study is not required for quantitation.
The data of Lin et al. (86) show nearly comparable results for floor SUV versus percentage decline in terms of ability to separate those with a good response from those with a less good response to treatment for non-Hodgkin lymphoma. However, several papers have shown that in lung cancer, for example, a decline in a tumor SUV to below 4–6 after treatment separates groups of patients with longer and shorter survival reasonably well (72,127). The differing cutoffs suggest possible differences in SUV calculation approaches. Reproducing absolute SUV across centers can be difficult, however, and although such absolute cutoffs may be valuable for determining prognosis, they are viewed as more suitable in single-center studies or in well-controlled multicenter approaches using careful standardization methods (31). It may be possible to determine a simple floor for PET through the use of normalization to structures such as the normal liver or blood pool, for example, as has been done qualitatively in the IWC + PET criteria (33).
SUVs in normal tissues are not stable with time, because blood-pool and liver uptake fall with increasing delays from injection, whereas uptake in tumor typically rises (20,128). Thus, normalization is difficult if scan uptake times vary. However, a threshold for posttreatment PET is an attractive concept and may be more important in the future as standardization for PET performance improves.
Methods of assessing response to treatment with total lesion glycolysis are still evolving. It appears that percentage declines in total lesion glycolysis are sometimes greater than declines in SUVand that total lesion glycolysis gives a larger range of changes after treatment than does SUV (111). This would suggest that larger changes in total lesion glycolysis would be required to have a meaningful response than are required for SUValone. Francis has found total lesion glycolysis to be superior to SUVmax in mesothelioma response assessment. However, SUVmax is also a potent predictor of outcomes in other studies of mesothelioma (52,129) and is quite strong in the data of Francis et al., as well (111). In studies of colorectal cancer neoadjuvant response, SUVmax appeared to perform somewhat better than total lesion glycolysis, though it depended on the specific task involved (57). Total lesion glycolysis has performed well in studies of colorectal cancer and brain tumor response (109,112, 119,120). In studies of sarcoma response, total lesion glycolysis performed less well than SUV peak (122). Thus, the total lesion glycolysis parameter appears promising in some, though not all, cancers. The method by which it is calculated can be quite variable, however.
The EORTC PET response criteria were proposed in 1999 (36). Given the limited data available on treatment response at that time, the criteria were useful and prescient. They recognized that the subclinical metabolic response seen early after treatment on PET, but not seen anatomically, was likely to be important. The group made several important points in its report regarding the 18F-FDG PET response: careful methods and patient preparation are essential; early declines in SUV with effective therapy will be smaller than later ones; with ineffective treatment, tumors can progress not only by increasing their SUV but also by physically growing; accurate and reproducible methods are essential for accurate reporting; and as the literature matures, updates will be needed (36).
Drawing from their work and the maturing literature on treatment response assessment over the intervening decade, some additional suggestions regarding treatment response criteria are in order.
Introduction to PERCIST 1.0
Based on the extensive literature now supporting the use of 18F-FDG PET to assess early treatment response as well as the known limitations of anatomic imaging, updated draft PET criteria are proposed that may be useful for consideration in clinical trials and possibly clinical practice. We have called these draft criteria “PERCIST”—Positron Emission tomography Response Criteria In Solid Tumors. The RECIST committee did not have a role in developing these criteria, but while we were developing them we acknowledged and appreciated the careful work and approaches of the RECIST committee. We also recognized that, as with RECIST, criteria such as PERCIST will need updates and validation in differing settings. With apologies to the RECIST group, we believed that the name PERCIST seemed quite appropriate as a complement to the well-developed anatomic criteria now in widespread use and recently updated.
The premise of the PERCIST 1.0 criteria is that cancer response as assessed by PET is a continuous and time-dependent variable. A tumor may be evaluated at any number of times during treatment, and glucose use may rise or fall from baseline values. SUV will likely vary for the same tumor and the same treatment at different times. For example, tracer uptake by a tumor is expected to decline over time with effective treatment. Thus, capturing and reporting the fractional change in SUV from the starting value and when the scan was obtained are important.
The optimal number of chemotherapy cycles before obtaining an 18F-FDG PET scan and the optimal interval between the last treatment and the scan are matters of debate and may be treatment-specific. Our assessment of the literature and the conceptual framework in Figure 1 suggest that early after treatment (i.e., after 1 cycle, just before the next cycle) may be a reasonable time for monitoring response, to determine whether the tumor shows no primary resistance to the treatment. Indeed, several studies, including one by Avril et al. on ovarian cancer, show that 60%–70% of the total SUV decline occurs after just 1 cycle of effective treatment (130). By contrast, waiting until the end of treatment can provide evidence that resistance to treatment was present throughout the treatment or evolved during treatment. End-of-therapy PET scans are quite commonly performed as restaging examinations to determine whether additional treatment or possibly surgery should be performed.
After chemotherapy, waiting a minimum of 10 d before performing 18F-FDG PET is advised. This time permits bypassing of the chemotherapeutic effect and of transient fluctuations in 18F-FDG uptake that may occur early after treatment—stunning or flare of tumor uptake (131–133). The guidelines of the IWC + PET criteria for lymphoma recommend waiting at least 3 wk between the last chemotherapy session and 18F-FDG PET, but we recognize that this longer waiting period might not be feasible for all cases. Longer and more variable times after external-beam radiation, 8–12 wk, have been recommended (134).
The basics of PERCIST 1.0 are shown in Table 7, where they are contrasted with the EORTC criteria. Key elements of PERCIST include performance of PET scans in a method consistent with the National Cancer Institute recommendations and those of The Netherlands multicenter trial group (30) on well-calibrated and well-maintained scanners. Patients should have been fasting for at least 4–6 h before undergoing scanning, and the measured serum glucose level (no correction) must be less than 200 mg/dL. The patients may be on oral hypoglycemics but not on insulin. A baseline PET scan should be obtained at 50–70 min after tracer injection. The follow-up scan should be obtained within 15 min (but always 50 min or later) of the baseline scan. All scans should be performed on the same PET scanner with the same injected dose ± 20% of radioactivity. Appropriate attenuation correction along with evaluation for proper PET and CT registration of the quantitated areas should be performed.
TABLE 7.
Comparison of EORTC and PERCIST 1.0 (36)
| Characteristic | EORTC | PERCIST 1.0 |
|---|---|---|
| Measurability of lesions at baseline | 1. Tumor regions defined on pretreatment scan should be drawn on region of high 18F-FDG uptake representing viable tumor. Whole tumor uptake should also be recorded. | 1. Measurable target lesion is hottest single tumor lesion SUL of “maximal 1.2-cm diameter volume ROI in tumor” (SUL peak). SUL peak is at least 1.5-fold greater than liver SUL mean + 2 SDs (in 3-cm spherical ROI in normal right lobe of liver). If liver is abnormal, primary tumor should have uptake > 2.0 × SUL mean of blood pool in 1-cm-diameter ROI in descending thoracic aorta extended over 2-cm z-axis. |
| 2. Same ROI volumes should be sampled on subsequent scans and positioned as close to original tumor volume as possible. Coregistration method should be recorded. | 2. Tumor with maximal SUL peak is assessed after treatment. Although typically this is in same region of tumor as that with highest SUL peak at baseline, it need not be. | |
| 3. Uptake measurements should be made for mean and maximal tumor ROI counts per pixel per second calibrated as MBq/L. | 3. Uptake measurements should be made for peak and maximal single-voxel tumor SUL. Other SUV metrics, including SUL mean at 50% or 70% of SUV peak, can be collected as exploratory data; TLG can be collected ideally on basis of voxels more intense than 2 SDs above liver mean SUL (see below). | |
| 4. Alterations in extent of 18F-FDG uptake should be documented, i.e., increase in orthogonal tumor dimensions including longest tumor dimension. | 4. These parameters can be recorded as exploratory data on up to 5 measurable target lesions, typically the 5 hottest lesions, which are typically the largest, and no more than 2 per organ. Tumor size of these lesions can be determined per RECIST 1.1. | |
| 5. Partial volume may affect measurement of 18F-FDG uptake. Tumor size from anatomic imaging in relation to PET scanner resolution should be documented where possible. | ||
| Normalization of uptake | Scanners should provide reproducible data. Reporting would need to be accompanied by adequate and disclosed reproducibility measurements from each center. An empiric 25% was found to be a useful cutoff point, but reproducibility analysis is needed to determine appropriate cutoffs for statistical significance. | Normal liver SUL must be within 20% (and <0.3 SUL mean units) for baseline and follow-up study to be assessable. If liver is abnormal, blood-pool SUL must be within 20% (and <0.3 SUL mean units) for baseline and follow-up study to be assessable. Uptake time of baseline study and follow-up study 2 must be within 15 min of each other to be assessable. Typically, these are at mean of 60 min after injection but no less than 50 min after injection. Same scanner, or same scanner model at same site, injected dose, acquisition protocol (2- vs. 3-dimensional), and software for reconstruction, should be used. Scanners should provide reproducible data and be properly calibrated. |
| Objective response | CMR: complete resolution of 18F-FDG uptake within tumor volume so that it was indistinguishable from surrounding normal tissue. | CMR: complete resolution of 18F-FDG uptake within measurable target lesion so that it is less than mean liver activity and indistinguishable from surrounding background blood-pool levels. Disappearance of all other lesions to background blood-pool levels. Percentage decline in SUL should be recorded from measurable region, as well as (ideally) time in weeks after treatment was begun (i.e., CMR −90, 4). No new 18F-FDG-avid lesions in pattern typical of cancer. If progression by RECIST, must verify with follow-up. |
| PMR: reduction of minimum of 15% ± 25% in tumor 18F-FDG SUV after 1 cycle of chemotherapy, and >25% after more than 1 treatment cycle; reduction in extent of tumor 18F-FDG uptake is not a requirement for PMR. | PMR: reduction of minimum of 30% in target measurable tumor 18F-FDG SUL peak. Absolute drop in SUL must be at least 0.8 SUL units, as well. Measurement is commonly in same lesion as baseline but can be another lesion if that lesion was previously present and is the most active lesion after treatment. ROI does not have to be in precisely same area as baseline scan, though typically it is. No increase, >30% in SUL or size of target or nontarget lesions (i.e., no PD by RECIST or IWC) (if PD anatomically, must verify with follow-up). Reduction in extent of tumor 18F-FDG uptake is not requirement for PMR. Percentage decline in SUL should be recorded, as well as (ideally) time in weeks after treatment was begun (i.e., PMR −40, 3). No new lesions. | |
