


Abstract
The chemical synthesis of various acylaminoacylated mononucleotides is described and their activities as donor substrates for the ribosomal peptide synthesis were investigated using PhetRNAPhe as an acceptor. This minimal reaction was characterized in detail and was shown to be stimulated by CMP, cytidine and cytosine. By using several cytidine and cytosine analogs evidence is provided that this enhancement is rather caused by base pairing to rRNA, followed by a structural change, than by a base mediated general acid/base catalysis. Only derivatives of AMP proved active as P-site substrates. Further, a significant contribution of the 2′-OH to activity was indicated by the finding that AcLeu-dAMP was inactive as donor substrate, although it is a good inhibitor of peptide bond formation and thus, is presumably bound to the P-site. However, Di(AcLeu)-2′-OCH3-Ade and DiAcLeu-AMP were moderately active in this assay suggesting that the reactivity of the 3′-acylaminoacid ester is stimulated by the presence of the 2′-oxygen group. A model is discussed how further interactions of the 2′-OH in the transition state might influence peptidyl transferase activity.
INTRODUCTION
Peptide synthesis is accomplished on the ribosome by binding of peptidyl- and aminoacyl-tRNA in the P-site and A-site, respectively. The substrate specificity of the peptidyl transferase activity is conveniently investigated by using simple substrate analogs based on the charged CCA termini of either aminoacyl- or peptidyl-tRNAs. It was established early on by Monro and coworkers (1,2) that 2′(3′)-O-(N-acylaminoacyl) derivatives of CCA oligonucleotides react as peptide donors on the ribosome with puromycin as an acceptor substrate. This fragment reaction was carried out by 50S subunits and was dependent on monovalent and divalent cations and alcohol. The alcohol apparently promotes the interaction of the donor substrate with the ribosome since the fragments do not bind to the ribosome under normal conditions (1). As these small analogs allow resolving peptidyl transferase from other steps of translation, exhaustive studies on the interaction of these tRNA analogs in the A-site and P-site of the ribosome have been undertaken (3–7). One of the results obtained already in these early studies was the importance of at least one cytidine residue in addition to the terminal adenosine for binding to the ribosome in both sites (7,8). These findings were further corroborated by resourceful genetic experiments (9,10) and recent X-ray analysis of tRNA analogs soaked into 50S subunits (11,12), which demonstrated Watson–Crick base pairing of the penultimate C of the 3′-CCA end of tRNA in both sites. However, the minimal substrate requirement for peptidyl transferase activity was found to be a 2′(3′)-O-(N-acylaminoacyl)adenosine 5′-phosphate at the P-site (13) and a 2′(3′)-O-aminoacyladenosine 5′-phosphate at the A-site (14), provided high concentrations of substrates were used. The activity of these simple P-site substrates could be significantly enhanced by addition of CMP in trans (8).
In our effort to characterize ribosomal peptidyl transferase, we used this simple system to investigate the influence of particular chemical groups of the P-site terminal adenosine and its attached acylamino acid. The chemical synthesis of these 2′(3′)-O-(N-acylaminoacyl)adenosine 5′-phosphate substrates yielded reasonable amounts and is therefore more straightforward than the synthesis of equivalently modified amino acyl-tRNAs. In this paper we describe the synthesis of substrates and optimization of their reaction with [14C]Phe-tRNAPhe on 50S subunits. By using various cytosine and cytidine analogs for enhancing the activity, we provide evidence that the stimulatory effect of these substances is caused by base pairing to rRNA rather than by general acid/base catalysis. Furthermore, the analysis of modified 2′-OH P-site substrates indicates the importance of this chemical group for the reactivity of the vicinal 3′-acylaminoacylester. During peptide bond formation this reactivity might be altered by various interactions of the 2′-OH. Taking theoretical considerations and recent X-ray structures of substrates bound to ribosomes into account, a model is discussed how the 2′-OH might influence the peptidyl transferase reaction.
MATERIALS AND METHODS
50S ribosomes were prepared from Escherichia coli strain D10-CAN20-12E according to protocols from (15). Yeast tRNAPhe (Boehringer Mannheim) was charged with purified yeast Phe-tRNA synthetase, which was kindly provided by Dr Hans Sternbach (Max-Planck-Institute, Göttingen, Germany) and Dr Franco Fasiolo (CNRS, Strasbourg, France). N-Acetylation of PhetRNAPhe and HPLC purification of PhetRNAPhe and AcPhetRNAPhe was done as described (15). The internal standard, Ac[14C]Phe, was prepared by acetylation of [14C]Phe (∼1100 d.p.m./pmol) with acetic anhydride (16). Cytosine, cytidine and their derivatives were purchased from Sigma. Stock solutions (100 mM) of cytidine, 3-methylcytidine and 5-methylcytidine were adjusted to pH 7.0. For cytosine, isocytosine and 5-fluorocytosine stocks, nucleobases were dissolved in 75% DMSO and the pH adjusted to 7.0; final concentration of DMSO in reactions was 7.5%. 2′-OCH3-Adenosine was purchased from Sigma or RI Chemicals. Sparsomycin was obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, Maryland, USA.
