


Abstract
The tumor suppressor protein p53 and the human DNA topoisomerase I (htopoI) interact with each other, which leads to a stimulation of the catalytic activity of htopoI. Moreover, p53 stimulates the topoisomerase I-induced recombination repair (TIRR) reaction. However, little was known about how p53 stimulates this topoisomerase I activity. Here we demonstrate that monomeric p53 is sufficient for the stimulation of the topoisomerase I-catalyzed relaxation activity, but the tetrameric form of p53 is required for the stimulation of TIRR. We also show that p53 stimulates topoisomerase I activity by increasing the dissociation of htopoI from DNA. Since htopoI forms a closed ring structure around the DNA, our results suggest that p53 induces a conformational change within htopoI that results in an opening of the clamp, and thereby releases htopoI from DNA.
INTRODUCTION
The tumor suppressor protein p53 is essential for the integrity of the genomic information of the cell and therefore is known as the ‘guardian of the genome’. Following genotoxic stress, p53 induces a cell cycle arrest and/or apoptosis by the transcriptional activation of several genes, such as p21CIP1/WAF1 and the proapoptotic PUMA gene [for a recent review see Vousden and Lu (1)]. Beside this role as a transcription factor, there is increasing evidence that p53 can directly influence some enzymatic activities within the cell.
Human topoisomerase I (htopoI) is an enzyme involved in several aspects of DNA metabolism, such as DNA replication and transcription, where it releases positive as well as negative supercoils [reviewed in Wang (2)]. During this process, a transient covalent 3′-phosphotyrosyl enzyme–DNA intermediate is formed. Interestingly, the stability of this intermediate is increased in the presence of various DNA lesions. This results in long-lived covalent protein–DNA complexes, so-called topoisomerase I cleavage complexes (TCCs), which also form on damaged DNA (3–6). The in vivo formation of TCCs has been shown to be regulated by p53 (7). Also, topoisomerase I cleavage complexes induced after UV irradiation were suggested to consist of two closely associated TCCs (7). Recently, we have identified a novel repair reaction for human TCCs in vitro, topoisomerase I-induced recombination repair (TIRR). TIRR is a reaction in which a second htopoI molecule recognizes a TCC and generates a so-called htopoI double cleavage complex (8). The second bound htopoI molecule renders another incision ∼13 nt upstream of the TCC. This results in the release of the TCC and is followed by a recombination-like reaction that eventually leads to complete repair of the TCC. We found that p53 strongly stimulates TIRR (9). Since TCCs occur in vivo (as mentioned above), it is reasonable to assume that TIRR-like reactions remove TCCs. Hence, several groups have investigated how TCCs are repaired, and some repair pathways for TCCs have been suggested (10–13). Nevertheless, an efficient repair pathway for TCCs has not yet been identified.
In the present study, we have investigated how p53 stimulates htopoI in general and TIRR in particular. We show that tetrameric p53 is essential for the stimulation of TIRR whereas this is not the case for the htopoI relaxation activity. Furthermore, we show that p53 stimulates the activity of htopoI by accelerating the dissociation of htopoI from the DNA. By this mechanism, p53 stimulates both DNA relaxation and TIRR.
MATERIALS AND METHODS
Materials
pUC19 plasmid was expressed and purified as described (14). The TIRR suicide substrate L193s and OL26 were used as described previously (9). The modified form of L193s, L193s–biotin, as well as OL25 were synthesized by Purimex, Staufenberg, Germany. L193s–biotin: A (145 nt), 5′-biotin– GGGGCCGAATTCACTCCGGGGATCCTTTTTTTTTTTT TTTTTTCTAAGTCTTTTTTTGCCTTCGCCCGGATCCC CGCCAAGCTTACCTGCCCTTTGGGCAGGTAAGCTT AAAAAGGATCCCCGGAGTGAATTCGGCCCC-3′. These two oligonucleotides were hybridized by heating to 95°C and slowly cooling down to room temperature. OL25: 5′-CAAAAAAAGACTTAGAAAAAAAAAA-3′. The human p53302–321 peptide, GSTKRALPNNISSSPQPKKK, was synthesized by Jenabioscience, Jena, Germany.
Recombinant proteins
HtopoI, human p53, murine His-p53 as well as murine His-p531–320 were expressed and purified as previously described (14).
DNA relaxation assay
The indicated amount of htopoI was incubated with 1 µg of pUC19 and different forms of p53 (as indicated) in a total reaction volume of 20 µl at 30°C for the indicated times. The reaction conditions were as follows: Figures 1A and 2A, buffer A: 12.5 mM HEPES-KOH pH 7.9, 50 mM KCl, 6 mM MgCl2, 8.5% glycerol, 1 mM dithiothreitol (DTT); Figure 3A–D, buffer B: 12.5 mM HEPES-KOH pH 7.9, 6 mM MgCl2, 8.5% glycerol, 1 mM DTT and the indicated concentrations of NaCl.
Figure 1.

