


Abstract
Hepatocyte Nuclear Factor 4α (HNF4α, NR2A1) is central to hepatocyte and pancreatic β-cell functions. Along with retinoid X receptor α (RXRα), HNF4α belongs to the nuclear receptor subfamily 2 (NR2), characterised by a conserved arginyl residue and a glutamate residue insert in helix 7 (H7) of the ligand binding domain (LBD). Crystallographic studies indicate that R348 and E352 residues in RXRα H7 are involved in charge-driven interactions that improve dimerisation. Consistent with these findings, we showed that removing the charge of the corresponding residues in HNF4α H7, R258 and E262, impaired dimerisation in solution. Moreover, our results provide a new concept according to which helices of the HNF4α LBD dimerisation interface contribute differently to dimerisation required for DNA binding; unlike H9 and H10, H7 is not involved in DNA binding. Substitutions of E262 decreased the repression of HNF4α transcriptional activity by a dominant-negative HNF4α mutant, highlighting the importance of this residue for dimerisation in the cell context. The E262 insert is crucial for HNF4α function since its deletion abolished HNF4α transcriptional activity and coactivator recruitment. The glutamate residue insert and the conserved arginyl residue in H7 most probably represent a signature of the NR2 subfamily of nuclear receptors.
INTRODUCTION
Hepatocyte Nuclear Factor 4 is a transcription factor encoded by two genes, HNF4α and HNF4γ, leading to two subtypes of proteins: HNF4α (NR2A1) and HNF4γ (NR2A2) (1). HNF4α is central to embryogenesis (2,3) and is required for the normal function of hepatocytes and pancreatic β-cells (4,5). HNF4α occupies a key position in a complex transcription factor network and directly regulates the expression of genes involved in the transport and metabolism of various nutrients, as well as that of genes coding for serum proteins (1). HNF4α is linked to human diseases: mutations in HNF4α response elements in promoters of factors VII and IX are correlated with haemophilia, while mutations in the HNF4α gene have been found in patients carrying the syndrome of maturity onset diabetes of the young 1 (MODY1) (1,6).
Like other members of the nuclear receptor superfamily, HNF4α has a modular structure consisting of functional domains (7). Two of these domains, the DNA binding domain (DBD) and the ligand binding domain (LBD) are involved in dimerisation of HNF4α, which behaves as a homodimer (8). Dimerisation via the DBD is involved in HNF4α dimerisation on DNA, whereas dimerisation via the LBD is essential for dimerisation in solution and strongly stabilises the HNF4α–DNA complex (9,10). The LBD is also involved in other functions, including transcriptional activation and interaction with transcriptional partners. Both domains are well conserved in HNF4γ (11). Crystal structures of the LBDs of HNF4α and HNF4γ have recently been resolved (12,13). These LBDs adopt the canonical fold of α-helices (10 helices numbered H1–H12 to follow the conventional nomenclature) and β-sheets arranged as an antiparallel α-helical ‘sandwich’ in a three-layer structure shared with the LBD of other nuclear receptors (14). The LBD structures of HNF4α and HNF4γ are very similar and closely resemble that of RXRα, another member of the nuclear receptor subfamily 2 (NR2). Indeed, 194 core α carbons of the LBD of HNF4α and RXRα superimpose with a root mean square deviation (rmsd) of 1.26 Å and 223 core α carbons of the LBD of HNF4γ and RXRα superimpose with a r.m.s.d. of 1.00 Å (12,13). In these structures, the LBD dimerisation interface is made of residues in helices 7 (H7), 9 (H9) and 10 (H10), the major part of the interface being made up of H10.
HNF4α and HNF4γ, which activate transcription in the absence of exogenous ligands, are considered to be constitutive transcriptional activators (1,11,13,15,16). Their LBDs are tightly associated with endogenous fatty acids, which do not act as classical ligands but are likely required for the stability of the protein conformation (12,13). HNF4α transcriptional activity can be enhanced by other transcription factors, including COUP-TF (17), Smad3 and Smad4 (18), or by coactivators, such as members of the p160 family (19,20), CBP/p300 (20–22) and PGC-1 (23). The HNF4α transcriptional activity can also be repressed by negative transcriptional partners such as SHP (24), p53 (25) and SMRT (26).
In the crystal structure of the RXRα LBD, H7 adopts an unusual π-helical geometry that forces the glutamic acid residue in position 352 to bulge outward from the H7 axis (27). This structure gives rise to the formation of a series of intramolecular and intermolecular hydrogen bonds that improve RXRα LBD homodimerisation (27). In particular, residues E352 and R348 are directly involved in the dimerisation interface by forming charge-driven interactions. The π-helical conformation near E352 in the RXRα homodimer and the resulting interactions were also observed in the PPARγ/RXRα LBD heterodimer (28). Sequence alignment indicates that a glutamic acid residue and an arginyl residue are also encountered at the equivalent positions in human and rat HNF4α (E262 and R258). Figure 1 shows that these residues are conserved in remote species such as Drosophila and in the Xenopus HNF4β and are specifically encountered in members of the NR2 subfamily (27–29). Interestingly, in the crystal structure of the HNF4α LBD, a bulge near E262 was also observed in H7 (12). Gampe et al. described this glutamate as a single residue E insert in H7 and hypothesised that it may play a crucial role in the function of nuclear receptors belonging to the NR2 subfamily, including HNF4α (27,28). We investigated the role of the E262 and R258 amino acid residues in HNF4α function using biochemical and mutagenesis studies.
Figure 1.

Structure-based amino acid sequence alignment of H7 of nuclear receptor LBDs (adapted from references 27–29). Usual names of nuclear receptors are indicated on the left side whereas names proposed by the Nuclear Receptor Nomenclature Committee are indicated on the right side. The arrow and boxed R indicate the glutamate and arginyl residues specifically found in members of the NR2 subfamily. Positions of the functional domains, of the I-box and of activation function 2 (AF-2) are shown. *, position in isoform 2 of HNF4α (HNF4α2), which was used in this study.
