

Abstract
Guidance from the Food and Drug Administration on drug interaction studies does not include a specific section on contributions of metabolites to observed inhibitory drug-drug interactions, and the quantitative role of drug metabolites in inhibitory drug-drug interactions is not presently known. The current work was undertaken to evaluate what fraction of inhibitors of common drug metabolizing enzymes (cytochrome P450 (P450) 1A2, 2E1, 2D6, 2C9, 2C19, 2C8, 2B6 and 3A4) have circulating metabolites that may contribute to observed in vivo interactions. A literature analysis was conducted using the Metabolism and Transport Drug Interaction Database to identify all precipitants (i.e. inhibitors) that cause more than 20 percent increase in the area under plasma concentration-time curve (AUC) of marker substrates. The database, PubMed and product labels were then used to determine whether circulating metabolites were present after administration of these inhibitors. Of the total of 129 inhibitors identified, 106 were confirmed to have metabolites that circulate in plasma. An additional 14 inhibitors were identified that are extensively metabolized but whose metabolites have either not been identified or investigated. Hence, only 7% of the inhibitors did not have circulating metabolites. Of the 21 potent inhibitors (≥ 5-fold increase in AUC) currently known, 17 had circulating metabolites and the remaining 4 were all extensively metabolized. Based on available in vitro data, 24 of all the inhibitors are mechanism-based inactivators of P450 enzymes while 105 were characterized as reversible inhibitors. In vitro evaluation of inhibition potential was conducted for only 32% of the circulating metabolites of the inhibitors. In conclusion, circulating metabolites are often present with inhibitors of P450 enzymes suggesting a need for increased efforts to characterize the inhibitory potency of metabolites of candidate drugs, and for newer models for in vitro to in vivo extrapolations.
Introduction
The FDA currently recommends that the development of new drug entities include the evaluation of both the parent compound as well as major metabolites (>10% of parent drug systemic exposure at steady state) to assess their potential to cause general toxicity, genotoxicity, alterations in embryo-fetal development, and carcinogenicity (1). This guidance is based on the possibility that certain metabolites may be formed in humans but not in animals and hence toxicities caused by these metabolites could be missed in preclinical studies. In contrast to toxicity studies, evaluation of drug-drug interactions does not rely on preclinical animal studies and the FDA guidance on drug interaction studies does not specifically address evaluation of the potential of metabolites to cause drug-drug interactions except in the case of prodrugs (2). This situation may be related to the fact that the FDA guidance recommends a sequential in vitro-in vivo approach. The potential of new entities to inhibit drug metabolizing enzymes or transporters is first characterized in vitro (using microsomal or recombinant systems or hepatocytes) and this information is used to optimize the design of the most relevant in vivo studies. The problem is that knowledge about relevant circulating plasma metabolites is not available in the early (in vitro) stages of evaluation of drug interaction liability of candidate compounds. Also, information about circulating plasma metabolites is not generated in a systematic fashion during phase I studies. In many cases, it becomes available in the later clinical stages of drug evaluation. It appears that there is a need for a consensus on a rational approach to the characterization of the roles of circulating metabolites in relation to the conduct of in vivo inhibition studies. Considering that metabolites are, in essence, novel chemical entities, it seems prudent that major metabolites be evaluated for interactions with both the pharmacologic target, as well as off-target interactions that may result in unpredicted toxicity or drug-drug interactions (3).
Another dimension of this issue concerns various predictions based on in vitro inhibition measurements. As mentioned, during early development, prediction of the in vivo inhibitory potential of a new candidate compound with the goal of understanding the magnitude of in vivo interaction tends to be based solely on parent drug behavior. Even in large in vitro-in vivo correlation studies focused on the development of theoretical prediction methods, approaches that include the contributions on inhibitory circulating metabolites have not been developed (4, 5) although theory for effect of multiple inhibitors has been presented briefly (6).