p53302–321 stimulates the htopoI relaxation activity but not TIRR. (A) Ethidium bromide staining of a 0.8% agarose gel. The reactions were performed at 30°C for 20 min. Lane 1, control; lane 2, 34 pmol p53302–321; lane 3, 0.4 pmol htopoI; lanes 4–13, 0.4 pmol htopoI and 68, 34, 17, 8.5, 4.3, 2, 1, 0.5, 0.26 or 0.13 pmol p53302–321, respectively. (B) Schematic representation of TIRR. A suicide cleavage complex is formed at the centrally located recognition sequence. This complex is recognized by an additional htopoI molecule and a double cleavage is generated. The suicide cleavage complex is released and the 3′-32P-labeled (open circle) OL26 can hybridize to the gap. OL26 is ligated to L193s by the second htopoI molecule and htopoI is released. HtopoI can once again cleave the DNA. Therefore, after BamHI cleavage, two main products are generated. The long 32P-labeled fragment R1 or the shorter 32P-labeled fragments (R2 and R3) (which were cleaved again by htopoI) are the end-products detected on the autoradiogram [see also Stephan et al. (9)]. (C) Autoradiogram of a 14% denaturing polyacrylamide gel. The reaction mixtures were incubated for 30 min at 30°C. L193s and OL26 were incubated with: lane 1, control; lane 2, 748 pmol p53302–321; lane 3, 8.8 pmol htopoI; lanes 4–9, 8.8 pmol htopoI and 2690, 750, 375, 187, 94 and 47 pmol p53302–321, respectively. R1, R2, R3: see (B). (D) Average effect of p53302–321 on TIRR from three independent measurements.
Figure 2.
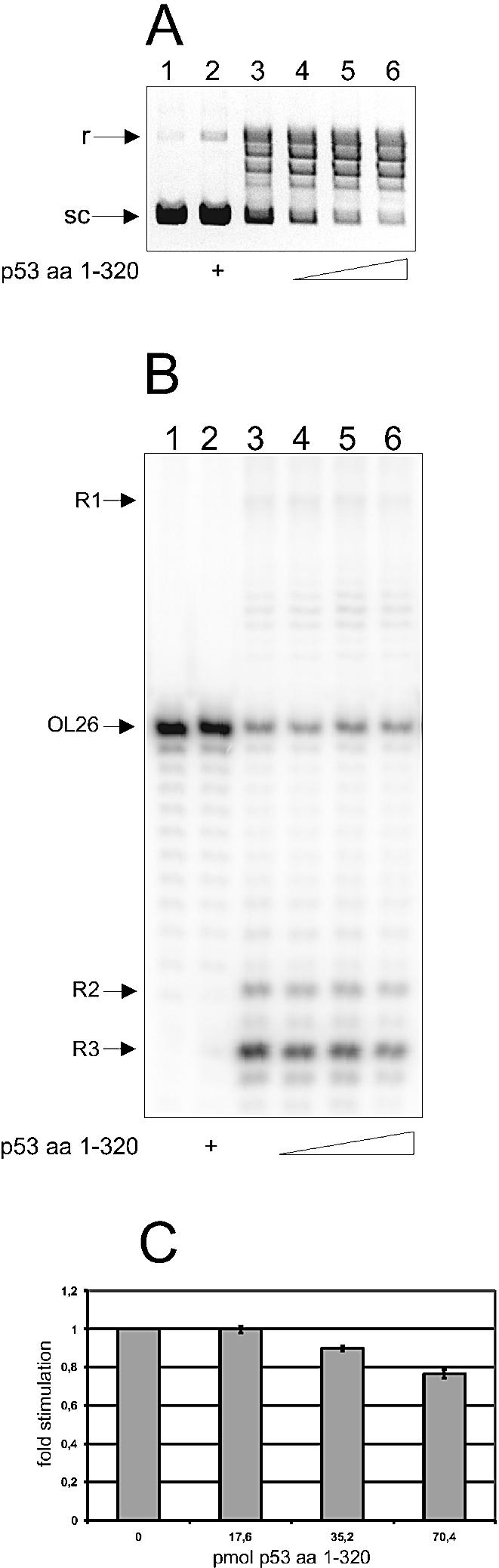
p53 without a tetramerization domain can no longer stimulate TIRR. (A) Ethidium bromide staining of a 0.8% agarose gel. The reactions were performed at 30°C for 10 min. Lane 1, control; lane 2, 2 pmol of p531–320; lane 3, 0.1 pmol htopoI, lanes 4–6, 0.1 pmol htopoI and 0.7, 1.4 and 2 pmol p531–320, respectively. (B) Autoradiogram of a 14% denaturing polyacrylamide gel. The reaction mixtures were incubated at 30°C for 30 min. Lane 1, control; lane 2, 35 pmol p531–320; lane 3, 8.8 pmol htopoI; lanes 4–6, 8.8 pmol htopoI and 18, 35 and 70 pmol p531–320, respectively. (C) Average effect of p531–320 on TIRR from three independent measurements.
Figure 3.