MATERIALS AND METHODS
DNA constructs
Plasmid pcDNA3 HNF4α2, described in Suaud et al. (30) was used as a template to create pcDNA3 HNF4α2-R258M, -E262A, -E262M, -E262K, -ΔE262, -ΔD261 and E327M constructs by site-directed mutagenesis using the QuickChange™ kit from Stratagene according to the supplier’s recommendations. Vectors pcDNA3 HNF4α-ΔAF-2 (residues 1–358) and pSG5 HNF4α3 were described previously (31,32). Vector pcDNA3 HNF4α-ΔAF-2-E262A was generated by site-directed mutagenesis. Plasmid pGBKT7 HNF4α2, used to express HNF4α2 fused to a c-myc tag in vitro, was obtained by inserting a PCR fragment encompassing human HNF4α2 cDNA into the EcoRI and BamHI sites of pGBKT7 (Clontech). This vector was then used as a template to create pGBKT7 HNF4α2-E262A and -ΔE262 by site-directed mutagenesis. Plasmids pCMVβ-NHA p300 and pGEX2TK p300(340–528) were kindly provided by S.R.Grossman. Plasmids pMT2 COUP-TFII, pTL1 myc-COUP-TFII ΔAB, pGEX5X2 PGC-1(36–797) and pGEX2TK SRC-1a(570–780) were gifts from S.K.Karathanasis, M.Leid, B.M.Spiegelman and M.Tsai, respectively. Plasmid pGEX2TK HNF4α2 was prepared by a strategy identical to that used for cloning pGEX2TK COUP-TFII (30) by inserting a PCR fragment encompassing the human HNF4α2 cDNA. This pGEX2TK HNF4α2 construct was then used as a template to generate pGEX2TK HNF4α2-E262A and -ΔE262 by site-directed mutagenesis. The human HNF1α promoter (–341/+183) cloned in pGL3 was a gift from G.Bell. All constructs were verified by DNA sequencing analysis.
Co-immunoprecipitation assays
Wild-type and mutated HNF4α were in vitro synthesised in reticulocyte lysates (Promega). In co-immunoprecipitation assays performed with an anti-c-myc tag antibody, 5 µl of non-radiolabelled wild-type or mutated c-myc-HNF4α2 was incubated with 5 µl of [35S]methionine-labelled wild-type or mutated HNF4α2 for 1 h at room temperature in 30 µl (final volume) of Ip buffer (50 mM Tris–HCl pH 7.0, 100 mM KCl, 1 mM EDTA, 1 mM DTT, 0.1% Nonidet P40, 0.2% bovine serum albumin, 0.5 mM phenylmethylsulphonyl fluoride and 0.1 mg/ml each of leupeptin, aprotinin and pepstatin). Then, 2 µg of anti-c-myc tag antibody (clone 9E10; Upstate Biotechnology) was added and incubation was continued for 1 h. An aliquot of 150 µl of Ip buffer containing 3 mg of hydrated protein A–Sepharose CL-4B beads (Sigma) was added and samples were incubated under constant agitation for an additional 1 h. After extensive washing of the beads with Ip buffer, bound proteins were resolved by SDS–PAGE. Co-immunoprecipitation assays performed with the α455 antiserum (33) were conducted similarly using 5 µl of non-radiolabelled wild-type or mutated HNF4α2, 5 µl of [35S]methionine-labelled HNF4α3 and 0.25 µl of α455 antiserum. Interactions were quantified using ImageQuant software on a PhosphorImager (Molecular Dynamics).
GST pull-down assays
GST pull-down assays were performed as described previously (30) using [35S]methionine-labelled, in vitro synthesised HNF4α and bacterially expressed GST fusion proteins. Interactions were quantified using ImageQuant software on a PhosphorImager (Molecular Dynamics).
Electrophoretic mobility shift assays (EMSA)
EMSA were performed using in vitro synthesised wild-type or mutated HNF4α2 and 32P-labelled oligonucleotides (0.2 ng) encompassing the HNF4α response element of the human HNF1α promoter (positions –66/–48) (1) or a mutated version of this response element (denoted HNF1 and HNF1 mt, respectively) or site B (position –67/–85) (1) of the human apolipoprotein CIII promoter (denoted CIIIB). Mutations in the HNF1 mt site are underlined in the following sequence, where half-sites are in upper case: tgaACTCCCaAGTTCAgtc. Complexes were formed in 20 µl (final volume) of binding buffer (100 mM KCl, 20 mM HEPES pH 7.9, 5 mM MgCl2, 0.1 mM EDTA, 20% glycerol, 1 mM DTT) in the presence of 1.25 µg of poly(dI·dC)–poly(dI·dC). Complexes were resolved by non-denaturing PAGE in 1× TEA. DNA binding was quantified using a PhosphorImager. In supershift assays, HNF4α proteins were incubated with 0.25 µl of the α455 HNF4α antiserum (33) for 15 min prior to adding the labelled probe.
Cell culture and transient transfection assays
Human embryonic kidney HEK 293 (1.5 × 105 cells per 24-well dishes), COS-1 (4 × 104 cells per 24-well dishes) and HeLa cells (5.5 × 104 cells per 24-well dishes) were grown and transfected as in Suaud et al. (31) with plasmid amounts indicated in the figure legends. Luciferase activities were measured using the Bright-Glo Luciferase assay system (Promega).
Western blot assays
Aliquots of 2.3 × 106 HeLa cells were transfected with 2 µg of wild-type or mutated HNF4α2 expression vector and whole-cell extracts were prepared as in Wang et al. (34). Western blot assays were carried out as in Suaud et al. (30).
Data analysis
Statistical analyses were based on Student’s t-test for unpaired data using Prism software. The statistical significance of differences between values obtained for mutant and wild-type HNF4α (P) is indicated in the legends to the figures.
RESULTS
Residues E262 and R258 are important for HNF4α2 dimerisation in solution
Figure 1 depicts the amino acid sequence alignment of H7 in nuclear receptor LBDs. According to Gampe et al. (27,28), the inserted glutamate residue (indicated by the arrow in Fig. 1) may facilitate RXRα dimerisation (i) because of the π-helix conformation its insertion generates and (ii) because of its carboxylic group, which forms a salt bridge and hydrogen bonds (27,28). A similar conformation was observed near the corresponding E262 residue in the HNF4α LBD (12). We investigated the role of this glutamate residue in HNF4α2 dimerisation. To this end, the HNF4α2 E262 residue was deleted in construct HNF4α2-ΔE262 and its charge was inverted in construct HNF4α2-E262K and removed in constructs HNF4α2-E262A and HNF4α2-E262M, the bulk of the residue being conserved in the latter mutation.