Synthesis of P-site substrates and reaction conditions
The chemical monoaminoacylation of AMP, dAMP, CMP, IMP and GMP was accomplished by reacting the nucleotide 5′-phosphate with the cyanomethyl ester of the appropriate N-acetyl-amino acid according to (17) yielding the desired 2′(3′)-O-(N-acylaminoacyl) nucleotide, which was purified by reversed phase HPLC as described therein. Diaminoacyla tion was achieved by using 1,1′-carbonyldiimidazole and N-acyl-amino acid yielding 5′-phospho-2′,3′-Di-O-(N-acylaminoacyl)-adenosine. For solubility reasons the tetrabutylammonium salt of AMP (TBA.AMP) was prepared by ion exchange chromatography (Dowex 50Wx4) (17). In a typical reaction, 0.2 mmol TBA.AMP were suspended in 0.7 ml anhydrous DMSO and kept at room temperature during amino acid activation. A solution containing 1.9 mmol N-acetylated amino acid and 2.2 mmol 1,1′-carbonyldiimidazole in 1 ml anhydrous DMSO was stirred at room temperature for 1 h and then treated with 0.2 mmol TBA.AMP (in 0.7 ml anhydrous DMSO). The reaction mixture was stirred for 2–3 h at 55°C. The clear yellow solution was directly subjected to reversed phase HPLC chromatography. The product was purified by two subsequent acetonitrile gradients in ammonium acetate, followed by two gradients in MilliQ-water. Gradients were run as for monoamino acylation reactions above. After each ammonium acetate gradient the product was passed over a Dowex 50Wx4 (K+) column to remove tetrabutylammonium ions. All products were checked for high purity by NMR and MALDI-TOF analysis. 2′-OCH3-Adenosine was aminoacylated in the same procedure as described for the diaminoacylation of mononucleotides except that no tetrabutylammonium was used in the reaction.
Fragment reaction with acylaminoacylated mononucleotides
50S ribosomes (6 pmol) were pre-incubated for 15 min at 37°C in reaction buffer containing 50 mM HEPES/KOH, 100 mM KCl (pH of the buffer was adjusted to pH 7.5 at 0°C in presence of KCl), 20 mM magnesium acetate and 10 mM cytidine if not otherwise indicated. For some reactions HEPES buffer was substituted by imidazole at concentrations indicated in the figures. After cooling for 10 min to 0°C, acylaminoacylated mononucleotide substrate at 1 mM final concentration and 6 pmol [14C]PhetRNAPhe (∼1000 d.p.m./pmol) pre-mixed with 0.3 pmol internal standard (Ac[14C]Phe, ∼1100 d.p.m./pmol) were added (final volume 25 µl). The reaction was initiated by addition of 25 µl cold methanol; however, all concentrations were calculated for the reaction mixture prior to addition of methanol. In competition experiments AcLeu-dAMP (4) was added to the reaction after addition of AcLeu-AMP (1) or DiAcLeu-AMP (3), but prior to addition of PhetRNAPhe. After 2 h at 0°C the reaction was terminated by addition of 25 µl 3 M NaOH (final concentration 1 M) and incubated for 30 min at 37°C to hydrolyze the ester bonds. The mixture was acidified with 100 µl 32% HCl and AcLeuPhe product was extracted with 1 ml ethyl acetate. The ethyl acetate was evaporated in the speedvac, the remaining substance was dissolved in ethyl acetate and subjected to thin layer chromatography on silica gel 60 TLC plates (Merck). The TLC system was chosen to ensure separation of the methyl ester of phenylalanine from the dipeptide. This ester is formed by hydrolysis of [14C]PhetRNAPhe with methanol, which is present in the reaction mixture. The TLC plates were developed for 20 min in CHCl3/MeOH/AcOH 96% = 10/2/1 and exposed overnight with tritium sensitive PhosphorImager screens. Screens were scanned with a Molecular Dynamics Storm 840 PhosphorImager (Amersham Pharmacia) and quantitative data were obtained in the ImageQuant software version 5.0.