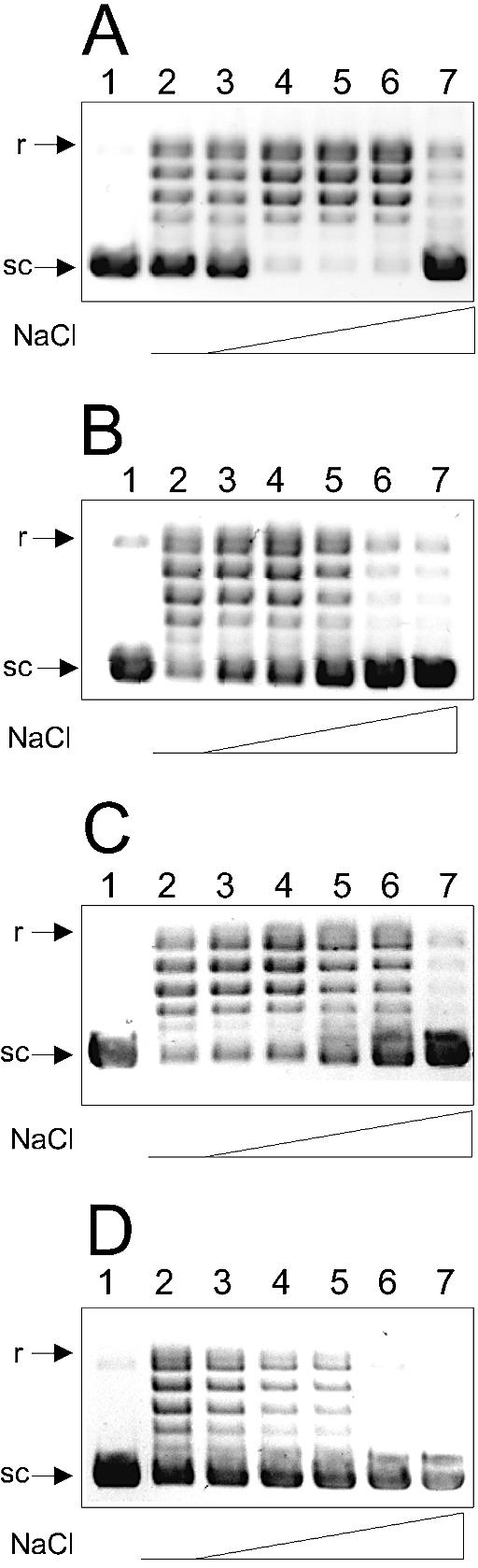
p53 sensitizes htopoI to NaCl. Ethidium bromide staining of a 0.8% agarose gel. The reactions were performed at 30°C for 15 min (A, C and D) or 10 min (B). (A) Lane 1, control; 0.4 pmol htopoI (lanes 2–7) was incubated in the presence of 0, 50, 100, 150, 200 or 300 mM NaCl (lanes 3–7), respectively. (B) Lane 1, 4.6 pmol p53; lanes 2–7, 0.05 pmol htopoI and 4.6 pmol p53 were incubated in the presence of 0, 50, 100, 150, 200 or 300 mM NaCl, respectively. (C) Lane 1, 2.2 pmol p531–320; lanes 2–7, 0.05 pmol htopoI and 2.2 pmol p531–320 were incubated in the presence of NaCl as in (B). (D) Lane 1, 34 pmol p53302–321; lanes 2–7, 0.05 pmol htopoI and 34 pmol p53302–321 were incubated in the presence of NaCl as in (B).
TIRR assays
HtopoI (8.8 pmol) was incubated with 50 pmol L193s, ∼0.1 pmol 3′-32P-labeled OL26 and the indicated amounts of murine His-p531–320, human p53302–321 and human wild-type p53 in a total reaction volume of 50 µl for 30 min at 30°C. Buffer A was used for the results shown in Figures 1B and 2B, and buffer B was used for those shown in Figure 4A. The experiments were performed as previously described (9).
Figure 4.

p53 neutralizes the stimulatory effect of NaCl on TIRR. (A) Autoradiogram of a 14% denaturing polyacrylamide gel. The reaction mixtures were incubated at 30°C for 30 min. L193s and OL26 were incubated with: lane 1, control; lanes 2–8, 5.5 pmol htopoI in the presence of 0, 50, 100, 150, 200, 300 or 400 mM NaCl, respectively; lane 9, 73 pmol p53; lanes 10–16, 73 pmol p53 and 5.5 pmol htopoI in the presence of 0, 50, 100, 150, 200, 300 or 400 mM NaCl, respectively. (B) Average of the NaCl stimulation of TIRR from 3–5 independent experiments in the presence or absence of p53.
Release of htopoI
For the result shown in Figure 5A, the DNA substrate L193s with a 32P label placed at position 15 (counting from the 3′ end) was used as described previously (8). Labeled L193s (20 000 c.p.m.) was incubated with 8.8 pmol htopoI for 30 min at 30°C in 50 µl of buffer B containing the indicated concentrations of NaCl. The reactions were stopped by the addition of loading buffer and the protein was separated by 7% SDS–PAGE. The gel was dried and analyzed with a phosphorimager (Storm 860, Molecular Dynamics, AmershamPharmacia).
Figure 5.