| SMD: increase in tumor 18F-FDG SUV < 25% or decrease of <15% and no visible increase in extent of 18F-FDG tumor uptake (20% in longest dimension). | SMD: not CMR, PMR, or PMD. SUL peak in metabolic target lesion should be recorded, as well as (ideally) time from start of most recent therapy, in weeks (i.e., SMD −15, 7). | |
| PMD: increase in 18F-FDG tumor SUV of >25% within tumor region defined on baseline scan; visible increase in extent of 18F-FDG tumor uptake (20% in longest dimension) or appearance of new 18F-FDG uptake in metastatic lesions. | PMD: >30% increase in 18F-FDG SUL peak, with >0.8 SUL unit increase in tumor SUV peak from baseline scan in pattern typical of tumor and not of infection/treatment effect. OR: Visible increase in extent of 18F-FDG tumor uptake (75% in TLG volume with no decline in SUL. OR: New 18F-FDG-avid lesions that are typical of cancer and not related to treatment effect or infection. PMD other than new visceral lesions should be confirmed on follow-up study within 1 mo unless PMD also is clearly associated with progressive disease by RECIST 1.1. PMD should be reported to include percentage change in SUV peak, (ideally, time after treatment, in weeks) and whether new lesions are present/absent and their number (i.e., PMD, +35, 4, new: 5). Because SUL is continuous variable, dividing response criteria into limited number of somewhat arbitrary response categories loses much data. For this reason, PERCIST preserves percentage declines in SUV peak in each reported category. Because rapidity with which scan normalizes is important (faster appears better), PERCIST asks for time from start of treatment as part of reporting. For example, CMR 90, 1, is probably superior to CMR 90, 10, especially if latter patient were SMD 20, 1. More than one measurement of PET response may be needed at differing times, and it may be treatment type–dependent. PERCIST 1.0 evaluates SUL peak of only hottest tumor. This is possible limitation of approach, but lesions and their responses are highly correlated in general. Additional data are required to determine how many lesions should be assessed over 1. A suggested option is to include the 5 hottest lesions, or the 5 observed on RECIST 1.1 that are most measurable. Percentage change in SUL can be reported for single lesion with largest increase in uptake or smallest decline in uptake. Additional studies will be needed to define how many lesions are optimal for assessment. | |
| Nonmeasurable disease: CR, disappearance of all known disease, confirmed at ≥4 wk; PR, estimated decrease of ≥50%, confirmed at 4 wk; PD, estimated increase of ≥25% in existent lesions; NC, neither PR nor PD criteria met. | Nontarget lesions: CMR, disappearance of all 18F-FDG–avid lesions: PMD, unequivocal progression of 18F-FDG–avid nontarget lesions or appearance of new 18F-FDG–avid lesions typical of cancer; non-PMD: persistence of one or more nontarget lesions or tumor markers above normal limits. | |
| Overall response | 1. Best response recorded in measurable disease from treatment start to disease progression or recurrence. | |
| 2. Non-PMD in measurable or nonmeasurable nontarget lesions will reduce CR in target lesion to overall PMR. | ||
| 3. Non-PMD in nontarget lesions will not reduce PR in target lesions. | ||
| Duration of response | 1. Overall CMR: from date CMR criteria are first met; to date recurrent disease is first noted. | |
| 2. Overall response: from date CMR or PMR criteria are first met (whichever status came first); to date recurrent disease is first noted. | ||
| 3. SMD: from date of treatment start to date PMD is first noted. | ||
TLG = total lesion glycolysis; CMR = complete metabolic response; PMR = partial metabolic response; PD = progressive disease; SMD = stable metabolic disease; PMD = progressive metabolic disease; CR = complete remission; PR = partial remission; NC = no change.
For PERCIST: Single-voxel SUL is commonly used but has been reported to be less reproducible than SUL peak, especially with very small single-voxel values. It is suggested, but not required, that lesions assessed on PERCIST be larger than the 1.5-cm-diameter volume ROI used to minimize partial-volume effects. Percentage changes are proposed to deal with SUL peak changes. Use of maximal SUL could be explored. If 5 lesions are used as exploratory approach, it is suggested that sum of SULs of baseline 5 lesions serve as baseline for study. After treatment, sum of same 5 lesions should be used. Percentage change in SUL is based on change in these sums from study 1 to study 2. Exploratory analysis can include calculating percentage change in SUL in individual lesions and averaging them. This may produce different result. We believe summed SUL approach will be less prone to minor errors in measurements.
For total lesion glycolysis: Exploratory analysis can include either all foci of tumor with maximal SUL > 2 SDs above normal liver, 5 lesions with highest SUL, or lesion with highest SUL. It is suggested that threshold approach, typically at 2 SDs above normal liver SUL, be used to generate lower bounds of ROI (3 SDs could be used for very active tumors). We believe this approach will be less variable than methods based on maximal SUL with percentage of maximal cutoff. Criteria for progression include 75% growth in TLG for SUL and are conservatively placed at 75% increase. Because 20% increase in EORTC linear size scales to 73% volume increase, the figures are comparable. Progression is judged from best response if being assessed after first scan was performed. For response by TLG, we propose 45% reduction as useful starting point, but more data are needed to make firm recommendations. If TLG is determined, explicit methodologic details should be provided. It should not be a primary metric, but a secondary endpoint at this time.
SUV should be corrected for lean body mass (SUL) and should not be corrected for serum glucose levels (glucose corrections have been variably useful, and errors in glucometer measurements are well known and may add errors (135)). Normal background 18F-FDG activity is determined in the right hepatic lobe and consists of mean SUL and SD in a 3-cm-diameter spherical ROI. Typically, liver uptake should not vary by more than 0.3 SUL unit from study to study.
The SUL is determined for up to 5 tumors (up to 2 per organ) with the most intense 18F-FDG uptake. These will typically be the lesions identified on RECIST 1.1. The SUV peak (this is a sphere with a diameter of approximately 1.2 cm—to produce a 1-cm3-volume spheric ROI) centering around the hottest point in the tumor foci should be determined, and the image planes and coordinates should be noted (SUL peak). This SUL peak ROI will typically include the maximal SUL pixel (which should also be recorded) but is not necessarily centered on the maximal SUL pixel. Automated methods for searching for this peak region have been described (20). Tumor sizes should be noted and should be 2 cm or larger in diameter for accurate measurement, though smaller lesions of sufficient 18F-FDG uptake, including those not well seen anatomically, can be assessed. Each baseline (pretreatment) tumor SUL peak must be 1.5 × mean liver SUL + 2 SDs of mean SUL. If the liver is diseased, 2.0 × blood-pool 18F-FDG activity + 2 SDs in the mediastinum is suggested as minimal metabolically measurable tumor activity.
In PERCIST, response to therapy is assessed as a continuous variable and expressed as percentage change in SUL peak (or sum of lesion SULs) between the pre- and posttreatment scans. Briefly, a complete metabolic response is defined as visual disappearance of all metabolically active tumor. A partial response is considered more than a 30% and a 0.8-unit decline in SUL peak between the most intense lesion before treatment and the most intense lesion after treatment, although not necessarily the same lesion. More than a 30% and 0.8-unit increase in SUL peak or new lesions, if confirmed, is classified as progressive disease. A greater than 75% increase in total lesion glycolysis is proposed as another metric of progression. Further details of the proposed PERCIST criteria for monitoring therapy response and comparison to EORTC are shown in Table 7.
Rationale for the Proposed Percist Criteria
Why PERCIST?
PET assessments of treatment response with 18F-FDG appear to have substantial biologic relevance when obtained at the end of treatment, at mid treatment, or soon after treatment is started. Indeed, the biologic predictive value of PET appears to be greater than that of anatomic studies, including for lymphoma, lung cancer, mesothelioma, and esophageal cancer. Although currently accepted response criteria are anatomic, it is quite possible that an approach using purely metabolic response criteria may ultimately be more predictive of outcomes. Given that some tumors do not have high uptake of 18F-FDG, or may be too small to be reliably quantified, it is likely that both anatomic and functional criteria will be important for the foreseeable future. Although it would be possible to propose an integrated CT + PET approach akin to that of the IWC + PET (i.e., that a PET scan only be interpreted as positive or negative and be used to trump anatomic imaging if the studies are disparate), this approach would seem to lose some of the advantages of the continuous output of the PET data through forced dichotomization. The inclusion of an 18F-FDG PET observation into the RECIST 1.1 criteria as a sign of disease recurrence is a step in this direction.
In preparing the PERCIST 1.0 criteria, at the request of The Journal of Nuclear Medicine editors (after the lead author had lectured on this topic), it was clear that many of the answers regarding the use of PET for assessing treatment response are not yet in. What is clear is that unless more precisely defined response criteria are in place and used by varying groups, it will be difficult to compare PET treatment response studies across centers or even to include PET in such studies. The Imaging Response Assessment Team at Johns Hopkins reviews clinical oncologic protocols at the Sidney Kimmel Comprehensive Cancer Center weekly. In nearly all of these, RECIST criteria are used for solid tumor evaluations. Only a few studies include PET. Although some use the EORTC criteria, methods for PET performance and interpretation are typically highly variable across studies and typically only exploratory. With over 30 ways to assess tumor response quantitatively and many articles using differing ROI selection techniques, arriving at a common approach, even if not proven ultimately to be the best in each case, will help generate more data on treatment response and allow a larger database to be developed for testing analytic tools retrospectively as has been done by the RECIST group.
Why the ROI?
Several points in the PERCIST 1.0 criteria are notable and may be controversial. ROI size is important and has varied from study to study. Larger ROIs give better precision but a lower SUL than do smaller ROIs (20,115). Despite its widespread use, maximal SUL was not selected as the primary metric of response because the size of the maximal voxel sampling ROI varies considerably by scanner, matrix size, slice thickness, and scanner diameter, resulting in various noise levels in the metric. Thus, the precision of maximal SUL is not well established. All but one of the studies examining the precision of SUV used larger regions of interest than the volume assessed to determine the current single-pixel SUVmax provided by modern high-resolution scanners. When tested, the small single-pixel SUVmax is more variable than the somewhat larger ROIs.
The maximal pixel value is possibly most advantageous in small tumors, as it would be somewhat less dependent on partial-volume effects. However, noise effects are substantial. Although correcting for partial volume is attractive conceptually, the PERCIST criteria have avoided partial-volume corrections. Measuring tumor or node size with CT from PET/CT is feasible, but slight errors in those measurements can have major effects on quantitation if used to correct for partial-volume effects. Studies in which complex partial-volume corrections have been performed in addition to corrections for background spillover from nearby tissues have sometimes, but not consistently, demonstrated quantitation to be superior to visual assessments for predicting response and outcome (136). We believe such corrections will be to too difficult to effect in routine practice because of the obvious challenges of measuring small lesions accurately. The maximal SUL should be recorded, however, for selected 18F-FDG–avid tumors.