Modification of rRNA with 1-cyclohexyl-2-morpholino-carbodiimidemetho-p-toluensulfonat (CMCT)
50S ribosomal subunits (6 pmol) were activated for 10 min at 37°C in 25 µl 80 mM K–borate (pH 8.0), 25 mM MgCl2 and 100 mM NH4Cl. When indicated, 10 mM cytidine was added together with P-site substrate [either AcLeuAMP (1) or 6 pmol Ac[14C]Phe-tRNAPhe], incubated for 5 min at 37°C and cooled to 0°C. Sparsomycin (100 µM) and 33% ethanol were added and incubated additional 20 min on ice. Ribosomes were modified by addition of an equivolume of 84 mg/ml CMCT in reaction buffer with ethanol and incubation for 10 min at 10°C. Ribosomes were precipitated, ribosomal RNA was purified and used as template for primer extension analysis as described in (18). Gels were scanned with Molecular Dynamics Storm 840 PhosphorImager (Amersham Pharmacia) and quantitative data were obtained in the ImageQuant software version 5.0.
RESULTS AND DISCUSSION
Synthesis of P-site substrates and reaction conditions
The chemical aminoacylation of the various mononucleotides was usually accomplished by reacting a nucleotide 5′-phosphate with the cyanomethyl ester of the appropriate N-acetyl-amino acid (see Material and Methods) yielding the desired 2′(3′)-O-(N-acylaminoacyl) nucleotide (Fig. 1). In case of diaminoacylation, a route with 1,1′-carbonyldiimidazole was followed yielding 5′-phospho-2′,3′-Di-O-(N-acylaminoacyl)-adenosine, which was extensively HPLC purified to avoid contamination with the monoaminoacylated product. All products were checked for high purity by NMR and mass spectrometry analysis. The amino acids used in this study were derivatives of leucine and phenylalanine as they were expected to give high intrinsic donor activity (2,6).
Figure 1.

Structure of acylaminoacylated mononucleotides used as P-site substrates.
Our assay conditions were modified from previously published procedures (13,19) and optimized to achieve a linear dependency of the reaction on ribosome concentration. 50S ribosomes and [14C]PhetRNAPhe as A-site substrate were used for the reaction. Routinely, 20 mM of magnesium ions were used as the optimum ranged from 20 to 60 mM (data not shown) and the reaction was stimulated with 10 mM cytidine (discussed in the following paragraph). All reactions were performed at 0°C as the competing PhePhe synthesis was reduced at this temperature. The reactions contained Ac[14C]Phe as internal standard to control for the purification steps and to allow quantification of the dipeptide product. The reaction conditions were optimized for either AcLeu-AMP (1) or AcPheAMP (2) (Fig. 1) as P-site substrate. For these substrates standard concentration was 1 mM as a further increase did not yield significantly more reaction product (Fig. 2B). The reaction time was routinely set to 2 h as time course experiments showed that the amount of product did not increase significantly after that time point (Fig. 2A). These parameters are very similar to the ones established for fMet-AMP (8,13) but the efficiency in our system is much higher (30–40% of the [14C]PhetRNAPhe reacted to AcLeuPhe, compared with ∼2% published for the fMet-AMP). In part, this difference results from the much better stimulation of our reaction due to the addition of 10 mM cytidine (discussed below).
Figure 2.

Time course (A) and concentration dependence (B) of AcLeu[14C]Phe formation. AcLeu-AMP (1) (squares) or DiAcLeu-AMP (3) (circles) was used as P-site substrate to react with [14C]Phe-tRNAPhe in a modified fragment reaction. Substrate concentration in (A) is 1 mM, time in (B) is 2 h. Formation of AcLeu[14C]Phe was analyzed by thin layer chromatography. Data represent the average of three to five independent series.
To establish correct binding of our minimal P-site substrate, we took advantage of an RNA footprint diagnostic for P-site substrates. Both U2584 and U2585 in the peptidyl transferase loop of 23S RNA are reactive to the chemical CMCT but are specifically protected by A76 of tRNAs bound to the P-site (16). Figure 3 shows a primer extension analysis of 23S RNA which was extracted from untreated 50S subunits (lane 1) or modified by CMCT (lane 2). Even though addition of 10 mM cytidine showed a moderate effect on modification of U2584 and U2585 (lane 3, 10–20% inhibition, average of four independent experiments), further addition of 1 or 4 mM AcLeu-AMP (1) inhibited the modification to ∼30–35 and 40–50%, respectively (lanes 4 and 5). As expected binding of Ac[14C]PhetRNAPhe footprinted to ∼85–90%. The moderate inhibition by 10 mM cytidine might indicate a structural change at the peptidyl transferase center (as discussed below); however, the concentration dependent footprinting shows that active AcLeu-AMP (1) is bound to the ribosomal P-site in a manner comparable with A76 of tRNA.