We analysed whether these mutations affected the ability of labelled HNF4α2 mutants to form dimers in solution with wild-type HNF4α2 (HNF4α2 WT). Dimerisation was studied by either co-immunoprecipitation assays using c-myc-HNF4α2 WT or GST pull-down assays using GST-HNF4α2 WT. As expected, HNF4α2 WT efficiently bound to both c-myc-HNF4α2 WT (Fig. 2A) and GST-HNF4α2 WT (Fig. 2B). Conversely, all mutants failed to bind efficiently to c-myc-HNF4α2 WT (Fig. 2A) or GST-HNF4α2 WT (Fig. 2B). Next, we analysed the effects of mutations on dimerisation of a given mutant with itself. For these assays, we focused on mutants E262A and ΔE262. In co-immunoprecipitation assays, no dimer was detectable with either E262A or ΔE262 (Fig. 2C). This result was not due to a lower expression of mutated c-myc-HNF4α2 (Fig. 2C, insert). Impaired dimerisation of mutants was confirmed by pull-down experiments performed in increasingly stringent conditions. HNF4α2 WT dimerisation was unaltered at 300 mM KCl and decreased by only 25% at 600 mM KCl (Fig. 2D). In sharp contrast, dimerisation dropped dramatically at 300 and 600 mM KCl for both mutants (Fig. 2D). Thus, removing the carboxylic group at position 262 is sufficient to strongly impair HNF4α2 dimerisation in solution.
Figure 2.

E262 and R258 are involved in HNF4α2 dimerisation in solution. (A and B) Analyses by co-immunoprecipitation assays and GST pull-down assays, respectively, of dimerisation between immobilised wild-type HNF4α2 fused to c-myc or GST (c-myc-HNF4α2 WT or GST-HNF4α2 WT) and wild-type or mutated [35S]methionine-labelled HNF4α2. Graphs in (A) and (B) indicate means ± SE of HNF4α2 mutant binding relative to that of the wild-type protein from three independent experiments. Inputs were taken into account for binding quantifications. (C) Dimerisation, analysed by co-immunoprecipitation assays of HNF4α2 WT, -ΔE262 or -E262A. For each assay, [35S]methionine-labelled HNF4α2 was incubated with the same protein fused to the c-myc tag. Control of synthesis of c-myc-HNF4α2 WT and mutated proteins is shown in the insert. (D) Dimerisation, analysed by GST pull-down assays of HNF4α2 WT, -ΔE262 and -E262A. For each assay, [35S]methionine-labelled HNF4α2 was incubated with the same protein fused to GST. Pull-down assays were performed in the indicated ionic strength conditions. The graph indicates means ± SE of HNF4α binding at 300 or 600 mM KCl relative to binding at 100 mM KCl (set to 100%) from three independent experiments. (E) Dimerisation, analysed by co-immunoprecipitation assays, between immobilised c-myc-HNF4α2 WT and [35S]methionine-labelled HNF4α2 WT or -R258M. The graph indicates mean ± SE of HNF4α2-R258M binding relative to that of the wild-type protein from four independent experiments. Inputs were taken into account for binding quantifications.
In RXRα, R348 is also involved in dimerisation (27,28). Interestingly, this arginyl residue is specifically conserved in all members of the NR2 subfamily (Fig. 1). This led us to investigate the role of the corresponding R258 residue of HNF4α2 in dimerisation. When the positive charge of this residue was removed in the R258M mutant, HNF4α dimerisation dropped (Fig. 2E), although less markedly than when elicited by E262 mutations (compare decrease in dimerisation in Fig. 2A and E). Together, these results show the involvement of the charged groups of residues E262 and R258 in the stabilisation of HNF4α2 dimerisation in solution.
Mutations of residues E262 and R258 do not impair DNA binding
Since HNF4α is known to bind DNA as a homodimer (8), we analysed the effect of E262 and R258 mutations on HNF4α2 DNA binding. Surprisingly, none of the mutations altered HNF4α2 binding to the HNF4α response element of the HNF1α promoter (HNF1 site, Fig. 3A). The specificity of the band shift was ascertained by supershifting it with HNF4α antiserum α455 (33) (Fig. 3B). Similar results were obtained on another HNF4 response element, site B of the apoCIII promoter (CIIIB site), which is also a direct repeat 1 (DR1) (Fig. 3E, results in the absence of competitor). Results obtained with increasing amounts of labelled probe exclude that lack of detection of DNA binding impairment by E262A mutation was due to saturated binding (Fig. 3C). Similar results were obtained with other mutants (data not shown). Mutated and wild-type HNF4α yielded a retarded band of identical intensity and electrophoretic mobility. More specifically, we did not observe a band of higher mobility corresponding to a HNF4α monomer bound to DNA as observed for the RAR monomer (15). In addition, to rule out the possibility that mutants bound to DNA as two adjacent monomers, we performed EMSA with the HNF1 mt site containing one mutated half-site and obtained no binding of the mutants (Fig. 3D). Thus, HNF4α mutants, like wild-type HNF4α, bind DNA as homodimers. To evidence possibly diminished interactions with DNA, we performed EMSA in less-favorable conditions. First, we increased the ionic strength (300 versus 100 mM KCl in Fig. 3A) in EMSA but found no difference in DNA binding between wild-type and mutated HNF4α (data not shown). Second, we carried out competition experiments with COUP-TFII, which binds to several HNF4α response elements. The rationale here was to check whether this nuclear receptor impinges on DNA binding of HNF4α2-E262A more efficiently than on wild-type HNF4α2. In these assays, the truncated COUP-TFII ΔAB was used to distinguish complexes formed with either HNF4α2 or COUP-TFII. EMSA was performed on the CIIIB site since COUP-TFII does not bind to the HNF1 site (17,30). COUP-TFII ΔAB similarly competed for HNF4α2 WT and -E262A DNA binding (Fig. 3E). Also, unlabelled DNA competed equally for DNA binding of both proteins (data not shown). In addition, we observed that mutations did not alter binding to a direct repeat 2 (DR2), to which HNF4α binds less efficiently than to a DR1 (9) (data not shown). It appears therefore that the E262 residue does not play a significant role in HNF4α2 DNA binding.
Figure 3.

Mutations of E262 and R258 residues do not impair HNF4α2 DNA binding. (A) DNA binding of HNF4α2 mutants to the 32P-labelled HNF4α response element of the HNF1α promoter (HNF1 site). Control of in vitro synthesis of wild-type and mutated HNF4α2, used in EMSA, is shown in the insert (values on the right end indicate molecular size markers). The graph indicates means ± SE of mutated HNF4α2 DNA binding relative to that of the wild-type protein from three independent experiments. (B) Specificity of binding. Unprogrammed reticulocyte lysate (mock) yielded no shifted band. Supershifting was performed in the presence of the specific α455 HNF4α antiserum. (C) EMSA performed with a constant amount of HNF4α2 WT or HNF4α2-E262A and increasing amounts of labelled HNF1 probe. (D) HNF4α2-E262A did not bind as a monomer to the half-site of the HNF4α response element (HNF1 mt). (E) Competition experiments with COUP-TFII ΔAB. EMSA were performed on the HNF4 response element of the apoCIII promoter (CIIIB site) using in vitro synthesised HNF4α2 WT or HNF4α2-E262A and increasing amounts of the competitor COUP-TFII ΔAB. The amount of reticulocyte lysate in each lane was held constant by the appropriate addition of unprogrammed lysate. The positions of HNF4α2 and COUP-TFII ΔAB homodimers bound to DNA are indicated.