Most studies of treatment response have focused on larger measurable tumors. We realize maximal SUL may be useful in small lesions and should be explored. Although imaging tumors larger than 2 cm is encouraged to minimize partial-volume effects, PERCIST 1.0 allows any tumor whose SUL peak is greater than 1.5 × liver mean + 2 SDs to be assessed quantitatively. This figure is based on cutoffs used by Weber and is used to ensure that the posttreatment lesion SUL can fall sufficiently to detect a response. Less avid tumors may be visualized and their disappearance can be noted, as well as their obvious progression. It is possible that a cutoff of 1.35 × hepatic uptake as was used by Weber may also be acceptable as a lower limit of measurable activity.
However, recording tumor size by RECIST criteria is suggested for measurable lesions larger than 1 cm. Because ROIs whose size is based on a 50%, or other, ROI threshold vary with the variability of the maximal pixel chosen, these were not chosen as the primary measurement metric. Rather, the SUL peak in a small volume of greatest metabolic activity in the tumor (approximately 1 cm3) is suggested for use. This size has been used in many studies and is statistically less subject to variance than is a small, single-pixel SUVmax.
Total lesion glycolysis is also attractive. PERCIST suggests that this be obtained but recommends that it be threshold-based, with an outer boundary equal to 3 SDs above normal-liver mean SUL determined in a standard-sized ROI of 3 cm in diameter. This should be relatively consistent, based on such factors as similar injection times for imaging on the baseline study and the follow-up study. However, the total lesion glycolysis metric is not proposed for primary response assessment. We suggested that it be routinely obtained for the 5 hottest lesions to estimate tumor burden, but it is optional for assessing all lesions. Collecting these data consistently should help us learn more about the best method to assess treatment response by disease type.
What Decline in SUV Is a Response?
Already, it is evident that the medically relevant cutoff for an SUL decline to represent response and predict outcomes may differ on the basis of the disease, the timing after treatment, the treatment itself, and the treatment goal. The 30% requirement for a tumor response (and the drop of 0.8 SUL unit) we propose in PERCIST (based on peak SUL) is more stringent than that proposed in the 1999 EORTC criteria (15% or 25% drops in SUV). The 15% decline in SUV in the original EORTC criteria for early response is probably too modest to reliably be discerned from variability in the study and likely is insufficient to be medically relevant based on data developed since that time.
For lymphomas, in which cure is feasible and a rapid drop in SUV is common, a higher cutoff for a medically relevant response (e.g., 65% at mid treatment) may be required (86). This cutoff is greater than that for the palliative or noncurative treatment of lung cancer (e.g., 30%–35%). Similarly, in sarcoma and gastric and ovarian carcinoma responses, a drop in SUV of more than 25% is associated with the best outcomes (43,87,137,138). When lower thresholds of, for example, 20%–30% are accepted as responses, limited data suggest that these patients are unlikely to have a medically relevant response, even if the response is statistically significant (87,130). For example, patients with GIST treated with imatinib who had only modest declines (∼30% decrease) in SUV early after therapy did not appear to have good outcomes, suggesting that a larger threshold may have been in order (87).
Although a decline of 25% or more is less likely to be due to chance than are smaller declines, this level of decline can occur in lesions with low SUVs and a rather modest change in total SUV. For this reason, a minimal level of tumor uptake is proposed in PERCIST 1.0 to be assessable. This minimal level is proposed as 1.5 × the liver SUV mean + 2 SDs. Because the typical SUL of the liver is around 1.6–1.8, the SUL peak of an assessable lesion is going to be approximately 2.5 or greater (Fig. 3). In addition to the requisite percentage change in SUL after treatment, PERCIST also requires a defined absolute change in SUL of 0.8 units in order to minimize overestimation of response or progression. Weber has proposed a 0.9 SUV change as the minimum to be significant (114); however, since SUL is typically somewhat less than SUV, we suggest a change of 0.8 SUL unit to be a reasonable absolute change. The 0.5 SUV unit change described as significant by Nahmias (115) may be too small with the ROI size proposed for PERCIST. We do not know what change in total lesion glycolysis is required for a response. Because the dynamic range is larger, a suggested figure of 40% for a response should be considered on the basis of the larger changes in total lesion glycolysis than SUVmax reported in mesothelioma, as well as a potentially, but not fully defined, lower precision for the volume × SUV figure, which would be expected because of measurement errors in both the volume and the SUV parameters (111).
FIGURE 3.
Example calculation of liver background for normalization of SUL. Images are displayed from Advantage Workstation (GE Healthcare). A 3-cm-diameter 3-dimensional ROI (ROI 1) is placed on normal inferior right lobe of liver (arrowhead). Average SUL and SD in ROI are displayed (arrows). Liver background is calculated as follows: (1.5 × average SUL liver) + (2 × SD average SUL liver). For this example, (1.5 × 1.4) + (2 × 0.2) = 2.5. Therefore, tumor SUL peak should be >2.5 in order to apply PERCIST criteria for this example.
It is also important in PERCIST to note how long into the therapy the response is obtained to take full advantage of the continuous nature of the SUV. Recording of the full continuous range of the percentage change in SUL allows for preservation of data that are otherwise lost by reducing the continuous variable to discrete bins of response.
Using continuous data, it should be possible to perform controlled trials in which experimental treatments are compared with standard treatments. In such trials, the expected change in SUL may not be known. However, the continuous readout of SUL change is expected to be quite helpful in detecting the activity of the therapeutic agent and to minimize sample sizes.
The PERCIST 1.0 criteria are designed to facilitate trials of drug development but, if sufficiently robust, could be applied to individual patients. In individual patients, determining what level of quantitative change in SUL is medically significant will depend on multiple factors, not just on what level of change exceeds that due to chance. Other factors will include the level of comfort the treating physician has in not treating with a regimen that may still have a small likelihood of being effective (i.e., of deciding to deny therapy to someone who may have a borderline response and a low, but possible, chance of benefit). Decisions to deny probably ineffective therapy depend on alternative therapeutic options and on the risks, cost, and benefits of the treatment and so are difficult to specifically address. If therapies are of low risk and there are no good alternatives, denial of treatment would seem unreasonable, even if benefit were quite improbable. By contrast, with a highly toxic treatment of high cost, denying treatment might be highly appropriate if the treatment is unlikely to be beneficial. As more data are generated on specific diseases with specific treatments, the development of likelihood ratios of probable benefit from treatment can be expected. An example of a partial metabolic response by PERCIST is shown in Figure 4, one in which the functional response exceeds the anatomic.
FIGURE 4.
PET/CT images obtained before (1) and after (2) treatment of pancreatic carcinoma with experimental therapy targeting mammalian target of rapamycin. Note profound decline in SUL (∼41%) despite stable pancreatic mass anatomically (arrows). This decline represents metabolic partial response by PERCIST (41% decline in marker lesion at 2 wk after therapy). Not all metabolic PMRs are clinically relevant; relevance will depend on the specific treatment.
What Decline in SUV Represents a Complete Response?
The PERCIST criteria do include the category “complete metabolic response.” It might seem logical that patients with a complete response would have a 100% SUV decline. However, in many studies the degree of SUV reduction associated with a complete metabolic response is less than 100% (139). PERCIST specifies that the SUL percentage reduction be noted from the pretreatment to the posttreatment PET scans, along with the time from the start of the most recent treatment regimen (in weeks), even for complete response in patients on active treatment. Because background rarely has an SUL of 0, declines in SUL to 0 are unlikely, as are 100% reductions in tumor SUL.
Drops in SUL of 100% could be achieved by subtracting the mean SUL of the liver + 2 SDs from the tumor activity and using the resultant dynamic range. However, after treatment, drops in SUL of over 100% are possible with such an approach. For small lesions after treatment, focal uptake may remain and may be less than liver uptake and visually detectable (32). Thus, the possibility of a incomplete response with over a 100% decline in background-corrected SUL exists. PERCIST 1.0 requires collection of the background SUL in the liver and the variance in SUL, which can allow for such post hoc calculations of background-corrected SUL changes if desired. For this PERCIST 1.0 version, we believe visual assessment is essential for determining the presence or absence of complete response, especially for small lesions after treatment. However, data collected from our approach should allow future studies of the best definition of complete response to help define whether a qualitative or quantitative metric is most robust at predicting outcomes. Quantitative metrics potentially may be developed to help in avoiding false-positive scans after treatment.
What About the Choice of Background?
Background tissues are important normal metrics for verifying that a PET study is performed properly from a technical standpoint. Many factors, including a poor intravenous injection, inaccurate dose calibration or camera calibration, or variable uptake times, can affect the SUL (30). We believe that the normal liver SUL is slightly more stable than determinations of blood-pool SUL. Practically, it is less effort to draw a 3-cm-diameter ROI on the right lobe of the liver than to repeatedly draw regions of interest on the aorta on multiple levels, taking care to avoid including uptake in the possibly diseased vessel wall (113,114,124,140). If the liver is diseased (most notably, full of cancer involvement), it is clearly unsuitable as a background area. An alternative in such a case is the blood-pool activity in the descending aorta. For either blood pool or liver, the SUL temporally depends on the time after injection. Thus, close similarity in uptake times is required for the baseline and follow-up studies to ensure the stability of background hepatic uptake.
How Many Lesions to Assess?
The number of lesions to evaluate when assessing response to therapy is a major issue, and the answer is uncertain for PET at this time. Most of the initial PET literature evaluated a single lesion, such as a primary lung, breast, or esophageal cancer. In such cases, n = 1 is obviously the appropriate number. In anatomic imaging assessments in which multiple tumors are present, the RECIST group has recently recommended evaluating the size of a maximum of 3–5 lesions (typically 5) anatomically to assess response, even if many more lesions are present. This does not mean other lesions are not assessed; rather, it means they are not measured. If tumors other than these 5 progress unequivocally, progression has occurred (39,40). RECIST separates between target and nontarget lesions (Tables 1 and 2).
TABLE 2.
| Nontarget lesions | New lesions | Overall response |
|---|---|---|
| CR | No | CR |
| Non-CR/non-PD | No | Non-CR/non-PD* |
| Not all evaluated | No | NE |
| Unequivocal PD | Yes or no | PD |
| Any | Yes | PD |
“Non-CR/non-PD” is preferred over “stable disease” for nontarget disease. Because stable disease is increasingly used as endpoint for assessment of efficacy in some trials, it is not advisable to assign this category when no lesions can be measured.
CR = complete response; PD = progressive disease; NE = not evaluable.
In the Hicks qualitative PET criteria (Table 5), multiple lesions are assessed (76,84,92,141). In quantitatively assessing treatment response in patients with disseminated ovarian cancer, Avril et al. assessed up to 4 lesions per patient, but an average of just 2.2 lesions were studied for response (130). They chose the lesion with the smallest percentage decline in SUV after therapy as representative (i.e., the worst responder), with a rationale that the metastatic tumor with the worst response would determine survival.