Figure 3.

CMCT footprinting of U2585 and U2584 with AcLeu-AMP (1) and AcPhe-tRNAPhe on 50S subunits. Primer extension analysis of 23S rRNA isolated from unmodified (CMCT–, lane 1) and modified (CMCT+, lane 2) 50S subunits. 50S subunits were further CMCT modified in the presence of 10 mM cytidine (lane 3), 10 mM cytidine and 1 mM AcLeu-AMP (lane 4), 10 mM cytidine and 4 mM AcLeu-AMP (lane 5) or AcPhe-tRNAPhe (lane 6).
Stimulation of the minimal P-site substrates by cytidine derivatives
An acylaminoacylated-AMP is the minimal donor for the ribosomal peptidyl transferase reaction, but its donor activity is significantly lower than that of an acylaminoacylated-CCA fragment (13). Intriguingly, in further studies the donor activity of fMet-AMP became 2–3-fold stimulated by 1 mM CMP, but not by other mononucleotides (8,19). Interestingly, by using AcPhe-AMP (2) as P-site substrate the reaction was stimulated to ∼15-fold in our system (Fig. 4A, compare lanes 1 and 2). To investigate whether the phosphate or ribose moiety contributes to this stimulation cytidine and cytosine were tested. As shown in Figure 4A, 1 mM cytidine enhanced the reaction ∼7-fold (lane 3), 1 mM cytosine 4-fold (lane 4), and 10 mM cytidine 33-fold (lane 5). Addition of CMP to a reaction mixture containing 10 mM cytidine did not further stimulate, thus this concentration was used in our standard reaction. As X-ray structure analysis of 50S complexes have shown that both cytosine residues of the CCA terminus of tRNA base pair with rRNA in the P-site, we argued that in case CMP stimulates by base pairing the use of CpC might further enhance the stimulation. However, as shown in Figure 4B, 1 mM CpC stimulated only about half as good as 1 mM CMP.
Figure 4.

Thin layer chromatography of fragment reactions in presence of various compounds. (A) Activity of AcPhe-AMP (2) with [14C]Phe-tRNAPhe (lane 1), in the presence of 1 mM CMP (lane 2), 1 mM cytidine (lane 2), 1 mM cytosine (lane 4) or 10 mM cytidine (lane 5). (B) Activity of AcLeu-AMP (1) with [14C]Phe-tRNAPhe in the presence of 1 mM CMP (lane 1), 10 mM cytidine (lane 2) or 1 mM CpC (lane 3). (C) Activity of AcLeu-AMP (1) with [14C]Phe-tRNAPhe in various buffer systems: HEPES/KOH (lane 1), 50 mM imidazole (lane 2), 200 mM imidazole (lane 3), 400 mM imidazole (lane 4) and HEPES/KOH with 10 mM cytidine (lane 5). (D) Activity of AcLeu-AMP (1) with [14C]Phe-tRNAPhe in the presence of cytidine (lane 1), 3-methylcytidine (lane 2), 5-methylcytidine (lane 3), cytosine (lane 4), isocytosine (lane 5) or 5-fluorocytosine (lane 6). Cytidine, cytosine and their derivatives were added to 10 mM final concentration.
So far the interpretation of the CMP stimulation was that binding of nucleotide C75 of the 3′ end of peptidyl-tRNA to ribosomal donor site is important for peptidyl transferase activity. Therefore, CMP might stimulate the donor activity of acylaminoacylated-AMP by base pairing to rRNA, similar to C75 thereby inducing conformational changes (8,19). However, another attractive interpretation is that the cytosine base acts as a general acid/base catalyst similar to the catalytic cytosine in the active site of the hepatitis delta virus ribozyme (20). In this case, an abasic mutant of this cytosine could be rescued by providing exogenous cytosine or imidazole analogs indicating that the N3 position of cytosine can act in acid/base catalysis (21,22). Consequently, we performed experiments with imidazole buffer (Fig. 4C). Fifty and 200 mM imidazole buffer resulted in a 3- and 5-fold stimulation of the reaction, respectively (compare lane 1 with lanes 2 and 3), whereas 400 mM already proved slightly inhibitory (compare lanes 3 and 4). Further increase of imidazole to 1 M resulted in a total inhibition of the reaction (data not shown). Although, the stimulation of the reaction with imidazole is interesting it is much less than with the cytosine derivatives. To investigate if the ability of the cytosine to form Watson–Crick base pair interactions is important for the stimulation or rather the pKa of the N3 position (4.6) of the cytosine, we performed experiments with several cytosine derivatives. 5-Fluoro cytosine with a pKa (N3) ∼3.6 should show a significant reduction of the reaction if the enhancement caused by the base would result from the ability of the N3 position to be involved in acid/base catalysis. However, as shown in Figure 4D, 5-fluorocytosine was as effective in stimulating the reaction as cytosine [pKa (N3) ∼4.6]. In addition, 5-methylcytidine [pKa (N3), 4.3] showed a marginally weaker stimulation (Fig. 4D, lanes 1 and 3) than cytidine [pKa (N3), 4.2]. These two derivatives are unaltered in their ability to engage in Watson–Crick base pairing. In contrast, 3-methylcytidine and isocytosine, that are impaired in Watson–Crick base pairing, showed no stimulation of the reaction (Fig. 4D, compare lanes 1 and 2, and lanes 4 and 5). Isocytosine is of particular interest as its N3 position has the same pKa as cytosine and hence should be able to stimulate if an N3 group would act in acid/base catalysis.