Differential effects on dimerisation in solution and DNA binding of mutations in H7 and H10 of HNF4α LBD
Within the LBD, the I-box has been shown to constitute a dimerisation interface that mediates cooperative binding to DNA of nuclear receptors (35). The I-box almost perfectly overlaps H9 and H10 that form the major portion of the dimer interface in RXRα and HNF4α homodimers (12,13,27,36). Mutagenesis studies also showed that the HNF4α I-box is an important interaction interface for homodimerisation in solution (37) and is likely involved in DNA binding (15). We therefore compared the effects of mutations in H7 and H10 on HNF4α dimerisation in solution and DNA binding. Until now, the role of the HNF4α I-box has been studied using multiple (double, triple or quadruple) mutations (15,37). For example, the K300E-E327K HNF4α double mutant was used to show the involvement of the salt bridge between these residues in the exclusive homodimerisation of HNF4α (15). For our comparative study, we chose a single mutation located in H10 E327M. In co-immunoprecipitation assays with c-myc-HNF4α2 WT, we observed that the E327M mutation decreased HNF4α2 dimerisation in solution by 25% (Fig. 4A). The extent of this decrease was similar to that observed for the R258M mutation (Fig. 2E). A similar drop in HNF4α2 dimerisation in solution with E327M and R258M mutations was also observed in GST pull-down assays (Fig. 4B). Note that the impairment of dimerisation by the E262 mutations was stronger, as observed in both co-immunoprecipitation and GST pull-down assays (Fig. 2A and B). The stronger effect of the E262A mutation on HNF4α2 dimerisation in solution was confirmed by a different co-immunoprecipitation assay, where unlabelled HNF4α2 WT, -R258M, -E262A or -E327M and the α455 antiserum raised against their common C-terminus were used to co-immunoprecipitate labelled HNF4α3, which contains a different C-terminus not recognised by this antiserum. R258M, E262A and E327M mutations strongly impaired the ability of HNF4α2 to interact with HNF4α3 (Fig. 4C), thus confirming the involvement of these three residues in HNF4α dimerisation. Quantification again indicated that the E262A mutation resulted in the strongest impairment of HNF4α2 dimerisation in solution (Fig. 4C).
Figure 4.

Differential effects on dimerisation in solution and DNA binding of mutations in H7 and H10 of the HNF4α LBD. (A) Dimerisation, analysed by co-immunoprecipitation assays, between immobilised c-myc-HNF4α2 WT and [35S]methionine-labelled HNF4α2 WT or -E327M. The graph indicates mean ± SE of HNF4α2-E327M binding relative to that of the wild-type protein from three independent experiments. Inputs were taken into account for binding quantifications. (B) Dimerisation, analysed by GST pull-down assays, between immobilised GST-HNF4α2 WT and [35S]methionine-labelled HNF4α2 WT, -R258M and -E327M. Pull-down assays were performed in various ionic strength conditions as in Figure 2D. The graph indicates means ± SE of mutant binding relative to that of the wild-type HNF4α from three independent experiments. Inputs were taken into account for binding quantifications. (C) Dimerisation, analysed by co-immunoprecipitation assays using the α455 antiserum, between immobilised HNF4α2 WT, -R258M, -E262A or -E327M and [35S]methionine-labelled HNF4α3, which is not recognised by the α455 antiserum. Control of HNF4α2 protein synthesis is shown in the insert. The graph indicates means ± SE of HNF4α3 retention by HNF4α2 mutants relative to HNF4α3 retention by HNF4α2 WT from four independent experiments. Control of protein synthesis was taken into account for binding quantifications. (D) DNA binding of HNF4α2-E327M on the HNF1 site, analysed by EMSA performed as in Figure 3A. Supershifts were obtained in the presence of the α455 HNF4α antiserum as indicated. Control of protein synthesis is shown in the insert. The graph indicates mean ± SE of HNF4α2-E327M DNA binding relative to that of the wild-type protein from three independent experiments.
In contrast to E262A and R258M mutations, the E327M mutation moderately but significantly decreased HNF4α2 DNA binding to the HNF1 site (Fig. 4D). The specificity of the band shift was ascertained by supershifting it with the α455 antiserum (Fig. 4D). Interestingly, the intensities of the supershifted bands obtained with wild-type and E327M HNF4α were similar. It has previously been shown that the impaired DNA binding of HNF4α mutants (i.e. K300E-E327K and R154X) due to decreased dimerisation could be rescued in the presence of antibodies, which facilitate dimerisation. This phenomenon is likely due to the bivalency of antigen recognition by antibodies (15,32). Conversely, when decreased DNA binding is not due to a loss of dimerisation, DNA binding is not recovered in the presence of antibodies, as observed for the D126Y and D126H HNF4α mutants (38). Therefore, the decreased DNA binding and impaired dimerisation of the E327M mutant are directly correlated. E327M mutation also impaired HNF4α2 DNA binding to the CIIIB site and to the synthetic DR2 site (data not shown). Our results not only confirm that H10 and H9 constitute a motif of the LBD dimerisation interface required for efficient DNA binding of HNF4α (15), but also show that another region of the LBD dimerisation interface, namely H7, is not required for strong DNA binding of this protein.
Deletion of the E262 residue dramatically impairs HNF4α2 transcriptional activity
Next we investigated the effects of mutations of E262 and R258 residues on HNF4α2 transcriptional activity. The HNF4α2-mediated activation of the HNF1α promoter was not altered by any of the substitution mutations in HeLa cells (Fig. 5A). However, HNF4α transcriptional activity was abolished by E262 deletion (Fig. 5A). In HEK 293 and COS-1 cells, transcriptional activity was unaffected by substitution mutations but was dramatically impaired by the deletion mutation ΔE262 (Fig. 5B and C, where only data with wild-type, E262A and ΔE262 are shown). The loss of activation by HNF4α2-ΔE262 was neither due to a lower protein expression, as controlled by western blotting (Fig. 6D), nor to an unfolding of the protein, as assessed by limited protease mapping assays using chymotrypsin or trypsin (data not shown). Deletion of the neighbouring residue, ΔD261, also abolished HNF4α transcriptional activity (Fig. 5A, last bar). The dramatic drop in HNF4α transcriptional activity caused by the E262 deletion prompted us to determine whether this mutant exhibits a dominant-negative effect on the wild-type protein. However, HNF4α2-ΔE262 was unable to repress the transcriptional activity of wild-type HNF4α (Fig. 5E).