The aim of this analysis was to evaluate the extent to which circulating metabolites are present in clinically observed drug-drug interactions. The analysis first identified inhibitors that have known circulating metabolites. The inhibitors were classified according to the FDA guidance and when possible the mechanism of inhibition based on in vitro data was determined. This analysis found that more than 80% of inhibitors including the large majority of potent inhibitors have circulating metabolites.
Literature search strategy
The Metabolism and Transport Drug Interaction Database® (MTDI database: http://www.druginteractioninfo.org) was queried to retrieve all reported in vivo interactions (defined as resulting in a ≥ 20% increase in the AUC or decrease in clearance of the victim drug). From the resulting list of in vivo inhibitors, the following were excluded: herbal and food products, biologicals, combination therapies (oral contraceptives, ritonavir boosted formulations), drugs withdrawn from the market and transporter inhibitors. This list of identified inhibitors was then examined for evidence of circulating metabolites. Additional searches of the MTDI database, product labels (recently approved drugs and those with no published pharmacokinetic studies) and PubMed were conducted to determine the major routes of elimination of the inhibitors, and to retrieve studies that had either detected the metabolites in vivo or had developed an appropriate assay for potential metabolites but did not detect the metabolites in vivo. The results of these three searches were combined and the resulting inhibitors were divided into three groups: 1) those for which known metabolites were quantified in plasma, 2) those that were extensively metabolized but plasma concentration data of metabolites was not available and 3) those that were mainly renally cleared or for which the absence of circulating metabolites was confirmed. In the next phase of the analysis, a list of circulating metabolites was compiled and the MTDI database was systematically searched for in vitro data on inhibition of any P450 enzymes by these metabolites. A search was also conducted to summarize all of the available in vitro inhibition data for the identified inhibitors with a focus on mechanism of inhibition. A drug was listed as a mechanism-based inhibitor if any studies showing enzyme inactivation were found, even if the majority of in vitro studies had characterized the inhibitor as a reversible inhibitor or if only one of multiple P450s inhibited was via irreversible inactivation.