Taken together, these experiments show that the stimulation of the peptidyl transferase reaction with cytosine derivatives does not result from a catalytic effect of the N3 position of the cytosine but might rather result from a structural change at the active site possibly due to Watson–Crick base pairing between cytosine and rRNA. Indeed, preliminary results from X-ray studies of 50S subunits from Deinococcus radiodurans with AcLeu-AMP (1) and CMP soaked into crystals show an AcLeu-AMP (1) binding close to the A76 of a P-site bound substrate analog, and a CMP at the same position as C75 (F.Schluenzen, A.Bashan, A.Yonath, personal communication).
Use of base modified mononucleotides as P-site substrate
In order to investigate the influence of the N6 amino group of the terminal adenosine we chemically acylaminoacylated inosine-5′-monophosphate thereby substituting the amino group with a keto group. Consequently, a potential proton donor is exchanged to a proton acceptor. When used as P-site substrate, AcPhe-IMP (8) showed a 10-fold reduction in AcPhePhe synthesis when compared with AcPhe-AMP (2) (Fig. 5A). The reduced activity of the IMP derivative indicates an important function for the amino group in binding or positioning the acylaminoacylated mononucleotide. However, the X-ray structures of P-site bound substrates do not indicate an interaction of the N6 amino group of adenosine with the surrounding rRNA (11).
Figure 5.
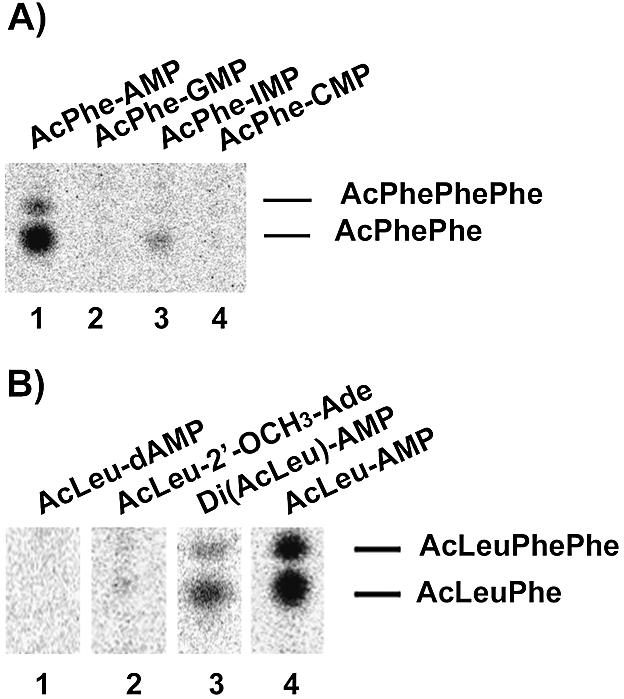
Thin layer chromatography of fragment reactions with [14C]Phe-tRNAPhe in the A-site and various P-site substrates. (A) Base modified substrates: AcPhe-AMP (2) (lane 1), AcPhe-GMP (7) (lane 2); AcPhe-IMP (8) (lane 3) or AcPhe-CMP (6) (lane 4). (B) Substrates with modified 2′-position: AcLeu-dAMP (4) (lane 1), AcLeu-2′-OCH3-Ade (5) (lane 2), DiAcLeu-AMP (3) (lane 3) or AcLeu-AMP (1) (lane 4).