Figure 5.

Deletion of E262 strongly affects HNF4α2 transcriptional activity. HeLa (A), HEK 293 (B) and COS-1 cells (C) were transiently transfected with 12.5 ng of expression vector for wild-type or mutated HNF4α2 or the corresponding empty vector (–) together with 250 ng of HNF1α promoter construct. Fold induction refers to the activity with no HNF4α2 derivative (–), which was set to 1. Results are means ± SE of three independent experiments performed in triplicate. **, P = 0.0015, 0.0060 and 0.0018 for the ΔE262 mutant in (A–C), respectively; ***, P < 0.0001 for the ΔD261 mutant in (A). (D) Western blotting of HeLa cell extracts. (E) HNF4α-ΔE262 does not exhibit a dominant-negative activity on wild-type HNF4α. COS-1 cells were transfected as in (C), except that equal amounts of wild-type HNF4α and HNF4α-ΔE262 or control vector (–) were co-transfected. (F and G) Effects of substitution mutations on the dominant-negative activity of HNF4α-ΔAF-2. COS-1 cells were transfected as in (C), except that in (F) plasmids expressing wild-type, E262A or E262K HNF4α were co-transfected with an equal amount of vector expressing HNF4α-ΔAF-2 or the control vector (–), whereas in (G) pcDNA3 HNF4α2 WT was co-transfected with an equal amount of vectors expressing HNF4α-ΔAF-2 or HNF4α-ΔAF-2-E262A or the control vector (–). Activation of the HNF1α promoter is expressed relative to that obtained when only full-length proteins were expressed. Results are means ± SE of three independent experiments performed in triplicate. **, P = 0.0040 in (F); ***, P < 0.0001 in (F); *, P = 0.0278 in (G).
Figure 6.
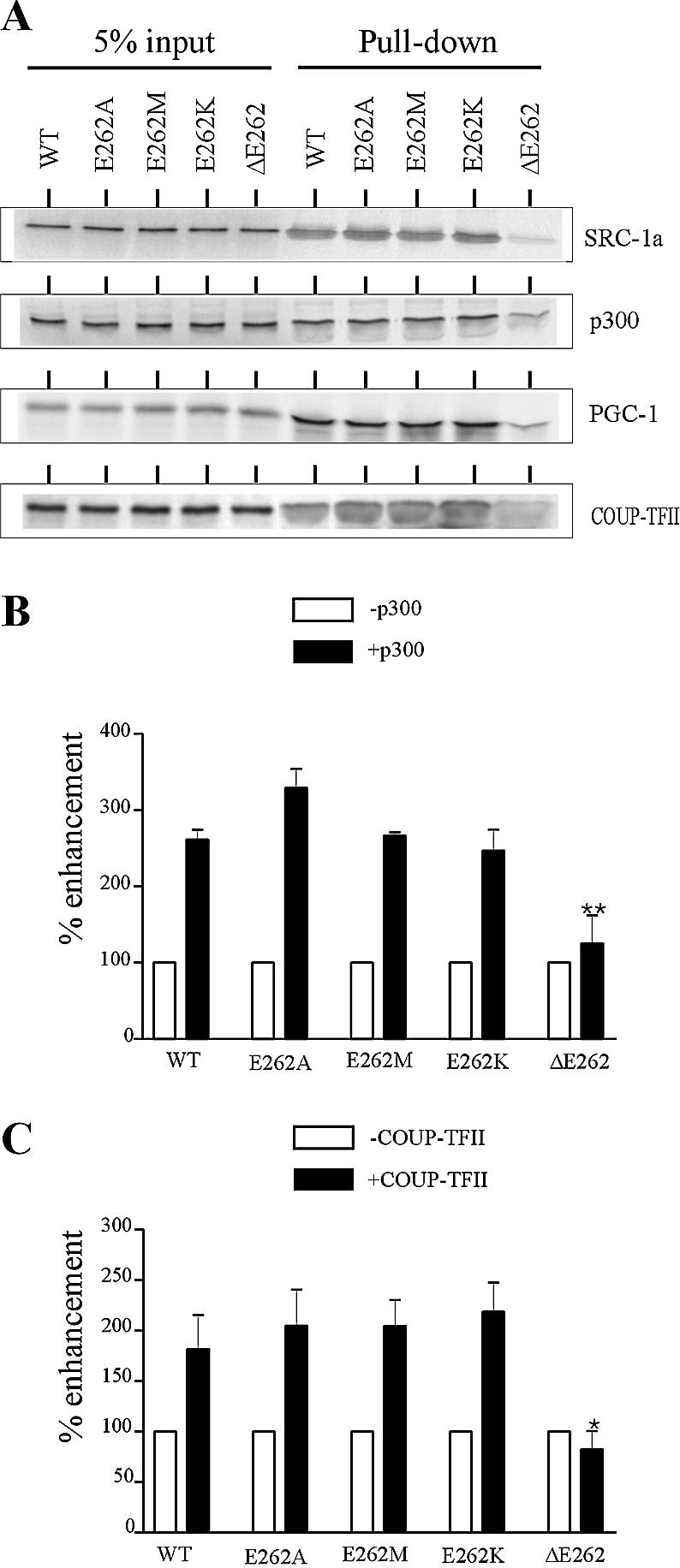
Deletion of E262 markedly decreases recruitment of transcriptional partners. (A) GST pull-down assays were performed using GST-SRC-1a (570–780), GST-p300 (340–528), GST-PGC-1 (36–797) or GST-COUP-TFII and [35S]methionine-labelled WT or mutated HNF4α2. Inputs correspond to 5 or 2% (for the experiment with GST-SRC-1a) of amounts of labelled proteins used in the assays. (B and C) Effects of mutations of the E262 residue on the enhancement of HNF4α2 transcriptional activity by p300 and COUP-TFII, respectively. HeLa cells were transiently transfected with 12.5 ng of wild-type or mutated HNF4α2 expression vector, 250 ng of HNF1α promoter construct and 250 ng of empty control vector (white bars) or expression vectors (black bars) for p300 or COUP-TFII. Shown are per cent enhancements of wild-type and mutated HNF4α activities. Results are means ± SE of three independent experiments performed in triplicate. *, P = 0.0109; **, P = 0.0037.