In another study of disseminated intraabdominal tumors, Stroobants et al. selected up to 3 foci of 18F-FDG uptake in GIST that were highest on baseline PET. All lesions had to decline by at least 25% to represent a partial response, and all had to disappear to background to represent a complete response (87).
Remarkably, several studies have shown that changes in the SUVof primary tumors can quite accurately predict the outcomes in their nodal metastases. Careful studies from Dooms et al. have shown that metastatic-tumor-involved mediastinal nodal pathology and clinical behavior are well predicted by changes in SUV and absolute SUV in the primary lung cancer and by qualitative visual assessments of nodal status (66,142). This is in part because “child” metastases biologically resemble their “parents” (143,144).
Several other interesting approaches have evaluated just a single lesion but considered the worst-case biologic behavior of the malignancy. Lin et al. found that the accuracy of predicting event-free survival in lymphoma response assessment was slightly better using the change in SUV from the hottest lesion on study 1 to the hottest lesion on study 2 (which was a different lesion in 18% of cases) than using the change in the hottest lesion on the baseline study (76.1% accuracy vs. 73.9% accuracy in outcome prediction) (86). Although comparable, there were slightly more false-negative scans when the same lesion was used for analysis. This approach is somewhat similar to that used by Wahl et al., in which the single hottest area in a primary breast cancer was used as the reference point on the pretreatment and posttreatment studies—often, but not necessarily, the same area (20).
Because the RECIST criteria examine a maximum of 5 lesions, we have proposed that PERCIST measure the SUL in no more than 5 lesions, as well (unless an automated total lesion glycolysis is determined as a corollary study). However, it is not known how to optimally combine the results of percentage change in SUL from multiple tumors to be predictive of outcome. For example, to have a response, does each metabolically assessable target tumor have to drop its uptake by 30%, or does the sum of the declines in SUL in the posttreatment group have to be 30% less than the sum of the SULs in the same lesions before treatment? Requiring each lesion to drop at least 30% is probably more stringent than the sum, but this is not clear. It is probable that combination methods of either summed SUL before and after treatment (sum of SUL for lesions 1–5 before treatment and sum of SUL of lesions 1–5 after treatment) or percentage decline in summed SUL between scans will be biased by the hottest lesion or largest percentage decline.
The uncertainty on how to precisely combine the SULs of 5 lesions, and evidence that a restricted dataset of fewer tumors is commonly adequate, along with simplicity of calculation are other reasons why, for this first-level analysis of PERCIST 1.0, it is suggested that only the percentage difference in SUL between the tumor with the most intense SUL on study 1 and the tumor with the most intense SUL on study 2 should be used as a classifier for response. This suggestion supposes that the most intense lesion on study 2 has not grossly progressed and that it was present at the time of study 1. As long as all other unmeasured lesions do not progress, this method would be used to determine whether a response had occurred. Given the uncertainty about the best metric, it is suggested that SUL peak data be determined and summed before and after treatment for up to the 5 hottest lesions and that the ratio of the sums before and after treatment be compared as a secondary analysis. Obvious progression of any tumor (i.e., >30% increase) or new lesions would negate a partial response.
Perhaps these findings that one or a few tumors predict outcome well are consistent with the clonality of metastases; that is, most are genetically comparable and most respond similarly to treatments. Thus, a good assessment of the most metabolically aggressive tumor before and after treatment may be reflective of the others in many instances. However, we all have observed cases in which new lesions appear and progress despite apparent control of a primary lesion (Fig. 5) (139). This observation may be related to the form of treatment but does occur. Thus, clearly progressive disease in any one lesion is disease progression, even if other tumor foci are responding.
FIGURE 5.
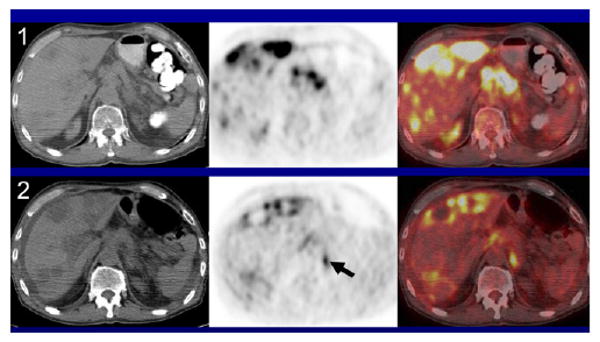
PET/CT image obtained before (1) and after (2) treatment of pancreatic carcinoma with experimental therapy targeting mammalian target of rapamycin. Glycolysis and apparent necrosis are profoundly reduced in intensely 18F-FDG–avid liver metastases. Although a reduction of more than 50% in SUL peak would suggest partial metabolic response, new lesion indicative of progressive metabolic disease is evident in left retroperitoneum (arrow).
Lack of Good Information for Progression
The precise optimal definition of tumor progression remains in evolution. The EORTC criteria defined progression as an increase in SUV of over 25%, an increase in the extent of 18F-FDG uptake by more than 20% in length, or new 18F-FDG–positive metastases. With PERCIST, we propose a more than 30% increase in SUL peak, new 18F-FDG–avid lesions, or growth in lesion total lesion glycolysis by more than 75%—somewhat more stringent criteria for progression.
New 18F-FDG–avid lesions associated with the CT abnormality most consistent with tumor and clearly not due to inflammation or infection can be considered progression. New 18F-FDG–avid foci unassociated with a CT finding may well represent progression but should typically be verified by a follow-up PET/CT scan, or by another verification method 1 mo after their initial presentation (Fig. 5). Sometimes, however, verification will not occur anatomically, such as in lesions in bone marrow or in the spleen. RECIST 1.1 has addressed these issues to some extent. Progression in the lungs, particularly in the presence of potential inflammation or infection while a patient is on treatment, should be viewed with great caution, as discussed in the revised response criteria in lymphoma (32,33). New pulmonary infiltrates after treatment are often due to inflammation or infection and should be excluded before progressive disease is classified.
The extent of increase in 18F-FDG uptake required to represent progression is unclear. It is also unclear if an increase in SUL of over 30% in a single lesion is truly progression if the lesion is not the hottest. It may be difficult for the most intense lesion to increase in uptake over 30%, as the lesion may be performing glycolysis at a rate that is the maximum possible for its blood supply. Thus, growth in lesion size or total glycolytic volume potentially may be more indicative of progression than a rise in SUL peak in some settings. We have proposed a 30% increase in maximal SUL of the most intense lesion, with an SUL of more than 1.5 mean liver + 2 SDs as progression and an absolute increase in SUL peak of 0.8 units. However, it is probable that a 30% increase may not be achieved in all cases of progression. Rising 30% is probably easier in less glycolytically active lesions. If 5 lesions are assessed, the increase in glycolysis would need to be a 30% increase in the summed SUL peaks for the 5 most active lesions after treatment, versus the summed SUL peak of the 5 most active lesions before treatment.
For this reason, an increase of 75% in total lesion glycolysis for the most active tumor is proposed. This metric is reportedly more variable (at least the volume component) than is SUL peak (104). Total lesion glycolysis of the up to 5 target metabolic lesions is recommended at a minimum. It is possible that total lesion glycolysis of all lesions of sufficient intensity will be a better metric of progression than that of a single lesion. Methods for delineating lesions for total lesion glycolysis based on threshold values have been developed and are entering practice (Fig. 6). Thus, PERCIST 1.0 recommends that these data be collected as part of trials including PET for treatment response assessment. It may also be reasonable to collect SUVmax data for a single pixel, though these data are not used in response determinations as presently configured.
FIGURE 6.
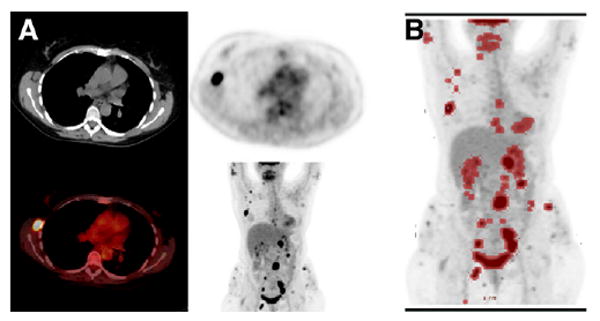
(A) Patient with extensive non-Hodgkin lymphoma before treatment. Tumor with most intense 18F-FDG activity is in abdomen. Transverse images of easily measurable right axillary lymph node on CT are shown for convenience. (B) Commercial software tool (PET Volume Computed Assisted Reading; GE Healthcare) was used to localize foci of 18F-FDG uptake greater than mean liver SUL + 2 SDs of normal liver background (red). Manual intervention is required to separate normal 18F-FDG–avid foci, including brain, heart, and excreted urine, from relevant tumor. This semiautomated segmentation can be used to estimate total lesion glycolysis.
It is rare for an 18F-FDG–avid tumor to progress in the fashion of a tumor that is not 18F-FDG–avid, at least for measurable lesions. Small metastases, such as in the lungs, could be falsely PET-negative early in their progression. However anatomic progression that is not 18F-FDG–avid by RECIST or IWC in a previously 18F-FDG–avid tumor and that does not otherwise meet PERCIST criteria for progression would need verification before being considered progression.
Conclusion
In the 15 years since quantitative monitoring of treatment effects with PET was introduced, there has been remarkable progress. It is clear that the biologic signal from 18F-FDG is important and often more predictive of histologic and survival outcomes than is anatomic imaging. Standardizing response assessment for PET in treatment monitoring is crucial to move the field forward and to allow comparisons from study to study. The considerable efforts of the WHO and RECIST groups on anatomic imaging and those of the EORTC PET response group a decade ago serve as a framework for the proposed PERCIST 1.0 criteria, which draw heavily from their efforts.
Although several, perhaps all, aspects of PERCIST 1.0 are likely to be controversial, PERCIST 1.0 is viewed as a starting point for studies and has pointed out several unanswered questions. Although PERCIST 1.0 has specific criteria for response based on a single marker lesion, collection of additional data on 5 tumors is strongly recommended so as to develop a database suitable for additional studies to refine the response metrics for a given tumor and therapy. Similarly, whereas SUL peak is the main chosen metric, collection of data on maximal single-voxel SUL and total lesion glycolysis is recommended as secondary for later analysis. The PERCIST 1.0 criteria are intended to represent a framework that can be used for clinical studies, for clinical care, and as a foundation for workshops to refine and validate quantitative approaches to monitoring PET tumor response—approaches that, it is hoped, can be improved and be accepted by the international community and regulatory agencies.