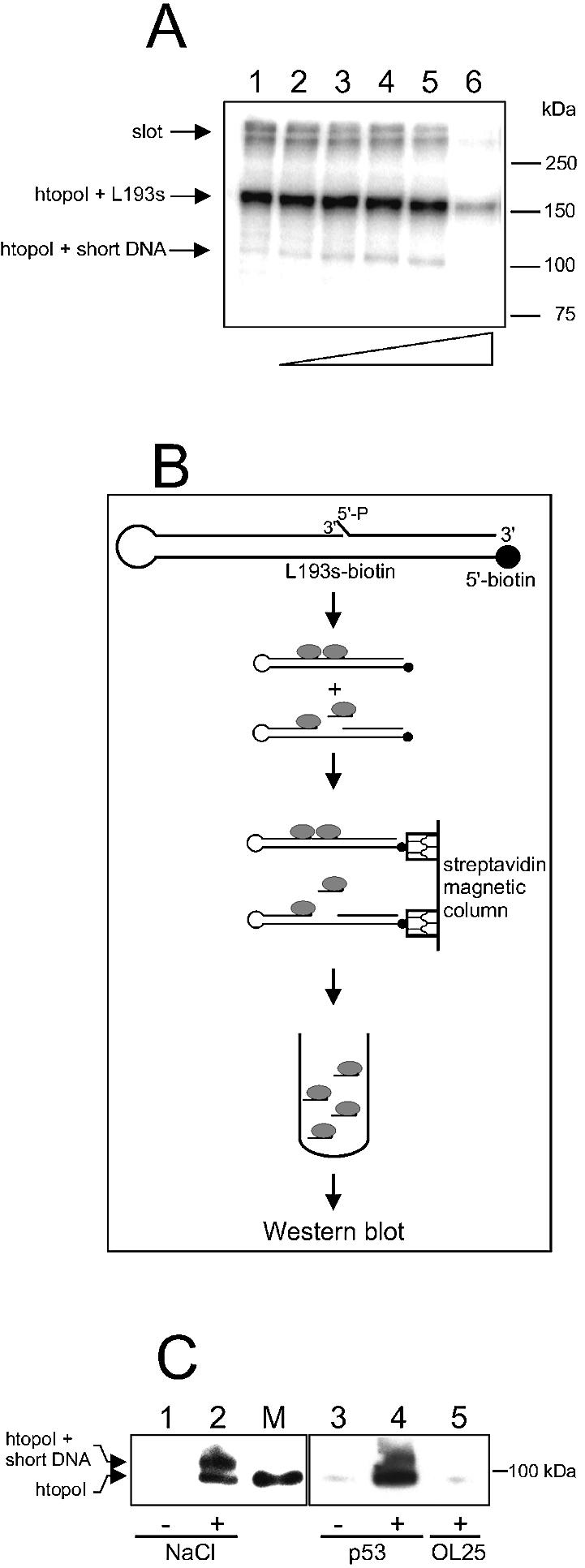
p53 stimulates the release of TCC from the DNA substrate under native conditions. (A) Autoradiogram of a 7% SDS–polyacrylamide gel. htopoI (8.8 pmol) was incubated with radioactively labeled L193s at 30°C for 30 min in the presence of 0, 50, 100, 150, 200 and 300 mM NaCl (lanes 1–6, respectively). The gel was dried and autoradiographed. (B) Experimental strategy for the detection of released htopoI molecules. A biotinylated form of the L193s substrate was incubated with htopoI for 30 min at 30°C and subsequently incubated with magnetic streptavidin microbeads. The solution was passed through a magnetic column. The biotinylated DNA was retained on the column including the bound protein. The run-through and the following wash fraction were collected, separated by 6% SDS–PAGE, and analyzed by western blotting using an anti-htopoI antibody. (C) Western blot of a 6% SDS–polyacrylamide gel. L193s–biotin (100 pmol) was incubated with: lanes 1 and 3, 18 pmol htopoI; lane 2, 18 pmol htopoI and 100 mM NaCl; lane 4, 18 pmol htopoI and 37 pmol p53; lane 5, 18 pmol htopoI and 5 pmol OL25. Subsequently, the reaction mixture was treated according to the procedure explained in (B) and analyzed by western blotting using an anti-htopoI antibody. M, htopoI loaded as marker.
The reactions leading to the results shown in Figure 5C were performed as follows: 100 pmol L193s–biotin was incubated with 18 pmol htopoI, murine His-p53 or OL25, as indicated, in 50 µl of reaction buffer C (30 mM HEPES-KOH pH 7.8, 6 mM MgCl2, 1 mM EDTA, 2 mM DTT and 15% glycerol) with or without NaCl. The reaction mixtures were incubated for 30 min at 30°C. Subsequently, a µMACS Streptavidin kit (Miltenyi Biotec, Bergisch Gladbach) was used according to the manufacturer’s protocol. The run-through and a 200 µl wash fraction (performed with buffer C) were collected and pooled. The protein in the collected fractions was precipitated with Strataclean beads (Stratagen, Kirkland, USA), mixed with loading buffer, incubated for 5 min at 95°C, and analyzed by 6% SDS–PAGE. Subsequently, the gel was subjected to western blotting. The protein was blotted onto a PVDF membrane using a semi-dry transfer apparatus (Bio-Rad, München, Germany). Thereafter, the membrane was incubated overnight at 4°C with the monoclonal murine anti-htopoI antibody (clone C-21.2) (BD Biosciences Pharmingen, San Diego, CA) in buffer D (82 mM NaCl, 10 mM Na2HPO4, 10 mM KH2PO4, 0.05% Tween-20 pH 7.5) supplemented with 5% skimmed milk powder. The membrane was washed with buffer D and subsequently incubated with horseradish peroxidase-coupled anti-mouse antibody (Promega, Madison, WI) in buffer D supplemented with 5% skimmed milk powder for 1 h at room temperature. Subsequently, the membrane was washed with buffer D and htopoI was detected by enhanced chemiluminescence (ECL).
RESULTS
p53 strongly stimulates the activity of htopoI through protein–protein interactions. Gobert et al. reported that a p53-derived peptide containing amino acids 302–321 (p53302–321) was sufficient to stimulate htopoI (15). We could confirm this result by showing that there is a clear stimulatory effect of p53302–321 on the relaxation activity of htopoI (Fig. 1A). However, an at least 21-fold molar excess of the peptide over htopoI was necessary to observe this stimulation (Fig. 1A, compare lanes 4–7 with lane 3).