To gain further insight into the consequences of altered dimerisation of HNF4α by substitution mutations, we took advantage of the fact that a truncated HNF4α lacking the AF-2 activation function module (HNF4α-ΔAF-2, left part of Fig. 5F) exhibits a dominant-negative effect through its ability to dimerise with the wild-type protein (7). In a first set of experiments, we compared HNF4α-ΔAF-2 repression on the transcriptional activities of wild-type HNF4α and of the two mutants E262A and E262K. The rationale was that the dominant-negative mutant would exhibit a weaker repression on mutant HNF4α, with impaired dimerisation, than on wild-type HNF4α. HNF4α-ΔAF-2 reduced the activity of wild-type HNF4α by 50% while it reduced that of E262A and E262K mutants by 29 and 26%, respectively (Fig. 5F). Accordingly, compared to HNF4α-ΔAF-2, HNF4α-ΔAF-2 bearing the E262A mutation (HNF4α-ΔAF-2-E262A) exhibited a weaker repressive activity on the wild-type protein. Indeed, HNF4α-ΔAF-2 reduced the wild-type HNF4α-mediated activation of transcription by 50% while HNF4α-ΔAF-2-E262A reduced it by 30% (Fig. 5G). By demonstrating that substitution mutations of E262 affect repression of HNF4α-mediated activation of transcription, our results highlight the importance of this residue for dimerisation in a cell context.
The E262 insert is required for efficient recruitment of coactivators by HNF4α
Next, we investigated the effects of the mutations on the physical interaction between HNF4α2 and its transcriptional coactivators. In this study we included interaction with COUP-TFII, which acts as an HNF4α transcriptional partner on the HNF1α promoter (17). GST pull-down assays showed that E262 deletion markedly impaired HNF4α2 interaction with SRC-1a, p300, PGC-1 and COUP-TFII, whereas substitution mutations of E262 did not alter these interactions (Fig. 6A). In line with these data, enhancement of HNF4α2 transcriptional activity on the HNF1α promoter by p300 or COUP-TFII was not altered by substitution mutations but was abolished by the E262 deletion, as evidenced by transient transfection assays (Fig. 6B and C). Deletion of the E262 residue also disrupted the functional cooperation between HNF4α2 and SRC-1a (data not shown).
DISCUSSION
We observed that two charged residues located in H7 of the HNF4α LBD, i.e. R258 and E262, are involved in HNF4α dimerisation in solution. We failed to detect significant impairment of DNA binding of their substitution mutants. At first glance, this may seem striking, since dimerisation via the LBD is thought to be crucial for strong HNF4α DNA binding. However, the accuracy of our findings is verified by the fact that (i) results obtained by various pull-down and co-immunoprecipitation assays all converge and (ii) the same methods and EMSA allowed us to detect the expected impairments due to the E327M mutation located in H10. In addition, our results are in agreement with recent crystallographic studies of the HNF4α LBD, which indicate that H7, together with H9 and H10, constitute the dimerisation interface of this nuclear receptor (12,13). The interest of our work was to describe the critical role of the charge of R258 and E262 residues in dimerisation in solution of HNF4α, a role suggested by Gampe et al. from the crystal structure of RXRα (27,28). Mutants of nuclear receptors that are deficient in dimerisation in solution after in vitro analysis but still able to efficiently bind DNA have already been described (39–41). To explain the behaviour of several of these mutants, it was suggested that dimers were stabilised on DNA via the DBD dimerisation interface. However, such a hypothesis is unlikely here since the E327M mutation impaired DNA binding in spite of its lower effect on dimerisation in solution than E262 mutations. A more probable explanation of the efficient DNA binding of H7 mutants is that the HNF4α dimerisation interface is improved by a DNA binding-induced conformational change of the LBD. An allosteric effect of DNA, which modulated HNF4α recruitment of co-repressors, has recently been documented (42). We also observed an allosteric effect of DNA that most likely occurs in the LBD and modulates coactivator recruitment (data not shown).
Like the substitution mutants in H7, several missense HNF4α mutants, including naturally occurring mutants associated with diabetes (6,30,38), also exhibit unaltered or slightly impaired transcriptional activities in transient transfection assays. Failure to detect a subtle loss of HNF4α function in these assays where the protein is overexpressed does not exclude that these mutations may have greater consequences on HNF4α function when the protein is expressed at normal levels. Interestingly, a significant effect of substitution mutants in H7 on HNF4α transcriptional activity could be detected by analysing the repression of this activity with an HNF4α mutant exhibiting a dominant-negative activity. These results demonstrate the importance of E262 for HNF4α dimerisation in a cell context. Moreover, it should be kept in mind that the activity of nuclear receptors can be repressed by proteins that prevent their dimerisation (43,44). HNF4α is a target of AMP-activated protein kinase, which inhibits HNF4α activity by decreasing its dimerisation (45). The ability of HNF4α to form a highly stable homodimer in solution (8) is likely crucial in this type of repressive mechanism that does not exclusively occur on DNA. Other pathways that do not necessarily require DNA binding include cross-regulatory mechanisms in which nuclear receptors are frequently involved. In these mechanisms, dimerisation can be of major importance (46). Since HNF4α is known to interact with numerous other transcription factors, including Smad proteins (18), it likely participates in cross-regulatory pathways. Therefore, involvement of H7 residues in dimerisation in solution may be crucial in HNF4α biological activities.
Results obtained with the deletion mutant HNF4α-ΔE262 indicate that the E insert is required for HNF4α transcriptional activity and recruitment of transcriptional partners. Note that, probably due in part to its impaired dimerisation, HNF4α-ΔE262 did not exhibit a dominant-negative activity. None of the MODY1-associated HNF4α mutants can repress wild-type HNF4α activity (6). Even single or double mutations in the AF-2 module, which is crucial for HNF4α transcriptional activity, do not result in a dominant-negative effect (J.Eeckhoute, unpublished results). Interestingly, HNF4α-ΔD261, a mutant having a deletion of the neighbouring residue, exhibited an abolished transcriptional activity but an unaltered DNA binding (EMSA data not shown), a behaviour that is very similar to that of the ΔE262 mutant. These findings may be explained by the loss of the special conformation (the π-helix) in H7, which was hypothesised to result from the presence of an additional residue, the ‘E insert’ (28). The D261 and E262 deletions most probably modify the orientation of the side chain of residues in H7. Several residues in H7 point towards the HNF4α ligand binding pocket and probably participate in fatty acid binding, which is required for the stability and function of HNF4α and HNF4γ (12,13). The marked decrease in HNF4α transcriptional activity and recruitment of transcriptional partners caused by the E262 deletion may reflect the alteration in fatty acid binding secondary to the loss of the π-helix conformation generated by this glutamate residue.