Acknowledgments
The thoughtful input of Dr. Wolfgang Weber and the encouragement of Drs. Johannes Czernin and Heinrich Schelbert are much appreciated. Without their respective efforts, this article would not have come to fruition. This work was supported in part by National Cancer Institute 3 P30 CA006973-43S2 and by the Imaging Response Assessment Teams in Cancer Center.
References
- 1.Karrison TG, Maitland ML, Stadler WM, Ratain MJ. Design of phase II cancer trials using a continuous endpoint of change in tumor size: application to a study of sorafenib and erlotinib in non small-cell lung cancer. J Natl Cancer Inst. 2007;99:1455–1461. doi: 10.1093/jnci/djm158. [DOI] [PubMed] [Google Scholar]
- 2.Ratain MJ, Eckhardt SG. Phase II studies of modern drugs directed against new targets: if you are fazed, too, then resist RECIST. J Clin Oncol. 2004;22:4442–4445. doi: 10.1200/JCO.2004.07.960. [DOI] [PubMed] [Google Scholar]
- 3.Rosner GL, Stadler W, Ratain MJ. Randomized discontinuation design: application to cytostatic antineoplastic agents. J Clin Oncol. 2002;20:4478–4484. doi: 10.1200/JCO.2002.11.126. [DOI] [PubMed] [Google Scholar]
- 4.Ratain MJ, Sargent DJ. Optimising the design of phase II oncology trials: the importance of randomisation. Eur J Cancer. 2009;45:275–280. doi: 10.1016/j.ejca.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 5.Schuetze SM, Baker LH, Benjamin RS, Canetta R. Selection of response criteria for clinical trials of sarcoma treatment. Oncologist. 2008;13(suppl 2):32–40. doi: 10.1634/theoncologist.13-S2-32. [DOI] [PubMed] [Google Scholar]
- 6.Moertel CG, Hanley JA. The effect of measuring error on the results of therapeutic trials in advanced cancer. Cancer. 1976;38:388–394. doi: 10.1002/1097-0142(197607)38:1<388::aid-cncr2820380156>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 7.Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer. 1981;47:207–214. doi: 10.1002/1097-0142(19810101)47:1<207::aid-cncr2820470134>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 9.Lavin PT. An alternative model for the evaluation of antitumor activity. Cancer Clin Trials. 1981;4:451–457. [PubMed] [Google Scholar]
- 10.Michaelis LC, Ratain MJ. Measuring response in a post-RECIST world: from black and white to shades of grey. Nat Rev Cancer. 2006 May;6:409–414. doi: 10.1038/nrc1883. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan WD, Jochelson MS, Herman TS, et al. Gallium-67 imaging: a predictor of residual tumor viability and clinical outcome in patients with diffuse large-cell lymphoma. J Clin Oncol. 1990;8:1966–1970. doi: 10.1200/JCO.1990.8.12.1966. [DOI] [PubMed] [Google Scholar]
- 12.Front D, Bar-Shalom R, Mor M, et al. Aggressive non-Hodgkin lymphoma: early prediction of outcome with 67Ga scintigraphy. Radiology. 2000;214:253–257. doi: 10.1148/radiology.214.1.r00ja03253. [DOI] [PubMed] [Google Scholar]
- 13.Front D, Bar-Shalom R, Mor M, et al. Hodgkin disease: prediction of outcome with 67Ga scintigraphy after one cycle of chemotherapy. Radiology. 1999;210:487–491. doi: 10.1148/radiology.210.2.r99fe07487. [DOI] [PubMed] [Google Scholar]
- 14.Israel O, Front D, Lam M, et al. Gallium 67 imaging in monitoring lymphoma response to treatment. Cancer. 1988;61:2439–2443. doi: 10.1002/1097-0142(19880615)61:12<2439::aid-cncr2820611208>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 15.Israel O, Mor M, Epelbaum R, et al. Clinical pretreatment risk factors and Ga-67 scintigraphy early during treatment for prediction of outcome of patients with aggressive non-Hodgkin lymphoma. Cancer. 2002;94:873–878. [PubMed] [Google Scholar]
- 16.Di Chiro G, Brooks RA. PET-FDG of untreated and treated cerebral gliomas. J Nucl Med. 1988;29:421–423. [PubMed] [Google Scholar]
- 17.Di Chiro G, Oldfield E, Wright DC, et al. Cerebral necrosis after radiotherapy and/or intraarterial chemotherapy for brain tumors: PET and neuropathologic studies. AJR. 1988;150:189–197. doi: 10.2214/ajr.150.1.189. [DOI] [PubMed] [Google Scholar]
- 18.Minn H, Soini I. [18F]fluorodeoxyglucose scintigraphy in diagnosis and follow up of treatment in advanced breast cancer. Eur J Nucl Med. 1989;15:61–66. doi: 10.1007/BF00702620. [DOI] [PubMed] [Google Scholar]
- 19.Hoekstra OS, van Lingen A, Ossenkoppele GJ, Golding R, Teule GJ. Early response monitoring in malignant lymphoma using fluorine-18 fluorodeoxyglucose single-photon emission tomography. Eur J Nucl Med. 1993;20:1214–1217. doi: 10.1007/BF00171020. [DOI] [PubMed] [Google Scholar]
- 20.Wahl RL, Zasadny K, Helvie M, Hutchins GD, Weber B, Cody R. Metabolic monitoring of breast cancer chemohormonotherapy using positron emission tomography: initial evaluation. J Clin Oncol. 1993;11:2101–2111. doi: 10.1200/JCO.1993.11.11.2101. [DOI] [PubMed] [Google Scholar]
- 21.Juweid ME, Cheson BD. Positron-emission tomography and assessment of cancer therapy. N Engl J Med. 2006;354:496–507. doi: 10.1056/NEJMra050276. [DOI] [PubMed] [Google Scholar]
- 22.Weber WA, Wieder H. Monitoring chemotherapy and radiotherapy of solid tumors. Eur J Nucl Med Mol Imaging. 2006;33(suppl 1):27–37. doi: 10.1007/s00259-006-0133-3. [DOI] [PubMed] [Google Scholar]
- 23.Kidd EA, Siegel BA, Dehdashti F, Grigsby PW. The standardized uptake value for F-18 fluorodeoxyglucose is a sensitive predictive biomarker for cervical cancer treatment response and survival. Cancer. 2007;110:1738–1744. doi: 10.1002/cncr.22974. [DOI] [PubMed] [Google Scholar]
- 24.Weber WA. Positron emission tomography as an imaging biomarker. J Clin Oncol. 2006;24:3282–3292. doi: 10.1200/JCO.2006.06.6068. [DOI] [PubMed] [Google Scholar]
- 25.Larson SM, Schwartz LH. 18F-FDG PET as a candidate for “qualified biomarker”: functional assessment of treatment response in oncology. J Nucl Med. 2006;47:901–903. [PubMed] [Google Scholar]
- 26.Kasamon YL, Wahl RL. FDG PET and risk-adapted therapy in Hodgkin's and non-Hodgkin's lymphoma. Curr Opin Oncol. 2008;20:206–219. doi: 10.1097/CCO.0b013e3282f5123d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasamon YL, Jones RJ, Wahl RL. Integrating PET and PET/CT into the risk-adapted therapy of lymphoma. J Nucl Med. 2007;48(suppl 1):19S–27S. [PubMed] [Google Scholar]
- 28.Kasamon YL, Wahl RL, Swinnen LJ. FDG PET and high-dose therapy for aggressive lymphomas: toward a risk-adapted strategy. Curr Opin Oncol. 2004;16:100–105. doi: 10.1097/00001622-200403000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Hutchings M, Loft A, Hansen M, et al. FDG-PET after two cycles of chemotherapy predicts treatment failure and progression-free survival in Hodgkin lymphoma. Blood. 2006;107:52–59. doi: 10.1182/blood-2005-06-2252. [DOI] [PubMed] [Google Scholar]
- 30.Shankar LK, Hoffman JM, Bacharach S, et al. Consensus recommendations for the use of 18F-FDG PET as an indicator of therapeutic response in patients in National Cancer Institute Trials. J Nucl Med. 2006;47:1059–1066. [PubMed] [Google Scholar]
- 31.Boellaard R, Oyen WJ, Hoekstra CJ, et al. The Netherlands protocol for standardisation and quantification of FDG whole body PET studies in multi-centre trials. Eur J Nucl Med Mol Imaging. 2008;35:2320–2333. doi: 10.1007/s00259-008-0874-2. [DOI] [PubMed] [Google Scholar]
- 32.Juweid ME, Stroobants S, Hoekstra OS, et al. Use of positron emission tomography for response assessment of lymphoma: consensus of the Imaging Subcommittee of International Harmonization Project in Lymphoma. J Clin Oncol. 2007;25:571–578. doi: 10.1200/JCO.2006.08.2305. [DOI] [PubMed] [Google Scholar]
- 33.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 34.Mijnhout GS, Riphagen II, Hoekstra OS. Update of the FDG PET search strategy. Nucl Med Commun. 2004;25:1187–1189. doi: 10.1097/00006231-200412000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Mijnhout GS, Hooft L, van Tulder MW, Deville WL, Teule GJ, Hoekstra OS. How to perform a comprehensive search for FDG-PET literature. Eur J Nucl Med. 2000;27:91–97. doi: 10.1007/pl00006669. [DOI] [PubMed] [Google Scholar]
- 36.Young H, Baum R, Cremerius U, et al. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35:1773–1782. doi: 10.1016/s0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
- 37.Therasse P, Eisenhauer EA, Verweij J. RECIST revisited: a review of validation studies on tumour assessment. Eur J Cancer. 2006;42:1031–1039. doi: 10.1016/j.ejca.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 38.Verweij J, Therasse P, Eisenhauer E. Cancer clinical trial outcomes: any progress in tumour-size assessment? Eur J Cancer. 2009;45:225–227. doi: 10.1016/j.ejca.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 39.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 40.Bogaerts J, Ford R, Sargent D, et al. Individual patient data analysis to assess modifications to the RECIST criteria. Eur J Cancer. 2009;45:248–260. doi: 10.1016/j.ejca.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 41.Schwartz LH, Bogaerts J, Ford R, et al. Evaluation of lymph nodes with RECIST 1.1. Eur J Cancer. 2009;45:261–267. doi: 10.1016/j.ejca.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 42.Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol. 2007;25:1760–1764. doi: 10.1200/JCO.2006.07.3411. [DOI] [PubMed] [Google Scholar]
- 43.Van den Abbeele AD. The lessons of GIST: PET and PET/CT—a new paradigm for imaging. Oncologist. 2008;13(suppl 2):8–13. doi: 10.1634/theoncologist.13-S2-8. [DOI] [PubMed] [Google Scholar]