The inhibitors that had available clearance or AUC data from marker substrates in the presence and absence of the inhibitor were classified according to the FDA recommended system (www.fda.gov/cder/drug/drugInteractions/) as potent (≥ 5-fold increase in AUC), moderate (≥ 2 but <5-fold increase in AUC), or weak (≥ 1.25 but <2-fold increase in AUC) inhibitors of the target enzyme.
Results
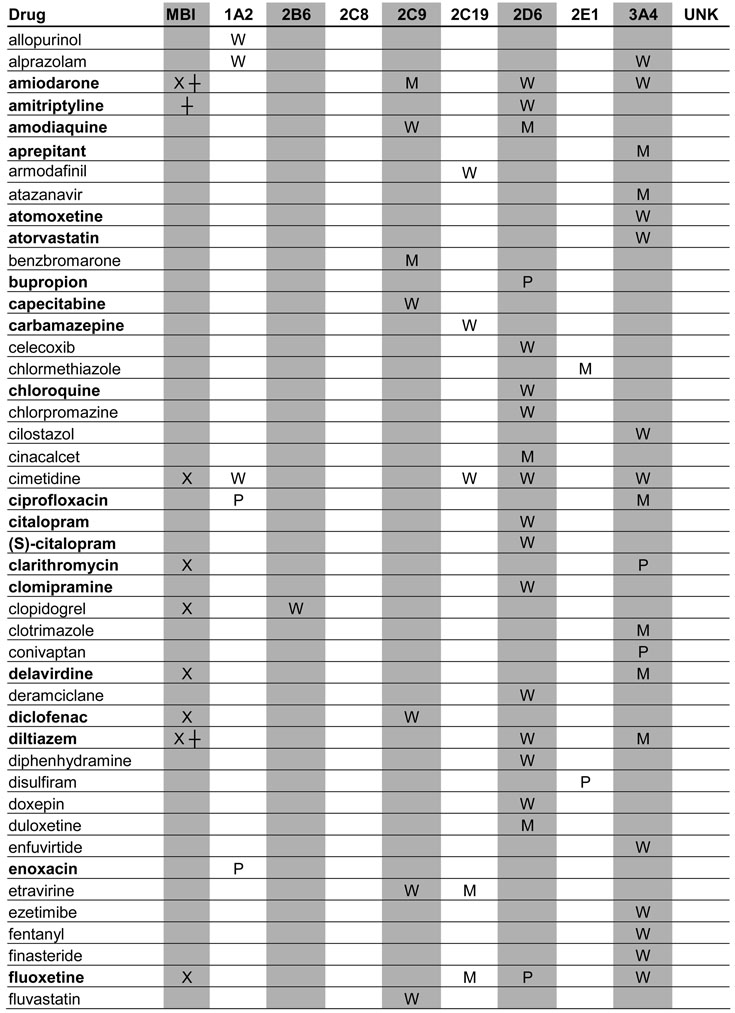
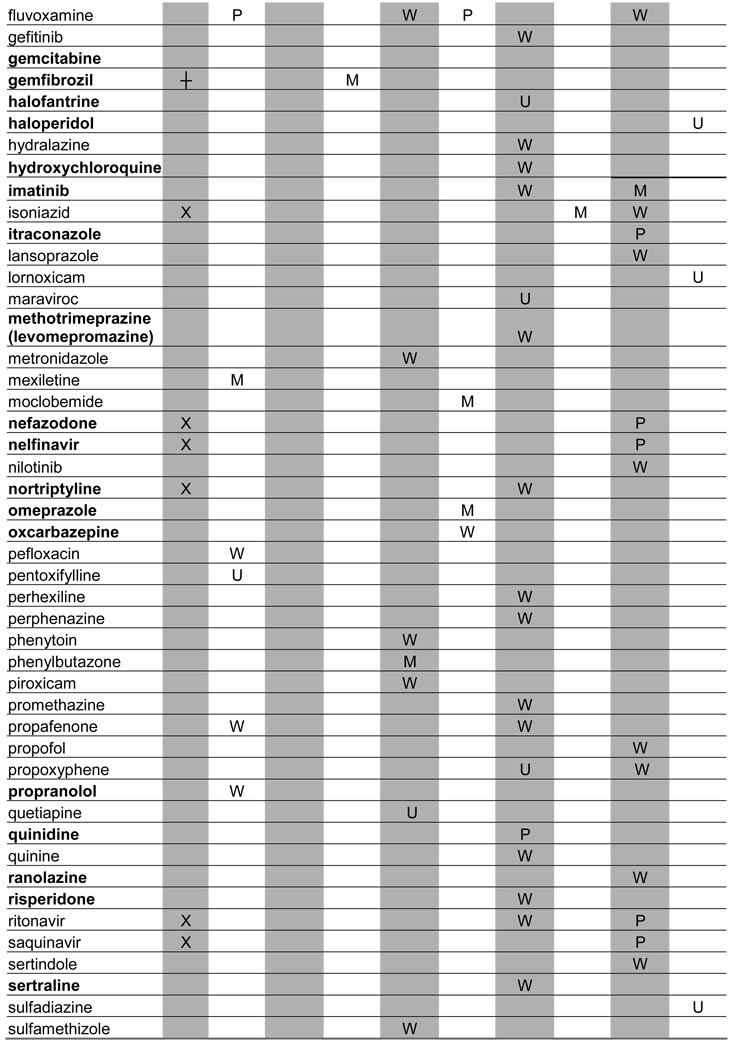
A total of 129 metabolic inhibitors were identified. The vast majority of these inhibitors, 106 (82%), were confirmed to have metabolites that circulate in plasma (Table 1). For the purposes of this study, the exact exposure for these metabolites in relation to the parent compound (AUC or Css ratio at steady state) was not recorded. An additional 14 inhibitors (11%) were considered likely to have circulating metabolites due to extensive metabolism, but no information was found regarding the presence or absence of circulating metabolites (Table 2). Lack of information was most often due to the fact that the main elimination pathways were not identified for these drugs. For example, the metabolites of ketoconazole and some of the protease inhibitors have not been structurally characterized even though these drugs are mainly cleared metabolically. In some cases, metabolites had been quantified in urine but not been analyzed in plasma. Interestingly, two classical mechanism-based inactivators of P450 3A4, troleandomycin and erythromycin, were also in the group of inhibitors with uncharacterized metabolites: despite the fact that metabolites are known to be involved in the formation of metabolic-intermediate (MI) complexes from these drugs (7), no data was found regarding the presence of circulating intermediate metabolites in vivo. Only 7% of all the identified inhibitors were confirmed to be devoid of circulating metabolites (Table 3). This group included those drugs that are predominantly renally cleared such as fluconazole, or drugs like posaconazole, which is metabolized (glucuronidated) but the metabolites are not detected in plasma.