In addition, AcPhe-CMP (6) and AcPhe-GMP (7) were tested for peptidyl transferase activity, however, both were inactive in this assay (Fig. 5A). This clear preference for adenosine as the terminal nucleotide is further supported by competition experiments, which showed that the reaction of AcLeu-AMP (1) can only be inhibited by increasing the amount of AMP or dAMP, but not with any of the other mononucleotides (data not shown).
Testing P-site substrates with a modified 2′-position
To investigate the influence of the vicinal 2′-OH group on peptidyl transferase activity we synthesized several P-site substrates with different 2′-positions (Fig. 1). AcLeu-dAMP (4) with a 2′-H proved unreactive under our assay conditions (Fig. 5B) and further increase of substrate even up to 10 mM did not result in any detectable activity. However, AcLeu-dAMP (4) is a good inhibitor of the activity of AcLeu-AMP (1) (Fig. 6) and hence presumably competes for the same binding site. This interpretation is supported by CMCT footprinting data on U2585, which showed a 30–35% inhibition by 4 mM AcLeu-dAMP (4) (data not shown). This is consistent with results obtained with another P-site substrate, AcLeu-C-2′-dA (6), which also proved inactive in the fragment reaction. However, donor activity, but not translational activity, of a terminal aminoacyl-2′-dA in the context of a complete tRNA could be demonstrated using an E.coli in vitro translation system (23). This discrepancy might result from the different systems used. From the sensitivity of our fragment assay we estimate that the reaction of the AcLeu-dAMP (4) must be down by at least 100-fold, a factor, which might go undetected in the much faster reaction of aa-tRNAs in a complete translational system. In addition to AcLeu-dAMP (4), a DiAcLeu-AMP (3) and AcLeu-2′-OCH3-AMP were synthesized; however, the latter substrate could not be obtained in sufficient purity to unambiguously interpret the results. Therefore, a Di(AcLeu)-2′OCH3-Ade (5) was synthesized and used as P-site substrate. Both substrates with a 2′-oxygen group but lacking the proton were active as donors but showed significantly reduced activity compared with the substrate with a 2′-OH (Fig. 5B). This indicates a favorable influence of the electrophilic 2′-oxygen group and underscores the importance of the 2′-OH of the terminal A for the activity of P-site substrates.
Figure 6.

Competitive inhibition of the activity of AcLeu-AMP (1) (squares) or DiAcLeu-AMP (3) (circles) with increasing amounts of inactive AcLeu-dAMP (4). Data represent average values of three independent experimental series.
DiAcLeu-AMP (3), which possesses only ∼5–10% of the activity of the AcLeu-AMP (1) was further characterized. Both time course (Fig. 2A) and concentration experiments (Fig. 2B) showed that the parameters chosen for the reaction (2 h incubation time and 1 mM substrate) were suitable for comparing the activities. Both, DiAcLeu-AMP (3) and AcLeu-AMP (1) were similarly inhibited by the inactive AcLeu-dAMP (4) (Fig. 6). In addition, DiAcLeu-AMP (3) footprinted CMCT modification of U2585 to the same extent as the other P-site substrates (data not shown) indicating correct binding to the P-site. However, we were surprised that such a large side chain was tolerated at the 2′-position. In order to exclude the possibility that the activity of DiAcLeu-AMP (3) is due to hydrolysis of one amino acid ester during the long incubation time and that in fact we were measuring AcLeu-AMP (1) activity, this substrate was pre-incubated under reaction conditions for 2 h prior to the addition of A-site substrate. However, this pre-incubation resulted in no statistically significant difference in activity (data not shown). This indicates that indeed the DiAcLeu-AMP (3) itself is the active substrate.
In conclusion, these results show that the 2′-position of the ribose influences the reactivity of the amino ester bond at the 3′-position. Although we cannot exclude subtle differences in binding of the modified substrates, it is likely that the loss of activity of the 2′-dAMP derivative is at least in part due to the reduced chemical reactivity of the 3′-amino acid ester. However, as we recently hypothesized, this 2′-OH group might additionally be directly involved in peptide bond formation (Fig. 7) (24). In this model, the nucleophilic attack of the amino group of A-site bound charged tRNA on the electrophilic carbon center at the P-site is supported by the 2′-proton, which helps to neutralize the developing negative charge at the 3′-oxygen in the transition state. Concomitantly, the 2′-oxygen accepts a proton from the amino group, which finally leads to a free tRNA and a peptidyl-tRNA on the A-site. That such a model is theoretically possible was shown by quantum mechanical calculations of the cyclic intermediate (25). In line with this hypothesis are recent results from model building including X-ray crystallography of various 50S substrate complexes, which showed that the 2′-OH of the P-site substrate could be within 2.5 Å from the attacking amino group (12). During preparation of this manuscript an interesting analysis of the possible catalytic power of RNA for amide synthesis was published (26). The authors show that a 2′-amino substitution on a nucleotide could easily react with an activated ester provided a 3′-hydroxyl or a 3′-phosphodiester group is adjacent. The importance of the vicinal hydroxyl group is supported by the fact that a 3′-OH positioned in trans to the 2′-amine is unreactive. From these data the authors also proposed that a properly positioned 2′-hydroxyl might aid peptide bond formation by donating a proton to the 3′-ester and accepting a hydrogen from the amino nucleophile.