Recently, we have shown that an in vitro induced TCC is recognized by a second htopoI molecule, giving rise to the formation of a so-called htopoI double cleavage complex (8). The formation of this complex eventually results in the removal and repair of both cleavage complexes by TIRR (9) (see Fig. 1B). This repair event is stimulated by p53 (9). Since a p53-derived peptide was sufficient to stimulate htopoI’s relaxation activity (Fig. 1A), we asked whether the same peptide could also stimulate the TIRR (9). A titration of p53302–321 into the TIRR reaction mixture revealed that the peptide alone could not increase the amount of the three expected recombination products (Fig. 1C). A quantification of the recombination frequency is given in Figure 1D. Thus, the interaction of htopoI with p53302–321 was sufficient to stimulate htopoI’s relaxation activity, but it was not sufficient to stimulate TIRR. Thus, for the stimulation of TIRR, either additional binding sites on p53 may be necessary or the tertiary or quaternary structure of p53 may play a role.
Indeed our earlier results indicated that tetrameric p53 may be necessary for an effective stimulation of TIRR (9,14). To further investigate this issue, we used a truncated form of p53 containing amino acids 1–320 (p531–320) lacking the tetramerization domain and the C-terminal unspecific DNA-binding domain. Figure 2A shows that p531–320 stimulated the relaxation activity of htopoI, demonstrating that neither the C-terminal domain nor tetrameric p53 is needed for this process. This result is in agreement with the peptide stimulation study shown in Figure 1A. However, with respect to TIRR, the C-terminally truncated p531–320 did not stimulate but rather inhibited the reaction (Fig. 2B, compare lanes 4–6 with lane 3). As shown in Figure 2C, an 8-fold molar excess of p531–320 over htopoI inhibited TIRR by ∼25%. Since the C-terminal domain of p53 is not essential for an interaction with htopoI (Figs 1A and 2A) (15), this result indicates that the relaxation activity is mechanistically different from the recombination activity and that a tetrameric form of p53 is obligatory for the stimulation of TIRR. Thus, stimulation of TIRR by p53 seems to be a more complicated process than the stimulation of DNA relaxation.
NaCl enhances the htopoI relaxation activity by lowering the affinity for DNA. This in turn stimulates the religation step and the release of htopoI from DNA (16). The stimulatory effect of NaCl on the relaxation activity of htopoI is shown in Figure 3A, where 150 mM NaCl was optimal for relaxation, whereas, at 300 mM, the activity was almost completely lost (lanes 5 and 7, respectively). However, when the effect of NaCl on htopoI activity was measured in the presence of p53, a different result was obtained. In the presence of p53, maximal relaxation was detected in the absence of salt (Fig. 3B, lane 2). When NaCl was added, the activity declined and was almost completely lost at 200 mM and no longer detectable at 300 mM NaCl (Fig. 3B). The same effect was obtained using p531–320 (Fig. 3C) as well as with p53302–321 (Fig. 3D). This is in striking contrast to the effect of NaCl in the absence of p53, where the optimal concentration was ∼150 mM NaCl (Fig. 3A, lane 5). These results support the view that p53 reduces the affinity of htopoI for DNA similarly to how NaCl reduces it in the absence of p53 (see also below).
To test this assumption, we investigated whether p53 stimulates TIRR by modulating the DNA-binding affinity of htopoI. Figure 4A shows that in the absence of p53, stimulation of TIRR was detected with increasing NaCl concentrations (lanes 2–6). NaCl stimulated TIRR ∼2.5-fold, again with an optimum at ∼150 mM NaCl (lane 5), while a dramatic reduction was seen at 300 and 400 mM NaCl (lanes 7 and 8, respectively). In the presence of p53, TIRR was clearly stimulated at low NaCl concentrations (compare lanes 2 and 3 with lanes 10 and 11), but NaCl was no longer stimulatory (Fig. 4B). The lack of stimulation despite increasing concentrations of NaCl supports the assumption that p53 lowers the affinity of htopoI for DNA. At first glance, however, the results of Figure 3 seem contradictory to the results of Figure 4. In Figure 3, the presence of p53 caused an increased sensitivity towards NaCl, whereas in Figure 4 NaCl showed no effect. The explanation may be that the htopoI-binding step shown in Figure 3 is reversible, whereas the htopoI-binding step of TIRR is irreversible. This will be further elucidated in the experiments described below.
The binding of htopoI to DNA is largely dependent on the negative charge of the DNA sugar–phosphate backbone (17). The Na+ ions neutralize the negative charge and thereby lower the affinity of htopoI for DNA. Increasing concentrations of Na+ are thought to open the clamp structure that holds htopoI on DNA; this in turn, promotes the dissociation of htopoI from its DNA substrate. Since p53 and NaCl seem to have similar effects on the htopoI activity, it is likely that p53 promotes dissociation of htopoI from DNA, but by a different mechanism. With respect to the recombination reaction, p53 might increase the release of the first bound htopoI that should contain a covalently attached oligonucleotide. This would result in a stabilization of the second htopoI cleavage complex and generate a gap, which could promote hybridization of an incoming complementary oligonucleotide and thereby stimulate the TIRR reaction in general. This would also explain our earlier data, where p53 stimulates both the formation of the second cleavage complex and the recombination reaction as a whole (9,14).