Table 1.
Profiles of in vivo P450 inhibition by drugs with confirmed circulating metabolites
 |
 |
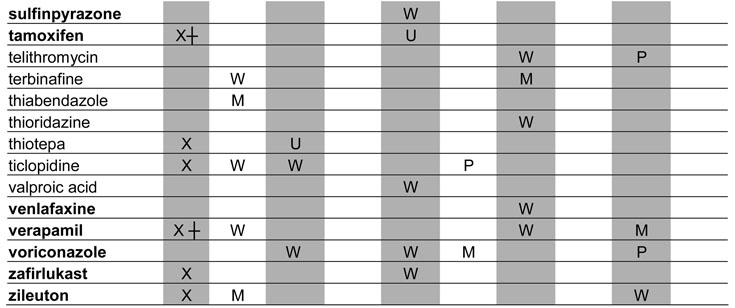 |
BOLD: Metabolites have been investigated in vitro
UNK: The identity of inhibited enzyme not specified due to lack of marker studies
X: Parent compound is an MBI
┼: Metabolite is an MBI
U: Unclassified
P: Potent inhibitor
M: Moderate inhibitor
W: Weak inhibitor
Note; percent of parent drug exposure associated with metabolites were not assessed.
Table 2.
Profiles of in vivo P450 inhibition for drugs that are extensively metabolized but whose metabolites were either not indentified or not studied.
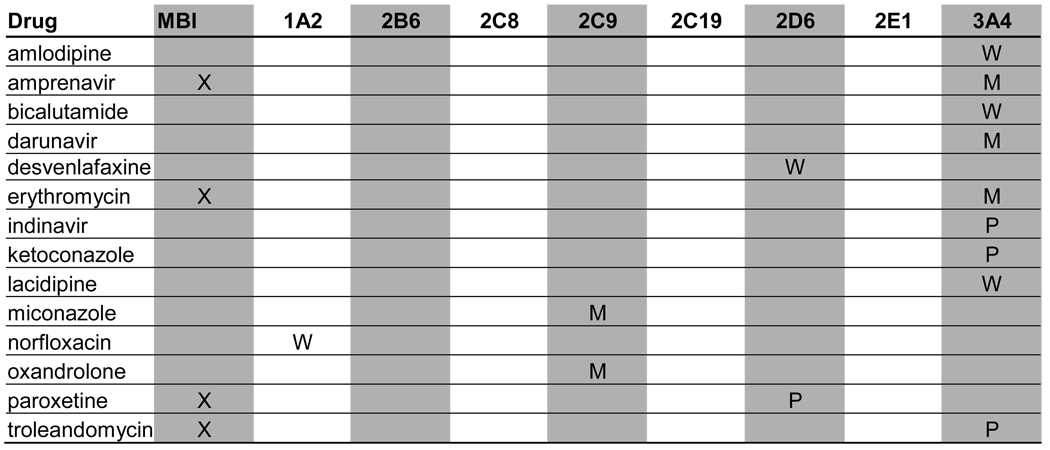 |
X: Parent compound is an MBI
M: Moderate inhibitor
P: Potent inhibitor
W: Weak inhibitor
Table 3.