Figure 7.

Model for a possible interaction of the P-site 2′-OH during peptide bond formation.
The model presented adds to the ongoing debate of whether and how peptide bond formation might be aided, however, it does not exclude other chemical groups of the ribosome from being involved in catalysis as has been proposed for metal ions (27) or nucleotides (11). Indeed, recent fast kinetic analysis revealed protonation of a ribosomal group in peptide synthesis (28). This protonation could be prevented by mutation of certain nucleotides, which was interpreted to be due to structural changes in rRNA. But aside from the contribution of rRNA to catalysis there is a clear effect of the 2′-OH of the terminal adenosine of P-site tRNA on the reactivity of the aminoacyl ester and on peptidyl transferase activity.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank W. Schmid’s group for technical help in organic chemistry matters, K. Nierhaus for ribosomes and F. Eckstein, and K. Katze for discussions. This work was supported by grant P-13651 and P-16523 from the Österreichischer Fonds zur Förderung der wissenschaftlichen Forschung (FWF) to A.B.
REFERENCES
- 1.Monro R.E. and Marcker,K.A. (1967) Ribosome-catalysed reaction of puromycin with a formylmethionine-containing oligonucleotide. J. Mol. Biol., 25, 347–350. [DOI] [PubMed] [Google Scholar]
- 2.Monro R.E., Cerna,J. and Marcker,K.A. (1968) Ribosome-catalyzed peptidyl transfer: substrate specificity at the P-site. Proc. Natl Acad. Sci. USA, 61, 1042–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourd S.B., Kukhanova,M.K., Gottikh,B.P. and Krayevsky,A.A. (1983) Cooperative effects in the peptidyltransferase center of Escherichia coli ribosomes. Eur. J. Biochem., 135, 465–470. [DOI] [PubMed] [Google Scholar]
- 4.Bhuta P. and Zemlicka,J. (1985) 2′(3′)-O-l-Phenylalanyl derivatives of N2,5′-anhydroformycin and N4,5′-anhydroformycin: new substrates for ribosomal peptidyltransferase with a fixed anti and syn conformation of the base. Biochim. Biophys. Acta, 841, 145–150. [DOI] [PubMed] [Google Scholar]
- 5.Tezuka M. and Chladek,S. (1990) Effect of nucleotide substitution on the peptidyltransferase activity of 2′(3′)-O-(aminoacyl) oligonucleotides. Biochemistry, 29, 667–670. [DOI] [PubMed] [Google Scholar]
- 6.Quiggle K., Kumar,G., Ott,T.W., Ryu,E.K. and Chladek,S. (1981) Donor site of ribosomal peptidyltransferase: investigation of substrate specificity using 2′(3′)-O-(N-acylaminoacyl)dinucleoside phosphates as models of the 3′ terminus of N-acylaminoacyl transfer ribonucleic acid. Biochemistry, 20, 3480–3485. [DOI] [PubMed] [Google Scholar]
- 7.Quiggle K. and Chladek,S. (1980) The role of the cytidine residues of the tRNA 3′-terminus at the peptidyltransferase A- and P-sites. FEBS Lett., 118, 172–175. [DOI] [PubMed] [Google Scholar]
- 8.Cerna J. (1975) Effect of cytidine-5′-monophosphate on peptidyl transferase activity. FEBS Lett., 58, 94–98. [DOI] [PubMed] [Google Scholar]
- 9.Samaha R.R., Green,R. and Noller,H.F. (1995) A base pair between tRNA and 23S rRNA in the peptidyl transferase centre of the ribosome. Nature, 377, 309–314. [DOI] [PubMed] [Google Scholar]
- 10.Kim D.F. and Green,R. (1999) Base-pairing between 23S rRNA and tRNA in the ribosomal A site. Mol. Cell, 4, 859–864. [DOI] [PubMed] [Google Scholar]
- 11.Nissen P., Hansen,J., Ban,N., Moore,P.B. and Steitz,T.A. (2000) The structural basis of ribosome activity in peptide bond synthesis. Science, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- 12.Schmeing T.M., Seila,A.C., Hansen,J.L., Freeborn,B., Soukup,J.K., Scaringe,S.A., Strobel,S.A., Moore,P.B. and Steitz,T.A. (2002) A pre-translocational intermediate in protein synthesis observed in crystals of enzymatically active 50S subunits. Nature Struct. Biol., 9, 225–230. [DOI] [PubMed] [Google Scholar]