From a mechanistic point of view, our results unravel distinct contributions of H7, H9 and H10 to HNF4α dimerisation and DNA binding. H9 and H10 constitute the core of the LBD dimerisation interface of several non-steroid nuclear receptors (27,36,47,48). This core is required for efficient DNA binding (35). In HNF4α, this core is also involved in dimerisation required for DNA binding, as evidenced using a double mutation in H9 and H10 (15). We observed that a single mutation in H10, E327M, was sufficient to alter HNF4α dimerisation and DNA binding. The role of H7 in nuclear receptors has been less extensively studied and only recent crystallographic data indicate that its contribution to dimerisation varies according to the receptor and, for RXRα, with its homodimeric or heterodimeric state (47). Concerning HNF4α, our results clearly show that H7 has a crucial role in dimerisation in solution but does not contribute to the dimerisation activity required for DNA binding.
In conclusion, our results demonstrate the key role of H7 in HNF4α dimerisation in solution and provide a new concept by which helices of the HNF4α LBD dimerisation interface contribute differently to the dimerisation required for DNA binding: whereas H9 and H10 are required for DNA binding, H7 is involved solely in dimerisation in solution. Our data also show the key role of the E262 insert that causes a special conformation in H7 observed in both RXRα and HNF4α. The key role of this glutamate is likely shared by other members of the NR2 subfamily, where it is specifically encountered, and most probably the E insert corresponds to a signature of this subfamily.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are indebted to Drs S. R. Grossman, S. K. Karathanasis, B. M. Spiegelman, M. Leid and M. Tsai for the generous gift of plasmids. They acknowledge Dr P. Lefebvre’s collaborators for helpful discussions, I. Briche for technical assistance and L. Hauffman-Touzet for proofreading.
REFERENCES
- 1.Sladek F.M. and Seidel,S.D. (2001) Hepatocyte Nuclear Factor 4alpha. In Burris,T.P. and McCabe,E. (eds), Nuclear Receptors and Genetic Disease. Academic Press, San Francisco, CA, pp. 309–361. [Google Scholar]
- 2.Duncan S.A., Nagy,A. and Chan,W. (1997) Murine gastrulation requires HNF-4 regulated gene expression in the visceral endoderm: tetraploid rescue of Hnf-4(–/–) embryos. Development, 124, 279–287. [DOI] [PubMed] [Google Scholar]
- 3.Nastos A., von Strandmann,E.P., Weber,H. and Ryffel,G.U. (1998) The embryonic expression of the tissue-specific transcription factor HNF1 alpha in Xenopus: rapid activation by HNF4 and delayed induction by mesoderm inducers. Nucleic Acids Res., 26, 5602–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayhurst G.P., Lee,Y.H., Lambert,G., Ward,J.M. and Gonzalez,F.J. (2001) Hepatocyte Nuclear Factor 4alpha (Nuclear Receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol., 21, 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang H., Maechler,P., Antinozzi,P.A., Hagenfeldt,K.A. and Wollheim,C.B. (2000) HNF4alpha regulates the expression of pancreatic beta-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J. Biol. Chem., 275, 35953–35959. [DOI] [PubMed] [Google Scholar]
- 6.Ryffel G.U. (2001) Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J. Mol. Endocrinol., 27, 11–29. [DOI] [PubMed] [Google Scholar]
- 7.Hadzopoulou-Cladaras M., Kistanova,E., Evagelopoulou,C., Zeng,S., Cladaras,C. and Ladias,J.A.A. (1997) Functional domains of the nuclear receptor Hepatocyte Nuclear Factor 4. J. Biol. Chem., 272, 539–550. [DOI] [PubMed] [Google Scholar]
- 8.Jiang G., Nepomuceno,L., Hopkins,K. and Sladek,F.M. (1995) Exclusive homodimerization of the orphan receptor hepatocyte nuclear factor 4 defines a new subclass of nuclear receptors. Mol. Cell. Biol., 15, 5131–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang G. and Sladek,F.M. (1997) The DNA binding domain of Hepatocyte Nuclear Factor 4 mediates cooperative, specific binding to DNA and heterodimerization with the retinoid X receptor α. J. Biol. Chem., 272, 1218–1225. [DOI] [PubMed] [Google Scholar]
- 10.Jiang G.Q., Lee,U. and Sladek,F.M. (1997) Proposed mechanism for the stabilization of nuclear receptor DNA binding via protein dimerization. Mol. Cell. Biol., 17, 6546–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drewes T., Senkel,S., Holewa,B. and Ryffel,G.U. (1996) Human hepatocyte nuclear factor 4 isoforms are encoded by distinct and differentially expressed genes. Mol. Cell. Biol., 16, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhe-Paganon S., Duda,K., Iwamoto,M., Chi,Y.I. and Shoelson,S.E. (2002) Crystal structure of the HNF4alpha ligand binding domain in complex with endogenous fatty acid ligand. J. Biol. Chem., 277, 37973–37976. [DOI] [PubMed] [Google Scholar]
- 13.Wisely G., Miller,A., Davis,R., Thornquest,A., Johnson,R., Spitzer,T., Sefler,A., Shearer,B., Moore,J., Miller,A. et al. (2002) Hepatocyte Nuclear Factor 4 is a transcription factor that constitutively binds fatty acids. Structure, 10, 1225–1234. [DOI] [PubMed] [Google Scholar]
- 14.Bourguet W., Germain,P. and Gronemeyer,H. (2000) Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci., 21, 381–388. [DOI] [PubMed] [Google Scholar]
- 15.Bogan A.A., Dallas-Yang,Q., Ruse,M.D.,Jr, Maeda,Y., Jiang,G., Nepomuceno,L., Scanlan,T.S., Cohen,F.E. and Sladek,F.M. (2000) Analysis of protein dimerization and ligand binding of orphan receptor HNF4alpha. J. Mol. Biol., 302, 831–851. [DOI] [PubMed] [Google Scholar]