- 44.Forner A, Ayuso C, Varela M, et al. Evaluation of tumor response after locoregional therapies in hepatocellular carcinoma: are response evaluation criteria in solid tumors reliable? Cancer. 2009;115:616–623. doi: 10.1002/cncr.24050. [DOI] [PubMed] [Google Scholar]
- 45.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 46.Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–1759. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 47.Choi H, Charnsangavej C, de Castro Faria S, et al. CT evaluation of the response of gastrointestinal stromal tumors after imatinib mesylate treatment: a quantitative analysis correlated with FDG PET findings. AJR. 2004;183:1619–1628. doi: 10.2214/ajr.183.6.01831619. [DOI] [PubMed] [Google Scholar]
- 48.Bensimhon D, Soyer P, Brouland JP, Boudiaf M, Fargeaudou Y, Rymer R. Gastrointestinal stromal tumors: role of computed tomography before and after treatment [in French] Gastroenterol Clin Biol. 2008;32:91–97. doi: 10.1016/j.gcb.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 49.Mabille M, Vanel D, Albiter M, et al. Follow-up of hepatic and peritoneal metastases of gastrointestinal tumors (GIST) under imatinib therapy requires different criteria of radiological evaluation (size is not everything!!!) Eur J Radiol. 2009;69:204–208. doi: 10.1016/j.ejrad.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Vossen JA, Buijs M, Kamel IR. Assessment of tumor response on MR imaging after locoregional therapy. Tech Vasc Interv Radiol. 2006;9:125–132. doi: 10.1053/j.tvir.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 51.Plathow C, Klopp M, Thieke C, et al. Therapy response in malignant pleural mesothelioma: role of MRI using RECIST, modified RECIST and volumetric approaches in comparison with CT. Eur Radiol. 2008;18:1635–1643. doi: 10.1007/s00330-008-0918-9. [DOI] [PubMed] [Google Scholar]
- 52.Ceresoli GL, Chiti A, Zucali PA, et al. Assessment of tumor response in malignant pleural mesothelioma. Cancer Treat Rev. 2007;33:533–541. doi: 10.1016/j.ctrv.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 53.van Klaveren RJ, Aerts JG, de Bruin H, Giaccone G, Manegold C, van Meerbeeck JP. Inadequacy of the RECIST criteria for response evaluation in patients with malignant pleural mesothelioma. Lung Cancer. 2004;43:63–69. doi: 10.1016/s0169-5002(03)00292-7. [DOI] [PubMed] [Google Scholar]
- 53A.Barnacle AM, McHugh K. Limitations with the response evaluation criteria in solid tumors (RECIST) guidance in disseminated pediatric malignancy. Pediatr Blood Cancer. 2006;46:127–134. doi: 10.1002/pbc.20344. [DOI] [PubMed] [Google Scholar]
- 54.Erasmus JJ, Gladish GW, Broemeling L, et al. Interobserver and intraobserver variability in measurement of non-small-cell carcinoma lung lesions: implications for assessment of tumor response. J Clin Oncol. 2003;21:2574–2582. doi: 10.1200/JCO.2003.01.144. [DOI] [PubMed] [Google Scholar]
- 55.Armitage JO, Weisenburger DD, Hutchins M, et al. Chemotherapy for diffuse large-cell lymphoma: rapidly responding patients have more durable remissions. J Clin Oncol. 1986;4:160–164. doi: 10.1200/JCO.1986.4.2.160. [DOI] [PubMed] [Google Scholar]
- 56.Birchard KR, Hoang JK, Herndon JE, Jr, Patz EF., Jr Early changes in tumor size in patients treated for advanced stage nonsmall cell lung cancer do not correlate with survival. Cancer. 2009;115:581–586. doi: 10.1002/cncr.24060. [DOI] [PubMed] [Google Scholar]
- 57.Melton GB, Lavely WC, Jacene HA, et al. Efficacy of preoperative combined 18-fluorodeoxyglucose positron emission tomography and computed tomography for assessing primary rectal cancer response to neoadjuvant therapy. J Gastrointest Surg. 2007;11:961–969. doi: 10.1007/s11605-007-0170-7. [DOI] [PubMed] [Google Scholar]
- 58.Jochelson M, Mauch P, Balikian J, Rosenthal D, Canellos G. The significance of the residual mediastinal mass in treated Hodgkin's disease. J Clin Oncol. 1985;3:637–640. doi: 10.1200/JCO.1985.3.5.637. [DOI] [PubMed] [Google Scholar]
- 59.Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 60.Reinhardt MJ, Herkel C, Altehoefer C, Finke J, Moser E. Computed tomography and 18F-FDG positron emission tomography for therapy control of Hodgkin's and non-Hodgkin's lymphoma patients: when do we really need FDG-PET? Ann Oncol. 2005;16:1524–1529. doi: 10.1093/annonc/mdi271. [DOI] [PubMed] [Google Scholar]
- 61.Juweid ME, Wiseman GA, Vose JM, et al. Response assessment of aggressive non-Hodgkin's lymphoma by integrated International Workshop Criteria and fluorine-18-fluorodeoxyglucose positron emission tomography. J Clin Oncol. 2005;23:4652–4661. doi: 10.1200/JCO.2005.01.891. [DOI] [PubMed] [Google Scholar]
- 62.Bos R, van Der Hoeven JJ, van Der Wall E, et al. Biologic correlates of 18fluorodeoxyglucose uptake in human breast cancer measured by positron emission tomography. J Clin Oncol. 2002;20:379–387. doi: 10.1200/JCO.2002.20.2.379. [DOI] [PubMed] [Google Scholar]
- 63.Brucher BL, Weber W, Bauer M, et al. Neoadjuvant therapy of esophageal squamous cell carcinoma: response evaluation by positron emission tomography. Ann Surg. 2001;233:300–309. doi: 10.1097/00000658-200103000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vansteenkiste JF, Stroobants SG, De Leyn PR, Dupont PJ, Verbeken EK. Potential use of FDG-PET scan after induction chemotherapy in surgically staged IIIa-N2 non-small-cell lung cancer: a prospective pilot study. The Leuven Lung Cancer Group. Ann Oncol. 1998;9:1193–1198. doi: 10.1023/a:1008437915860. [DOI] [PubMed] [Google Scholar]
- 65.Bryant AS, Cerfolio RJ, Klemm KM, Ojha B. Maximum standard uptake value of mediastinal lymph nodes on integrated FDG-PET-CT predicts pathology in patients with non-small cell lung cancer. Ann Thorac Surg. 2006;82:417–422. doi: 10.1016/j.athoracsur.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 66.Dooms C, Verbeken E, Stroobants S, Nackaerts K, De Leyn P, Vansteenkiste J. Prognostic stratification of stage IIIA-N2 non-small-cell lung cancer after induction chemotherapy: a model based on the combination of morphometricpathologic response in mediastinal nodes and primary tumor response on serial 18-fluoro-2-deoxy-glucose positron emission tomography. J Clin Oncol. 2008;26:1128–1134. doi: 10.1200/JCO.2007.13.9550. [DOI] [PubMed] [Google Scholar]
- 67.Humm JL, Rosenfeld A, Del Guerra A. From PET detectors to PET scanners. Eur J Nucl Med Mol Imaging. 2003;30:1574–1597. doi: 10.1007/s00259-003-1266-2. [DOI] [PubMed] [Google Scholar]
- 68.Tatsumi M, Cohade C, Nakamoto Y, Fishman EK, Wahl RL. Direct comparison of FDG PET and CT findings in patients with lymphoma: initial experience. Radiology. 2005;237:1038–1045. doi: 10.1148/radiol.2373040555. [DOI] [PubMed] [Google Scholar]
- 69.Kelloff GJ, Hoffman JM, Johnson B, et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res. 2005;11:2785–2808. doi: 10.1158/1078-0432.CCR-04-2626. [DOI] [PubMed] [Google Scholar]
- 70.Weber WA, Figlin R. Monitoring cancer treatment with PET/CT: does it make a difference? J Nucl Med. 2007;48(suppl 1):36S–44S. [PubMed] [Google Scholar]
- 71.Weber WA. Use of PET for monitoring cancer therapy and for predicting outcome. J Nucl Med. 2005;46:983–995. [PubMed] [Google Scholar]
- 72.Hellwig D, Graeter TP, Ukena D, Georg T, Kirsch CM, Schafers HJ. Value of F-18-fluorodeoxyglucose positron emission tomography after induction therapy of locally advanced bronchogenic carcinoma. J Thorac Cardiovasc Surg. 2004;128:892–899. doi: 10.1016/j.jtcvs.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 73.Port JL, Kent MS, Korst RJ, Keresztes R, Levin MA, Altorki NK. Positron emission tomography scanning poorly predicts response to preoperative chemotherapy in non-small cell lung cancer. Ann Thorac Surg. 2004;77:254–259. doi: 10.1016/s0003-4975(03)01457-7. [DOI] [PubMed] [Google Scholar]
- 74.Cerfolio RJ, Bryant AS, Winokur TS, Ohja B, Bartolucci AA. Repeat FDG-PET after neoadjuvant therapy is a predictor of pathologic response in patients with non-small cell lung cancer. Ann Thorac Surg. 2004;78:1903–1909. doi: 10.1016/j.athoracsur.2004.06.102. [DOI] [PubMed] [Google Scholar]
- 75.Akhurst T, Downey RJ, Ginsberg MS, et al. An initial experience with FDG-PET in the imaging of residual disease after induction therapy for lung cancer. Ann Thorac Surg. 2002;73:259–264. doi: 10.1016/s0003-4975(01)03257-x. [DOI] [PubMed] [Google Scholar]
- 76.Mac Manus MP, Hicks RJ, Matthews JP, Wirth A, Rischin D, Ball DL. Metabolic (FDG-PET) response after radical radiotherapy/chemoradiotherapy for non-small cell lung cancer correlates with patterns of failure. Lung Cancer. 2005;49:95–108. doi: 10.1016/j.lungcan.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 77.Flamen P, Van Cutsem E, Lerut A, et al. Positron emission tomography for assessment of the response to induction radiochemotherapy in locally advanced oesophageal cancer. Ann Oncol. 2002;13:361–368. doi: 10.1093/annonc/mdf081. [DOI] [PubMed] [Google Scholar]
- 78.Swisher SG, Maish M, Erasmus JJ, et al. Utility of PET, CT, and EUS to identify pathologic responders in esophageal cancer. Ann Thorac Surg. 2004;78:1152–1160. doi: 10.1016/j.athoracsur.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 79.Swisher SG, Erasmus J, Maish M, et al. 2-Fluoro-2-deoxy-D-glucose positron emission tomography imaging is predictive of pathologic response and survival after preoperative chemoradiation in patients with esophageal carcinoma. Cancer. 2004;101:1776–1785. doi: 10.1002/cncr.20585. [DOI] [PubMed] [Google Scholar]