Profiles of in vivoP450 inhibition for drugs that are renally cleared or metabolized but whose metabolites were not detected in plasma.
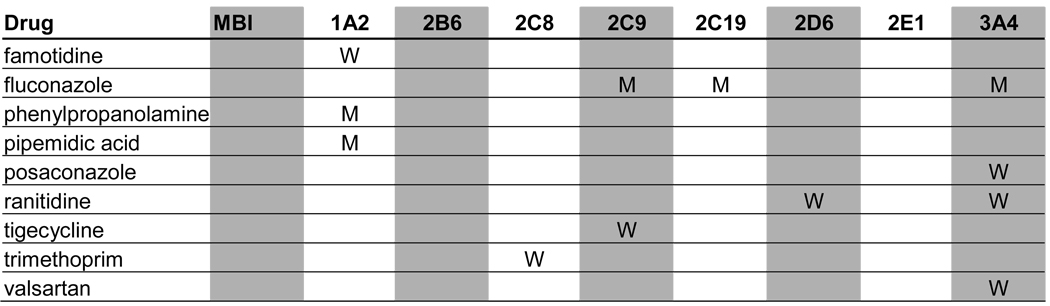 |
M: Moderate inhibitor
W: Weak inhibitor
When possible, the identified inhibitors were classified according to the FDA classification system as potent, moderate or weak. The target enzymes for each inhibitor and classification of their inhibitory potency in vivo are presented in Tables 1–3. Of the 21 potent inhibitors currently known, 17 were confirmed to have circulating metabolites. The remaining 4 potent inhibitors, ketoconazole, indinavir, paroxetine and troleandomycin, are all extensively metabolized. No potent inhibitor was found to be devoid of circulating metabolites.
Metabolic processes are, by definition, essential for mechanism-based inhibition of enzymes (7) and hence, mechanism-based inhibitors represent a special group of precipitants with possible circulating metabolites. For example, paroxetine and troleandomycin are known mechanism-based inactivators of P450 2D6 and P450 3A4, respectively, and it is clear that metabolism is necessary for the inactivation of P450s by these compounds. However, no data are available regarding circulating metabolites of these compounds and therefore, the role of metabolites in in vivo P450 inactivation is unknown.
To better determine the frequency of mechanism-based inactivation, the MTDI database was queried for in vitro inactivation data pertaining to the identified inhibitors. Based on these in vitro data, 24 mechanism-based inactivators of P450 enzymes were identified representing 19% of all inhibitors. Eight (33%) of these 24 mechanism based inhibitors caused potent interactions in vivo, and 38% of all the known potent inhibitors were mechanism-based inhibitors in in vitro studies. The mechanism-based inhibitors (inactivation by parent compound and/or metabolite) are denoted in Tables 1–3.
Finally, we investigated whether in vitro enzyme inhibition data were available for those metabolites of inhibitors that circulate in vivo. Despite the fact that these metabolites are known to circulate in plasma, only 34 of the metabolites (32%) had been investigated in vitro as potential inhibitors. The inhibitors whose metabolites have been studied in vitro are indicated in boldface in Table 1.
Discussion and Conclusion
The critical finding of this review is that the vast majority of clinically important inhibitors possess circulating metabolites. It is possible that only a fraction of these metabolites contribute to observed P450 inhibition in vivo and some of these metabolites may not circulate at concentrations comparable to the parent compound. However, the abundance of circulating metabolites provides a challenge to in vitro - in vivo correlation analyses and rationalization of in vivo interactions. Due to the lack of specific in vitro enzyme inhibition data and in vivo pharmacokinetic data concerning the metabolites, the overall quantitative role of these circulating metabolites cannot be accurately evaluated. To improve our understanding of the P450 inhibition caused by metabolites in vivo, more information is needed on the inhibitory potency of the metabolites towards the P450 that is subject to the clinical interaction, the unbound fraction of the metabolite, the elimination rate and mechanisms of elimination of these metabolites in vivo, the identity of predominant enzyme that form the metabolites and the impact of the metabolite on kinetics of the parent drug.