- 13.Cerna J., Rychlik,I., Krayevsky,A.A. and Gottikh,B.P. (1973) 2′ (3′)-O-N-(Formylmethionyl)-adenosine-5′-phosphate, a new donor substrate in peptidyl transferase catalyzed reactions. FEBS Lett., 37, 188–191. [DOI] [PubMed] [Google Scholar]
- 14.Rychlik I., Cerna,J., Chladek,S., Zemlicka,J. and Haladova,Z. (1969) Substrate specificity of ribosomal peptidyl transferase: 2′(3′)-O-aminoacyl nucleosides as acceptors of the peptide chain on the amino acid site. J. Mol. Biol., 43, 13–24. [DOI] [PubMed] [Google Scholar]
- 15.Rheinberger H.J., Geigenmuller,U., Wedde,M. and Nierhaus,K.H. (1988) Parameters for the preparation of Escherichia coli ribosomes and ribosomal subunits active in tRNA binding. Methods Enzymol., 164, 658–670. [DOI] [PubMed] [Google Scholar]
- 16.Moazed D. and Noller,H.F. (1989) Interaction of tRNA with 23S rRNA in the ribosomal A, P and E sites. Cell, 57, 585–597. [DOI] [PubMed] [Google Scholar]
- 17.Robertson S.A., Ellman,J.A. and Schultz,P.G. (1991) A general and efficient route for chemical aminoacylation of transfer RNAs. J. Am. Chem. Soc., 113, 2722–2729. [Google Scholar]
- 18.Polacek N. and Barta,A. (1998) Metal ion probing of rRNAs: evidence for evolutionarily conserved divalent cation binding pockets. RNA, 4, 1282–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spahn C.M., Schafer,M.A., Krayevsky,A.A. and Nierhaus,K.H. (1996) Conserved nucleotides of 23 S rRNA located at the ribosomal peptidyltransferase center. J. Biol. Chem., 271, 32857–32862. [DOI] [PubMed] [Google Scholar]
- 20.Nakano S., Chadalavada,D.M. and Bevilacqua,P.C. (2000) General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science, 287, 1493–1497. [DOI] [PubMed] [Google Scholar]
- 21.Perrotta A.T., Shih,I. and Been,M.D. (1999) Imidazole rescue of a cytosine mutation in a self-cleaving ribozyme. Science, 286, 123–126. [DOI] [PubMed] [Google Scholar]
- 22.Shih I.H. and Been,M.D. (2001) Involvement of a cytosine side chain in proton transfer in the rate-determining step of ribozyme self-cleavage. Proc. Natl Acad. Sci. USA, 98, 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner T., Cramer,F. and Sprinzl,M. (1982) Activity of the 2′ and 3′ isomers of aminoacyl transfer ribonucleic acid in the in vitro peptide elongation on E. coli ribosomes. Biochemistry, 21, 1521–1529. [DOI] [PubMed] [Google Scholar]
- 24.Dorner S., Polacek,N., Schulmeister,U., Panuschka,C. and Barta,A. (2002) Molecular aspects of the ribosomal peptidyl transferase. Biochem. Soc. Trans., 30, 1131–1136. [DOI] [PubMed] [Google Scholar]
- 25.Das G.K., Bhattacharyya,D. and Burma,D.P. (1999) A possible mechanism of peptide bond formation on ribosome without mediation of peptidyl transferase. J. Theor. Biol., 200, 193–205. [DOI] [PubMed] [Google Scholar]
- 26.Chamberlin S.I., Merino,E.J. and Weeks,K.M. (2002) Catalysis of amide synthesis by RNA phosphodiester and hydroxyl groups. Proc. Natl Acad. Sci. USA, 99, 14688–14693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barta A. and Halama,I. (1996) In Green,R. and Schroeder,R. (eds), Ribosomal RNA and Group I Introns. R.G. Landes Company, Chapman & Hall, New York, pp. 35–54. [Google Scholar]
- 28.Katunin V.I., Muth,G.W., Strobel,S.A., Wintermeyer,W. and Rodnina,M.V. (2002) Important contribution to catalysis of peptide bond formation by a single ionizing group within the ribosome. Mol. Cell, 10, 339–346. [DOI] [PubMed] [Google Scholar]