- 80.Levine EA, Farmer MR, Clark P, et al. Predictive value of 18-fluoro-deoxy-glucose-positron emission tomography (18F-FDG-PET) in the identification of responders to chemoradiation therapy for the treatment of locally advanced esophageal cancer. Ann Surg. 2006;243:472–478. doi: 10.1097/01.sla.0000208430.07050.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Downey RJ, Akhurst T, Ilson D, et al. Whole body 18FDG-PET and the response of esophageal cancer to induction therapy: results of a prospective trial. J Clin Oncol. 2003;21:428–432. doi: 10.1200/JCO.2003.04.013. [DOI] [PubMed] [Google Scholar]
- 82.Duong CP, Hicks RJ, Weih L, et al. FDG-PET status following chemoradiotherapy provides high management impact and powerful prognostic stratification in oesophageal cancer. Eur J Nucl Med Mol Imaging. 2006;33:770–778. doi: 10.1007/s00259-005-0040-z. [DOI] [PubMed] [Google Scholar]
- 83.Duong CP, Demitriou H, Weih L, et al. Significant clinical impact and prognostic stratification provided by FDG-PET in the staging of oesophageal cancer. Eur J Nucl Med Mol Imaging. 2006;33:759–769. doi: 10.1007/s00259-005-0028-8. [DOI] [PubMed] [Google Scholar]
- 84.Kalff V, Duong C, Drummond EG, Matthews JP, Hicks RJ. Findings on 18F-FDG PET scans after neoadjuvant chemoradiation provides prognostic stratification in patients with locally advanced rectal carcinoma subsequently treated by radical surgery. J Nucl Med. 2006;47:14–22. [PubMed] [Google Scholar]
- 85.Kim MK, Ryu JS, Kim SB, et al. Value of complete metabolic response by 18F-fluorodeoxyglucose-positron emission tomography in oesophageal cancer for prediction of pathologic response and survival after preoperative chemoradiotherapy. Eur J Cancer. 2007;43:1385–1391. doi: 10.1016/j.ejca.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 86.Lin C, Itti E, Haioun C, et al. Early 18F-FDG PET for prediction of prognosis in patients with diffuse large B-cell lymphoma: SUV-based assessment versus visual analysis. J Nucl Med. 2007;48:1626–1632. doi: 10.2967/jnumed.107.042093. [DOI] [PubMed] [Google Scholar]
- 87.Stroobants S, Goeminne J, Seegers M, et al. 18FDG-positron emission tomography for the early prediction of response in advanced soft tissue sarcoma treated with imatinib mesylate (Glivec) Eur J Cancer. 2003;39:2012–2020. doi: 10.1016/s0959-8049(03)00073-x. [DOI] [PubMed] [Google Scholar]
- 88.Kaplan WD. Residual mass and negative gallium scintigraphy in treated lymphoma: when is the gallium scan really negative? J Nucl Med. 1990;31:369–371. [PubMed] [Google Scholar]
- 89.Mikhaeel NG, Hutchings M, Fields PA, O'Doherty MJ, Timothy AR. FDG-PET after two to three cycles of chemotherapy predicts progression-free and overall survival in high-grade non-Hodgkin lymphoma. Ann Oncol. 2005;16:1514–1523. doi: 10.1093/annonc/mdi272. [DOI] [PubMed] [Google Scholar]
- 90.Kasamon YL, Wahl RL, Ziessman HA, et al. Phase II study of risk-adapted therapy of newly diagnosed, aggressive non-Hodgkin lymphoma based on midtreatment FDG-PET scanning. Biol Blood Marrow Transplant. 2009;15:242–248. doi: 10.1016/j.bbmt.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hicks RJ, Mac Manus MP, Matthews JP, et al. Early FDG-PET imaging after radical radiotherapy for non-small-cell lung cancer: inflammatory changes in normal tissues correlate with tumor response and do not confound therapeutic response evaluation. Int J Radiat Oncol Biol Phys. 2004;60:412–418. doi: 10.1016/j.ijrobp.2004.03.036. [DOI] [PubMed] [Google Scholar]
- 92.Mac Manus MP, Hicks RJ, Matthews JP, et al. Positron emission tomography is superior to computed tomography scanning for response-assessment after radical radiotherapy or chemoradiotherapy in patients with non-small-cell lung cancer. J Clin Oncol. 2003;21:1285–1292. doi: 10.1200/JCO.2003.07.054. [DOI] [PubMed] [Google Scholar]
- 93.Cachin F, Prince HM, Hogg A, Ware RE, Hicks RJ. Powerful prognostic stratification by [18F]fluorodeoxyglucose positron emission tomography in patients with metastatic breast cancer treated with high-dose chemotherapy. J Clin Oncol. 2006;24:3026–3031. doi: 10.1200/JCO.2005.04.6326. [DOI] [PubMed] [Google Scholar]
- 94.Hicks RJ. The role of PET in monitoring therapy. Cancer Imaging. 2005;5:51–57. doi: 10.1102/1470-7330.2005.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wahl RL, Siegel BA, Coleman RE, Gatsonis CG. Prospective multicenter study of axillary nodal staging by positron emission tomography in breast cancer: a report of the staging breast cancer with PET Study Group. J Clin Oncol. 2004;22:277–285. doi: 10.1200/JCO.2004.04.148. [DOI] [PubMed] [Google Scholar]
- 96.Smulders SA, Gundy CM, van Lingen A, Comans EF, Smeenk FW, Hoekstra OS, Study Group of Clinical PET Observer variation of 2-deoxy-2-[F-18]fluoro-D-glucose-positron emission tomography in mediastinal staging of non-small cell lung cancer as a function of experience, and its potential clinical impact. Mol Imaging Biol. 2007;9:318–322. doi: 10.1007/s11307-007-0108-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van der Putten L, Hoekstra OS, de Bree R, et al. 2-deoxy-2[F-18]FDG-PET for detection of recurrent laryngeal carcinoma after radiotherapy: interobserver variability in reporting. Mol Imaging Biol. 2008;10:294–303. doi: 10.1007/s11307-008-0154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hutchings M, Mikhaeel NG, Fields PA, Nunan T, Timothy AR. Prognostic value of interim FDG-PET after two or three cycles of chemotherapy in Hodgkin lymphoma. Ann Oncol. 2005;16:1160–1168. doi: 10.1093/annonc/mdi200. [DOI] [PubMed] [Google Scholar]
- 99.Zijlstra JM, Comans EF, van Lingen A, et al. FDG PET in lymphoma: the need for standardization of interpretation—an observer variation study. Nucl Med Commun. 2007;28:798–803. doi: 10.1097/MNM.0b013e3282eff2d5. [DOI] [PubMed] [Google Scholar]
- 100.Krak NC, van der Hoeven JJ, Hoekstra OS, Twisk JW, van der Wall E, Lammertsma AA. Measuring [18F]FDG uptake in breast cancer during chemotherapy: comparison of analytical methods. Eur J Nucl Med Mol Imaging. 2003;30:674–681. doi: 10.1007/s00259-003-1127-z. [DOI] [PubMed] [Google Scholar]
- 101.Graham MM, Peterson LM, Hayward RM. Comparison of simplified quantitative analyses of FDG uptake. Nucl Med Biol. 2000;27:647–655. doi: 10.1016/s0969-8051(00)00143-8. [DOI] [PubMed] [Google Scholar]
- 102.Sugawara Y, Zasadny KR, Neuhoff AW, Wahl RL. Reevaluation of the standardized uptake value for FDG: variations with body weight and methods for correction. Radiology. 1999;213:521–525. doi: 10.1148/radiology.213.2.r99nv37521. [DOI] [PubMed] [Google Scholar]
- 103.Zasadny KR, Wahl RL. Standardized uptake values of normal tissues at PET with 2-[fluorine-18]-fluoro-2-deoxy-D-glucose: variations with body weight and a method for correction. Radiology. 1993;189:847–850. doi: 10.1148/radiology.189.3.8234714. [DOI] [PubMed] [Google Scholar]
- 104.Boellaard R, Krak NC, Hoekstra OS, Lammertsma AA. Effects of noise, image resolution, and ROI definition on the accuracy of standard uptake values: a simulation study. J Nucl Med. 2004;45:1519–1527. [PubMed] [Google Scholar]
- 105.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 106.Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14:43–47. doi: 10.1016/j.gde.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 107.Matsui W, Wang Q, Barber JP, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68:190–197. doi: 10.1158/0008-5472.CAN-07-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huff CA, Matsui W, Smith BD, Jones RJ. The paradox of response and survival in cancer therapeutics. Blood. 2006;107:431–434. doi: 10.1182/blood-2005-06-2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Larson SM, Erdi Y, Akhurst T, et al. Tumor treatment response based on visual and quantitative changes in global tumor glycolysis using PET-FDG imaging: the visual response score and the change in total lesion glycolysis. Clin Positron Imaging. 1999;2:159–171. doi: 10.1016/s1095-0397(99)00016-3. [DOI] [PubMed] [Google Scholar]
- 110.Nakamoto Y, Zasadny KR, Minn H, Wahl RL. Reproducibility of common semi-quantitative parameters for evaluating lung cancer glucose metabolism with positron emission tomography using 2-deoxy-2-[18F]fluoro-D-glucose. Mol Imaging Biol. 2002;4:171–178. doi: 10.1016/s1536-1632(01)00004-x. [DOI] [PubMed] [Google Scholar]
- 111.Francis RJ, Byrne MJ, van der Schaaf AA, et al. Early prediction of response to chemotherapy and survival in malignant pleural mesothelioma using a novel semiautomated 3-dimensional volume-based analysis of serial 18F-FDG PET scans. J Nucl Med. 2007;48:1449–1458. doi: 10.2967/jnumed.107.042333. [DOI] [PubMed] [Google Scholar]
- 112.Guillem JG, Moore HG, Akhurst T, et al. Sequential preoperative fluorodeoxyglucose-positron emission tomography assessment of response to preoperative chemoradiation: a means for determining longterm outcomes of rectal cancer. J Am Coll Surg. 2004;199:1–7. doi: 10.1016/j.jamcollsurg.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 113.Minn H, Zasadny KR, Quint LE, Wahl RL. Lung cancer: reproducibility of quantitative measurements for evaluating 2-[F-18]-fluoro-2-deoxy-D-glucose uptake at PET. Radiology. 1995;196:167–173. doi: 10.1148/radiology.196.1.7784562. [DOI] [PubMed] [Google Scholar]