To address experimentally the described scenario, we investigated whether p53 increased the release of TCCs attached to a 13 nt oligomer. Figure 5A shows the covalent bonding of htopoI to the radioactively labeled DNA substrate, L193s, with increasing concentrations of NaCl. Free htopoI has a mobility of ∼100 kDa, and free L193s runs out of the gel. In Figure 5A, it is seen that the formation of a covalent bond between htopoI and L193s results in a mobility shift to a position slightly above 150 kDa. With increasing NaCl concentrations, a radioactively labeled protein band was revealed with a mobility of ∼104 kDa. Since htopoI alone has a mobility of 100 kDa, as indicated from a parallel Coomassie-stained gel (data not shown), a short radioactively labeled DNA fragment must have become covalently attached to the protein. Previously, we showed that under such reaction conditions, a htopoI double cleavage reaction takes place that results in an htopoI molecule attached to a radioactively labeled 13 nt oligomer (8). Thus, the 104 kDa band should represent the htopoI double cleavage product.
To further investigate this issue, we developed an assay to detect the release of htopoI molecules from the suicide substrate under non-denaturing conditions (see Fig. 5B). The assay is based on the use of a biotin-modified suicide substrate as described in Materials and Methods. The DNA substrate was incubated with htopoI in the absence or presence of either NaCl, p53 or OL25, and subsequently incubated with magnetic streptavidin microbeads. The reaction mixture was passed through a column placed in a strong magnetic field and the run-through and the following wash fraction were collected and subjected to western blot analysis. Without NaCl, hardly any htopoI was released from the column (Fig. 5C, lanes 1 and 3), demonstrating that virtually all htopoI molecules were retained on the immobilized DNA substrate. However, when the reaction was performed in the presence of 100 mM NaCl, two htopoI bands were revealed (Fig. 5C, lane 2). The lower band runs exactly to the position of the marker htopoI (Fig. 5C, lane M), while the 104 kDa band should represent htopoI molecules attached to a short piece of DNA. Since these bands were only visible in the presence of NaCl, it is reasonable to assume that NaCl reduced the affinity of htopoI for DNA and that this reduced affinity was necessary for the release of the htopoI cleavage complex. It can also explain why NaCl stimulates the TIRR reaction as shown in Figure 4A, since prior release of the cleavage complex is necessary for the subsequent ligation reaction to take place. Interestingly, p53 had a very similar effect to that of NaCl on the release of the cleavage complex (Fig. 5C, compare lanes 1 and 2, and lanes 3 and 4). This was not merely a protein effect since bovine serum albumin did not cause release of htopoI from the DNA substrate (data not shown). Furthermore, the gap-complementary oligonucleotide OL25 (the same one as OL26 but unlabeled) was not able to push the htopoI cleavage complex off the substrate (Fig. 5C, compare lanes 3 and 5). All these experiments support the view that htopoI needs to open its clamp structure in order to dissociate from its substrate DNA. This could be accomplished by both NaCl and p53. It should be noted that a difference can be observed in the efficiency of release of non-covalently bound htopoI between NaCl and p53. This may be an interesting finding, but further investigations will be needed to examine this more closely.
DISCUSSION
In the past few years, several research groups have reported that p53 and htopoI functionally interact (7,15,18,19). Gobert et al. (15) published their finding that a peptide of human p53 comprising amino acids 302–321 was sufficient to stimulate the htopoI relaxation activity. In the present study, we verified this result (see Fig. 1A) and also showed that the truncated p531–320 could stimulate the relaxation activity of htopoI (see Fig. 2A). Therefore, monomeric p53 is sufficient for the stimulation of the htopoI relaxation activity since neither the peptide nor p531–320 can tetramerize. In addition to the enhancement of the relaxation activity, p53 is also able to stimulate the htopoI-mediated recombination-like reaction called TIRR (9). TIRR is a reaction where a second htopoI molecule binds next to a TCC and releases the first bound htopoI molecule together with a covalently attached oligonucleotide by another nucleolytic step. In the generated gap, a recombination-like inter-strand ligation can take place that leads to the eventual repair of the TCC. Importantly, this reaction can also be stimulated by p53. Until now, it was not known whether this stimulation could also be achieved with monomeric p53 or a fragment thereof, as it is the case for the relaxation reaction. Therefore, we asked whether p53302–321 or p531–320 had a similar impact on TIRR. Unexpectedly, neither p53302–321 nor p531–320 had a positive influence on TIRR (see Figs 1C and 2B); rather p531–320 was inhibitory (Fig. 2B and C). These results indicate that in contrast to the relaxation reaction, tetrameric p53 is required for the stimulation of TIRR. Also, the C-terminus does not appear to play a role since p531–320 binds to htopoI (see Fig. 2A) but inhibits TIRR (see Fig. 2B). This finding is very interesting since it may imply that activated p53 is required for TIRR stimulation. This in turn suggests that a TIRR may only take place after genotoxic stress, as suggested previously (9).