For some parent-metabolite pairs, the formation of the metabolite is independent of the inhibited pathway and the metabolite may be the sole cause of the clinical interactions. For example, gemfibrozil glucuronide, generated by the UGT conjugation pathway, inactivates P450 2C8, an enzyme unrelated to the formation of the metabolite (8). In other cases the metabolites are formed by the inhibited enzyme and inhibit this same enzyme. For example, the circulating metabolites of itraconazole (hydroxy-, keto and N-desalkyl- itraconaozle) are all formed by P450 3A4, the enzyme that is the target of itraconazole inhibition in vivo (9). In a similar manner, norfluoxetine is formed by P450 2D6, the enzyme that is potently inhibited in vivo by fluoxetine, the parent drug (10). In both cases the metabolites inhibit the same enzyme as the parent drug, potentially increasing the total magnitude of interaction. Additionally, formation of these inhibitory metabolites is a major route of elimination of itraconazole and fluoxetine and the metabolites may inhibit the elimination of the parent drug and lead to nonlinear kinetics, thereby complicating analysis of the in vivo data and requiring complex models of in vitro to in vivo extrapolations.
It is tempting to use the metabolite-to-parent plasma concentration ratio as an indicator of the importance of metabolites in a specific interaction. However, without detailed in vitro and in vivo data, it is impossible to conclude that a metabolite is an inhibitor in vivo, even if it is formed by the same enzyme involved in the clinically observed inhibition. The relative contribution of a metabolite in a given interaction will depend on the free plasma concentration and the unbound Ki of the metabolite and the relationship of these values to those of the parent drug. Most metabolites have different unbound fractions than the parent drug in in vivo and in vitro systems impacting the predicted in vivo contribution of the metabolites (11). Thus, simple concentration or AUC ratios of metabolite-to-parent may be misleading. As a result, even if a circulating metabolite is inhibitory it does not necessarily contribute to interactions associated with its parent drug. A good example of this is atomoxetine, which possesses several inhibitory metabolites but these metabolites are not predicted to contribute to in vivo interactions with P450 3A4 (12).
The best evidence of the potentially important contribution of metabolites in clinical drug-drug interactions can be obtained in cases where the metabolite kinetics are characterized as elimination rate-limited instead of formation rate-limited. In such cases, the metabolite persists in circulation after the parent compound has been cleared and the inhibitory effects of the metabolite can be studied in the absence of the parent compound. The contribution of nor-fluoxetine to P450 2D6 mediated clearance of desipramine has been shown after washout of fluoxetine (13).
In conclusion, this review highlights the fact that circulating metabolites are often present with inhibitors of P450 enzymes. The fact that circulating metabolites are so common has at least two consequences: first, it suggests the need for more vigorous experimental research on the inhibitory potency of metabolites of candidate drugs, and second, it encourages the development of better mathematical models for in vitro to in vivo extrapolation that account for multi-inhibitor systems.
Acknowledgments
This work was partially supported by NIH grant P01 GM32165 (NI) and the Elmer J. and Joy B. Plein Fellowship for Excellence in Pharmacy Education (CKY).