- 114.Weber WA, Ziegler SI, Thodtmann R, Hanauske AR, Schwaiger M. Reproducibility of metabolic measurements in malignant tumors using FDG PET. J Nucl Med. 1999;40:1771–1777. [PubMed] [Google Scholar]
- 115.Nahmias C, Wahl LM. Reproducibility of standardized uptake value measurements determined by 18F-FDG PET in malignant tumors. J Nucl Med. 2008;49:1804–1808. doi: 10.2967/jnumed.108.054239. [DOI] [PubMed] [Google Scholar]
- 116.Minn H, Clavo AC, Grenman R, Wahl RL. In vitro comparison of cell proliferation kinetics and uptake of tritiated fluorodeoxyglucose and L-methionine in squamous-cell carcinoma of the head and neck. J Nucl Med. 1995;36:252–258. [PubMed] [Google Scholar]
- 117.Sugawara Y, Zasadny KR, Grossman HB, Francis IR, Clarke MF, Wahl RL. Germ cell tumor: differentiation of viable tumor, mature teratoma, and necrotic tissue with FDG PET and kinetic modeling. Radiology. 1999;211:249–256. doi: 10.1148/radiology.211.1.r99ap16249. [DOI] [PubMed] [Google Scholar]
- 118.Krak NC, Boellaard R, Hoekstra OS, Twisk JW, Hoekstra CJ, Lammertsma AA. Effects of ROI definition and reconstruction method on quantitative outcome and applicability in a response monitoring trial. Eur J Nucl Med Mol Imaging. 2005;32:294–301. doi: 10.1007/s00259-004-1566-1. [DOI] [PubMed] [Google Scholar]
- 119.Erdi YE, Macapinlac H, Rosenzweig KE, et al. Use of PET to monitor the response of lung cancer to radiation treatment. Eur J Nucl Med. 2000;27:861–866. doi: 10.1007/s002590000258. [DOI] [PubMed] [Google Scholar]
- 120.Akhurst T, Ng VV, Larson SM, et al. Tumor burden assessment with positron emission tomography with [18-F] 2-fluoro 2-deoxyglucose (FDG PET) modeled in metastatic renal cell cancer. Clin Positron Imaging. 2000;3:57–65. doi: 10.1016/s1095-0397(00)00041-8. [DOI] [PubMed] [Google Scholar]
- 121.Zasadny KR, Kison PV, Francis IR, Wahl RL. FDG-PET determination of metabolically active tumor volume and comparison with CT. Clin Positron Imaging. 1998;1:123–129. doi: 10.1016/s1095-0397(98)00007-7. [DOI] [PubMed] [Google Scholar]
- 122.Benz MR, Allen-Auerbach MS, Eilber FC, et al. Combined assessment of metabolic and volumetric changes for assessment of tumor response in patients with soft-tissue sarcomas. J Nucl Med. 2008;49:1579–1584. doi: 10.2967/jnumed.108.053694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Roedl JB, Halpern EF, Colen RR, Sahani DV, Fischman AJ, Blake MA. Metabolic tumor width parameters as determined on PET/CT predict disease-free survival and treatment response in squamous cell carcinoma of the esophagus. Mol Imaging Biol. 2009;11:54–60. doi: 10.1007/s11307-008-0169-9. [DOI] [PubMed] [Google Scholar]
- 124.Paquet N, Albert A, Foidart J, Hustinx R. Within-patient variability of 18F-FDG: standardized uptake values in normal tissues. J Nucl Med. 2004;45:784–788. [PubMed] [Google Scholar]
- 125.Weber WA, Ott K, Becker K, et al. Prediction of response to preoperative chemotherapy in adenocarcinomas of the esophagogastric junction by metabolic imaging. J Clin Oncol. 2001;19:3058–3065. doi: 10.1200/JCO.2001.19.12.3058. [DOI] [PubMed] [Google Scholar]
- 126.Ott K, Fink U, Becker K, et al. Prediction of response to preoperative chemotherapy in gastric carcinoma by metabolic imaging: results of a prospective trial. J Clin Oncol. 2003;21:4604–4610. doi: 10.1200/JCO.2003.06.574. [DOI] [PubMed] [Google Scholar]
- 127.Sasaki R, Komaki R, Macapinlac H, et al. [18F]fluorodeoxyglucose uptake by positron emission tomography predicts outcome of non-small-cell lung cancer. J Clin Oncol. 2005;23:1136–1143. doi: 10.1200/JCO.2005.06.129. [DOI] [PubMed] [Google Scholar]
- 128.Hamberg LM, Hunter GJ, Alpert NM, Choi NC, Babich JW, Fischman AJ. The dose uptake ratio as an index of glucose metabolism: useful parameter or oversimplification? J Nucl Med. 1994;35:1308–1312. [PubMed] [Google Scholar]
- 129.Ceresoli GL, Chiti A, Zucali PA, et al. Early response evaluation in malignant pleural mesothelioma by positron emission tomography with [18F]fluorodeoxyglucose. J Clin Oncol. 2006;24:4587–4593. doi: 10.1200/JCO.2006.06.8999. [DOI] [PubMed] [Google Scholar]
- 130.Avril N, Sassen S, Schmalfeldt B, et al. Prediction of response to neoadjuvant chemotherapy by sequential F-18-fluorodeoxyglucose positron emission tomography in patients with advanced-stage ovarian cancer. J Clin Oncol. 2005;23:7445–7453. doi: 10.1200/JCO.2005.06.965. [DOI] [PubMed] [Google Scholar]
- 131.Spaepen K, Stroobants S, Dupont P, et al. [18F]FDG PET monitoring of tumour response to chemotherapy: does [18F]FDG uptake correlate with the viable tumour cell fraction? Eur J Nucl Med Mol Imaging. 2003;30:682–688. doi: 10.1007/s00259-003-1120-6. [DOI] [PubMed] [Google Scholar]
- 132.Engles JM, Quarless SA, Mambo E, Ishimori T, Cho SY, Wahl RL. Stunning and its effect on 3H-FDG uptake and key gene expression in breast cancer cells undergoing chemotherapy. J Nucl Med. 2006;47:603–608. [PubMed] [Google Scholar]
- 133.Dehdashti F, Flanagan FL, Mortimer JE, Katzenellenbogen JA, Welch MJ, Siegel BA. Positron emission tomographic assessment of “metabolic flare” to predict response of metastatic breast cancer to antiestrogen therapy. Eur J Nucl Med. 1999;26:51–56. doi: 10.1007/s002590050359. [DOI] [PubMed] [Google Scholar]
- 134.Higashi K, Clavo AC, Wahl RL. In vitro assessment of 2-fluoro-2-deoxy-D-glucose, L-methionine and thymidine as agents to monitor the early response of a human adenocarcinoma cell line to radiotherapy. J Nucl Med. 1993;34:773–779. [PubMed] [Google Scholar]
- 135.Dai KS, Tai DY, Ho P, et al. Accuracy of the EasyTouch blood glucose self-monitoring system: a study of 516 cases. Clin Chim Acta. 2004;349:135–141. doi: 10.1016/j.cccn.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 136.Pottgen C, Levegrun S, Theegarten D, et al. Value of 18F-fluoro-2-deoxy-D-glucose-positron emission tomography/computed tomography in non-small-cell lung cancer for prediction of pathologic response and times to relapse after neoadjuvant chemoradiotherapy. Clin Cancer Res. 2006;12:97–106. doi: 10.1158/1078-0432.CCR-05-0510. [DOI] [PubMed] [Google Scholar]
- 137.Gayed I, Vu T, Iyer R, et al. The role of 18F-FDG PET in staging and early prediction of response to therapy of recurrent gastrointestinal stromal tumors. J Nucl Med. 2004;45:17–21. [PubMed] [Google Scholar]
- 138.Ott K, Herrmann K, Lordick F, et al. Early metabolic response evaluation by fluorine-18 fluorodeoxyglucose positron emission tomography allows in vivo testing of chemosensitivity in gastric cancer: long-term results of a prospective study. Clin Cancer Res. 2008;14:2012–2018. doi: 10.1158/1078-0432.CCR-07-0934. [DOI] [PubMed] [Google Scholar]
- 139.Jacene HA, Filice R, Kasecamp W, Wahl RL. 18F-FDG PET/CT for monitoring the response of lymphoma to radioimmunotherapy. J Nucl Med. 2009;50:8–17. doi: 10.2967/jnumed.108.055376. [DOI] [PubMed] [Google Scholar]
- 140.Tatsumi M, Cohade C, Nakamoto Y, Wahl RL. Fluorodeoxyglucose uptake in the aortic wall at PET/CT: possible finding for active atherosclerosis. Radiology. 2003;229:831–837. doi: 10.1148/radiol.2293021168. [DOI] [PubMed] [Google Scholar]
- 141.Hicks RJ, Kalff V, MacManus MP, et al. The utility of 18F-FDG PET for suspected recurrent non-small cell lung cancer after potentially curative therapy: impact on management and prognostic stratification. J Nucl Med. 2001;42:1605–1613. [PubMed] [Google Scholar]
- 142.Hoekstra CJ, Stroobants SG, Smit EF, et al. Prognostic relevance of response evaluation using [18F]-2-fluoro-2-deoxy-D-glucose positron emission tomography in patients with locally advanced non-small-cell lung cancer. J Clin Oncol. 2005;23:8362–8370. doi: 10.1200/JCO.2005.01.1189. [DOI] [PubMed] [Google Scholar]
- 143.Hicks RJ. Time and again, children resemble their parents. J Nucl Med. 2008;49:1577–1578. doi: 10.2967/jnumed.108.052142. [DOI] [PubMed] [Google Scholar]
- 144.Blum R, Prince HM, Hicks RJ, Patrikeos A, Seymour J. Discordant response to chemotherapy detected by PET scanning: unveiling of a second primary cancer. Am J Clin Oncol. 2002;25:368–370. doi: 10.1097/00000421-200208000-00010. [DOI] [PubMed] [Google Scholar]
- 145.Ford R, Schwartz L, Dancey J, et al. Lessons learned from independent central review. Eur J Cancer. 2009;45:268–274. doi: 10.1016/j.ejca.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 146.MacManus MP, Seymour JF, Hicks RJ. Overview of early response assessment in lymphoma with FDG-PET. Cancer Imaging. 2007;7:10–18. doi: 10.1102/1470-7330.2007.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ng AP, Wirth A, Seymour JF, et al. Early therapeutic response assessment by 18FDG-positron emission tomography during chemotherapy in patients with diffuse large B-cell lymphoma: isolated residual positivity involving bone is not usually a predictor of subsequent treatment failure. Leuk Lymphoma. 2007;48:596–600. doi: 10.1080/10428190601099965. [DOI] [PubMed] [Google Scholar]
- 148.Hicks RJ, Kalff V, MacManus MP, et al. 18F-FDG PET provides high-impact and powerful prognostic stratification in staging newly diagnosed non-small cell lung cancer. J Nucl Med. 2001;42:1596–1604. [PubMed] [Google Scholar]