Until now, it was virtually unknown how p53 might stimulate the htopoI activities. As one approach to a better mechanistical understanding, we systematically varied the salt concentrations and studied the influence on both the relaxation and the recombination reaction. We found a maximal relaxation activity at ∼150 mM NaCl, which confirms several published reports [see, for example, McConaughy et al. (20)]. However, when purified p53 was added to the reaction mixture, the optimal NaCl concentration shifted to 0 and the activity was almost completely lost at 200 mM NaCl, despite the high activity at this concentration in the absence of p53 (compare Fig. 3A with B, C and D). This shows that p53 sensitizes htopoI to NaCl. Na+ ions reduce the ionic interactions between htopoI and the DNA backbone (20). Since htopoI primarily binds DNA through ionic interactions with the sugar–phosphate backbone (17), Na+ should have a strong effect on the DNA affinity of htopoI. The binding of stoichiometric amounts of p53 to htopoI must somehow interfere with the DNA-binding ability of the DNA-relaxing enzyme. On the other hand, Na+ could not stimulate the TIRR reaction when p53 was present, but did so in its absence. The optimal NaCl concentration in the absence of p53 was 150 mM. Since both NaCl and p53 reduce the strength with which htopoI binds to DNA, it is reasonable to assume that TIRR is stimulated by the same mechanism. How can this lead to an increased TIRR activity? An important step during TIRR is the htopoI double cleavage reaction where the second htopoI molecule cleaves just upstream from the original cleavage complex. This double cleavage complex is highly unstable and can be reversed, at least in part, by high salt concentrations (8), but only as long as the first bound htopoI (together with the covalently attached 13mer) has not yet been dissociated from the template DNA. NaCl and p53 reduce the DNA binding of both htopoI complexes. In the case of the second bound TCC, this would lead to an increased frequency of dissociations and associations as long as the first TCC is still at its place. This has no influence on the overall reaction rate. In the case of the first TCC, high salt or p53 would again increase the dissociation of htopoI from double-stranded DNA, this time, however, with a covalently attached 13 nt oligomer. A religation by the second TCC would be impossible and thus the second bound htopoI molecule becomes trapped. Only when a complementary oligomer hybridizes to the gap (OL25/OL26 in our experiments) is religation possible. Therefore, the faster dissociation rate of the first bound htopoI in the presence of either NaCl or p53 is responsible for the increased product formation of the TIRR reaction. Obviously p53 is sufficient to optimally stimulate this dissociation reaction; therefore, NaCl has no additional effect (14).
In order to find out if increased release of the suicide complex was really taking place, we designed an assay to test our working hypothesis, i.e. that p53 binding to htopoI would catalyze the opening of the clamp and thereby lead to a dissociation of the complex from DNA. On the other hand, NaCl would not directly modulate the complex, but reduce the ionic interactions with DNA and thereby cause an effect similar to p53. In agreement with our prediction, we found that non-covalently bound htopoI as well as TCCs attached to a short oligomer were released from the substrate with both NaCl and p53. The complementary oligomer (OL25) did not have any effect, demonstrating that only after the htopoI–13mer complex has been dissociated can the 25mer enter the gapped DNA. Since htopoI forms a tightly closed clamp around the DNA (17), opening of this clamp is a prerequisite for the dissociation of htopoI from DNA. Since p53 causes a dissociation of htopoI from the DNA (Fig. 5C), this at least indirectly implicates that p53 must cause an opening of the clamp that is probably due to a conformational change of htopoI.
In contrast to our results, it was reported that p53 stimulates the cleavage step of htopoI (15,18). In these studies, a linearized plasmid was used and detergent was employed to detect stimulation. Here, we used a supercoiled plasmid instead of linear DNA. It is known that htopoI binds more strongly to supercoiled than to linear DNA (21). Therefore, a lowering of the binding affinity by p53 might not have been detectable on a linear substrate. On our linear DNA substrate, we show that p53 leads to a release of htopoI from the substrate under native conditions and therefore demonstrate that p53 reduces the affinity of htopoI for DNA. Our finding is also in agreement with a recent report which shows that another topoisomerase I-interacting protein, the protein-phosphorylating enzyme casein kinase II (CK II), through protein–protein interactions (22), stimulates htopoI relaxation activity by reducing the affinity of htopoI for DNA (23). Possibly p53 and protein kinase CK II have the same binding site on htopoI, which could explain why both proteins stimulate htopoI activity by reducing its affinity for DNA. The binding site of p53 on htopoI was suggested to comprise amino acids 156–170 (14); starting from amino acid 156 of htopoI, there is indeed a D-T-K-K-E-K consensus motif for a CK II phosphorylation site.
Radford (24) has suggested that incomplete TIRR-like reactions trigger apoptosis as a response to ionizing radiation. This may be of great interest combined with our finding that tetrameric p53 is required for the stimulation of TIRR. Tetrameric p53 is necessary for the transcriptional activation of apoptosis-inducing genes. Furthermore, we could show that UV light-induced TCC formation correlates with the onset of apoptosis (K.Søe, A.Rockstroh, P.Schache and F.Grosse, unpublished observations). Damage-induced TCCs may at least partially represent htopoI double cleavage complexes and therefore indicate incomplete TIRR reactions. This in turn would corroborate the model of Radford (24).
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Nicole Hartenstein, Annerose Schneider, and Simone Matthä for excellent technical assistance. This work was supported by the ‘Mildred Scheel Stiftung’, Deutsche Krebshilfe.