Abbreviations
- AUC
area under plasma concentration-time curve
- FDA
Food and Drug Administration
- MBI
Mechanism Based Inhibitor
- MTDI database
Metabolism and Transport Drug Interaction Database®
- P450
cytochrome P450
References
- 1.U.S. Food and Drug Administration. Guidance for Industry. Safety Testing of Drug Metabolites. 2008. [Google Scholar]
- 2.U.S. Food and Drug Adminstration. Guidance for Industry Drug Interaction Studies — Study Design, Data Analysis, and Implications for Dosing and Labeling. Draft Guidance. 2006. [Google Scholar]
- 3.Smith DA, Obach RS. Metabolites and Safety: What Are the Concerns, and How Should We Address Them? Chem. Res. Toxicol. 2006;19:1570–1579. doi: 10.1021/tx0602012. [DOI] [PubMed] [Google Scholar]
- 4.Brown HS, Galetin A, Hallifax D, Houston JB. Prediction of in Vivo Drug-Drug Interactions from in Vitro Data: Factors Affecting Prototypic Drug-Drug Interactions Involving CYP2C9, CYP2D6 and CYP3A4. Clin. Pharmacokinet. 2006;45:1035–1050. doi: 10.2165/00003088-200645100-00006. [DOI] [PubMed] [Google Scholar]
- 5.Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, Tremaine LM. The Utility of in Vitro Cytochrome P450 Inhibition Data in the Prediction of Drug-Drug Interactions. J. Pharmacol. Exp. Ther. 2006;316:336–348. doi: 10.1124/jpet.105.093229. [DOI] [PubMed] [Google Scholar]
- 6.Rostami-Hodjegan A, Tucker G. 'in Silico' Simulations to Assess the 'in Vivo' Consequences of 'in Vitro' Metabolic Drug-Drug Interaction. Drug Discovery Today: Technologies. 2004;1:441–448. doi: 10.1016/j.ddtec.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Kalgutkar AS, Obach RS, Maurer TS. Mechanism-Based Inactivation of Cytochrome P450 Enzymes: Chemical Mechanisms, Structure-Activity Relationships and Relationship to Clinical Drug-Drug Interactions and Idiosyncratic Adverse Drug Reactions. Curr. Drug. Metab. 2007;8:407–447. doi: 10.2174/138920007780866807. [DOI] [PubMed] [Google Scholar]
- 8.Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, Toren P, Parkinson A. Glucuronidation Converts Gemfibrozil to a Potent, Metabolism-Dependent Inhibitor of CYP2C8: Implications for Drug-Drug Interactions. Drug. Metab. Dispos. 2006;34:191–197. doi: 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- 9.Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. Role of Itraconazole Metabolites in CYP3A4 Inhibition. Drug. Metab. Dispos. 2004;32:1121–1131. doi: 10.1124/dmd.104.000315. [DOI] [PubMed] [Google Scholar]
- 10.Stevens JC, Wrighton SA. Interaction of the Enantiomers of Fluoxetine and Norfluoxetine with Human Liver Cytochromes P450. J. Pharmacol. Exp. Ther. 1993;266:964–971. [PubMed] [Google Scholar]
- 11.Templeton IE, Thummel KE, Kharasch ED, Kunze KL, Hoffer C, Nelson WL, Isoherranen N. Contribution of Itraconazole Metabolites to Inhibition of CYP3A4 in Vivo. Clin. Pharmacol. Ther. 2008;83:77–85. doi: 10.1038/sj.clpt.6100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sauer JM, Long AJ, Ring B, Gillespie JS, Sanburn NP, DeSante KA, Petullo D, VandenBranden MR, Jensen CB, Wrighton SA, Smith BP, Read HA, Witcher JW. Atomoxetine Hydrochloride: Clinical Drug-Drug Interaction Prediction and Outcome. J. Pharmacol. Exp. Ther. 2004;308:410–418. doi: 10.1124/jpet.103.058727. [DOI] [PubMed] [Google Scholar]
- 13.Preskorn SH, Alderman J, Chung M, Harrison W, Messig M, Harris S. Pharmacokinetics of desipramine coadministered with sertraline or fluoxetine. J. Clin. Psychopharmacol. 1994;14:90–98. [PubMed] [Google Scholar]