REFERENCES
- 1.Vousden K. and Lu,X. (2002) Live or let die: the cell’s response to p53. Nature Rev. Cancer, 2, 594–604. [DOI] [PubMed] [Google Scholar]
- 2.Wang J. (2002) Cellular roles of DNA topoisomerases: a molecular perspective. Nature Rev. Mol. Cell Biol., 3, 430–440. [DOI] [PubMed] [Google Scholar]
- 3.Subramanian D., Rosenstein,B. and Muller,M. (1998) Ultraviolet-induced DNA damage stimulates topoisomerase I–DNA complex formation in vivo: possible relationship with DNA repair. Cancer Res., 58, 976–984. [PubMed] [Google Scholar]
- 4.Pourquier P., Takebayashi,Y., Urasaki,Y., Gioffre,C., Kohlhagen,G. and Pommier,Y. (2000) Induction of topoisomerase I cleavage complexes by 1-β-d-arabinofuranosylcytosine (ara-C) in vitro and in ara-C-treated cells. Proc. Natl Acad. Sci. USA, 97, 1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pourquier P., Waltman,J., Urasaki,Y., Loktionova,N., Pegg,A., Nitiss,J. and Pommier,Y. (2001) Topoisomerase I-mediated cytotoxicity of N-methyl-N′-nitro-N-nitrosoguanidine: trapping of topoisomerase I by the O6-methylguanine. Cancer Res., 61, 53–58. [PubMed] [Google Scholar]
- 6.Pourquier P., Gioffre,C., Kohlhagen,G., Urasaki,Y., Goldwasser,F., Hertel,L., Yu,S., Pon,R., Gmeiner,W. and Pommier,Y. (2002) Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin. Cancer Res., 8, 2499–2504. [PubMed] [Google Scholar]
- 7.Mao Y., Okada,S., Chang,L. and Muller,M. (2000) p53 dependence of topoisomerase I recruitment in vivo. Cancer Res., 60, 4538–4543. [PubMed] [Google Scholar]
- 8.Soe K., Dianov,G., Nasheuer,H., Bohr,V., Grosse,F. and Stevnsner,T. (2001) A human topoisomerase I cleavage complex is recognized by an additional human topisomerase I molecule in vitro. Nucleic Acids Res., 29, 3195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stephan H., Grosse,F. and Soe,K. (2002) Human topoisomerase I cleavage complexes are repaired by a p53-stimulated recombination-like reaction in vitro. Nucleic Acids Res., 30, 5087–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao Y., Sun,M., Desai,S. and Liu,L. (2000) SUMO-1 conjugation to topoisomerase I: a possible repair response to topoisomerase-mediated DNA damage. Proc. Natl Acad. Sci. USA, 97, 4046–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vance J. and Wilson,T. (2002) Yeast Tdp1 and Rad1–Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc. Natl Acad. Sci. USA, 99, 13669–13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu C., Pouliot,J. and Nash,H. (2002) Repair of topoisomerase I covalent complexes in the absence of the tyrosyl-DNA phosphodiesterase Tdp1. Proc. Natl Acad. Sci. USA, 99, 14970–14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai S., Zhang,H., Rodriguez-Bauman,A., Yang,J., Wu,X., Gounder,M., Rubin,E. and Liu,L. (2003) Transcription-dependent degradation of topoisomerase I–DNA covalent complexes. Mol. Cell. Biol., 23, 2341–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soe K., Hartmann,H., Schlott,B., Stevnsner,T. and Grosse,F. (2002) The tumor suppressor protein p53 stimulates the formation of the human topoisomerase I double cleavage complex in vitro. Oncogene, 21, 6614–23. [DOI] [PubMed] [Google Scholar]
- 15.Gobert C., Skladanowski,A. and Larsen,A. (1999) The interaction between p53 and DNA topoisomerase I is regulated differently in cells with wild-type and mutant p53. Proc. Natl Acad. Sci. USA, 96, 10355–10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durnford J. and Champoux,J. (1978) The DNA untwisting enzyme from Saccharomyces cerevisiae. Partial purification and characterization. J. Biol. Chem., 253, 1086–1089. [PubMed] [Google Scholar]
- 17.Redinbo M., Stewart,L., Kuhn,P., Champoux,J. and Hol,W. (1998) Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science, 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- 18.Gobert C., Bracco,L., Rossi,F., Olivier,M., Tazi,J., Lavelle,F., Larsen,A. and Riou,J. (1996) Modulation of DNA topoisomerase I activity by p53. Biochemistry, 35, 5778–5786. [DOI] [PubMed] [Google Scholar]
- 19.Albor A., Kaku,S. and Kulesz-Martin,M. (1998) Wild-type and mutant forms of p53 activate human topoisomerase I: a possible mechanism for gain of function in mutants. Cancer Res., 58, 2091–2094. [PubMed] [Google Scholar]
- 20.McConaughy B., Young,L. and Champoux,J. (1981) The effect of salt on the binding of the eucaryotic DNA nicking–closing enzyme to DNA and chromatin. Biochim. Biophys. Acta, 655, 1–8. [DOI] [PubMed] [Google Scholar]
- 21.Krogh S., Mortensen,U., Westergaard,O. and Bonven,B. (1991) Eukaryotic topoisomerase I–DNA interaction is stabilized by helix curvature. Nucleic Acids Res., 19, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kordiyak G., Jakes,S., Ingebritsen,T. and Benbow,R. (1994) Casein kinase II stimulates Xenopus laevis DNA topoisomerase I by physical association. Biochemistry, 33, 13484–13491. [DOI] [PubMed] [Google Scholar]
- 23.Kowalska-Loth B., Girstun,A., Derlacz,R. and Staron,K. (2003) Activation of human topoisomerase I by protein kinase CK2. Mol. Biol. Rep., 30, 107–111. [DOI] [PubMed] [Google Scholar]
- 24.Radford I. (2002) Model for the initiation of ionizing radiation-induced apoptosis in lymphoid cells by complex DNA double-strand breaks. Int. J. Radiat. Biol., 78, 467–474. [DOI] [PubMed] [Google Scholar]