- 16.Peiler G., Bockmann,B., Nakhei,H. and Ryffel,G.U. (2000) Inhibitor of the tissue-specific transcription factor HNF4, a potential regulator in early Xenopus development. Mol. Cell. Biol., 20, 8676–8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ktistaki E. and Talianidis,I. (1997) Chicken ovalbumin upstream promoter transcription factors act as auxiliary cofactors for hepatocyte nuclear factor 4 and enhance hepatic gene expression. Mol. Cell. Biol., 17, 2790–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou W.C., Prokova,V., Shiraishi,K., Valcourt,U., Moustakas,A., Hadzopoulou-Cladaras,M., Zannis,V.I. and Kardassis,D. (2003) Mechanism of a transcriptional cross talk between transforming growth factor-beta-regulated Smad3 and Smad4 proteins and orphan nuclear receptor Hepatocyte Nuclear Factor-4. Mol. Biol. Cell, 14, 1279–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J.C., Stafford,J.M. and Granner,D.K. (1998) SRC-1 and GRIP1 coactivate transcription with hepatocyte nuclear factor 4. J. Biol. Chem., 273, 30847–30850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sladek F.M., Ruse,M.D., Nepomuceno,L., Huang,S.M. and Stallcup,M.R. (1999) Modulation of transcriptional activation and coactivator interaction by a splicing variation in the F domain of nuclear receptor hepatocyte nuclear factor 4 alpha 1. Mol. Cell. Biol., 19, 6509–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dell H. and Hadzopoulou-Cladaras,M. (1999) CREB-binding protein is a transcriptional coactivator for hepatocyte nuclear factor-4 and enhances apolipoprotein gene expression. J. Biol. Chem., 274, 9013–9021. [DOI] [PubMed] [Google Scholar]
- 22.Soutoglou E., Katrakili,N. and Talianidis,I. (2000) Acetylation regulates transcription factor activity at multiple levels. Mol. Cell, 5, 745–751. [DOI] [PubMed] [Google Scholar]
- 23.Yoon J.C., Puigserver,P., Chen,G., Donovan,J., Wu,Z., Rhee,J., Adelmant,G., Stafford,J., Kahn,C.R., Granner,D.K. et al. (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature, 413, 131–138. [DOI] [PubMed] [Google Scholar]
- 24.Lee Y.K., Dell,H., Dowhan,D.H., Hadzopoulou-Cladaras,M. and Moore,D.D. (2000) The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Mol. Cell. Biol., 20, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maeda Y., Seidel,S.D., Wei,G., Liu,X. and Sladek,F.M. (2002) Repression of hepatocyte nuclear factor 4alpha tumor suppressor p53: involvement of the ligand-binding domain and histone deacetylase activity. Mol. Endocrinol., 16, 402–410. [DOI] [PubMed] [Google Scholar]
- 26.Ruse M.D. Jr, Privalsky,M.L. and Sladek,F.M. (2002) Competitive cofactor recruitment by orphan receptor hepatocyte nuclear factor 4alpha1: modulation by the F domain. Mol. Cell. Biol., 22, 1626–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gampe R.T. Jr, Montana,V.G., Lambert,M.H., Wisely,G.B., Milburn,M.V. and Xu,H.E. (2000) Structural basis for autorepression of retinoid X receptor by tetramer formation and the AF-2 helix. Genes Dev., 14, 2229–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gampe R.T. Jr, Montana,V.G., Lambert,M.H., Miller,A.B., Bledsoe,R.K., Milburn,M.V., Kliewer,S.A., Willson,T.M. and Xu,H.E. (2000) Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol. Cell, 5, 545–555. [DOI] [PubMed] [Google Scholar]
- 29.Watkins R.E., Wisely,G.B., Moore,L.B., Collins,J.L., Lambert,M.H., Williams,S.P., Willson,T.M., Kliewer,S.A. and Redinbo,M.R. (2001) The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science, 292, 2329–2333. [DOI] [PubMed] [Google Scholar]
- 30.Suaud L., Hemimou,Y., Formstecher,P. and Laine,B. (1999) Functional study of the E276Q mutant Hepatocyte Nuclear Factor-4α found in type 1 maturity-onset diabetes of the young, impaired synergy with Chicken Ovalbumin Upstream Promoter Transcription Factor II on the Hepatocyte Nuclear Factor-1 promoter. Diabetes, 48, 1162–1167. [DOI] [PubMed] [Google Scholar]
- 31.Suaud L., Formstecher,P. and Laine,B. (1999) The activity of the activation function 2 of the human hepatocyte nuclear factor 4 (HNF-4) is differently modulated by F domains from various origins. Biochem. J., 340, 161–169. [PMC free article] [PubMed] [Google Scholar]
- 32.Laine B., Eeckhoute,J., Suaud,L., Briche,I., Furuta,H., Bell,G.I. and Formstecher,P. (2000) Functional properties of the R154X HNF-4alpha protein generated by a mutation associated with maturity-onset diabetes of the young, type 1. FEBS Lett., 479, 41–45. [DOI] [PubMed] [Google Scholar]
- 33.Sladek F.M., Zhong,W., Lai,E. and Darnell,J.E.,Jr (1990) Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev., 4, 2353–2365. [DOI] [PubMed] [Google Scholar]
- 34.Wang J.C., Stromstedt,P.E., Sugiyama,T. and Granner,D.K. (1999) The phosphoenolpyruvate carboxykinase gene glucocorticoid response unit: identification of the functional domains of accessory factors HNF3 beta (hepatic nuclear factor-3 beta) and HNF4 and the necessity of proper alignment of their cognate binding sites. Mol. Endocrinol., 13, 604–618. [DOI] [PubMed] [Google Scholar]
- 35.Perlmann T., Umesono,K., Rangarajan,P.N., Forman,B.M. and Evans,R.M. (1996) Two distinct dimerization interfaces differentially modulate target gene specificity of nuclear hormone receptors. Mol. Endocrinol., 10, 958–966. [DOI] [PubMed] [Google Scholar]
- 36.Bourguet W., Ruff,M., Chambon,P., Gronemeyer,H. and Moras,D. (1995) Crystal structure of the ligand-binding domain of the human nuclear receptor RXR alpha. Nature, 375, 377–382. [DOI] [PubMed] [Google Scholar]
- 37.Lee S.-K., Na,S.-Y., Kim,H.-J., Soh,J., Choi,H.-S. and Lee,J.W. (1998) Identification of critical residues for heterodimerization within the ligand-binding domain of retinoid X receptor. Mol. Endocrinol., 12, 325–332. [DOI] [PubMed] [Google Scholar]
- 38.Oxombre B., Moerman,E., Eeckhoute,J., Formstecher,P. and Laine,B. (2002) Mutations in hepatocyte nuclear factor 4alpha gene associated with diabetes result in greater loss of HNF4alpha function in pancreatic beta-cells than in nonpancreatic beta-cells and in reduced activation of the apolipoprotein CIII promoter in hepatic cells. J. Mol. Med., 80, 423–430. [DOI] [PubMed] [Google Scholar]
- 39.Gorla-Bajszczak A., Juge-Aubry,C., Pernin,A., Burger,A.G. and Meier,C.A. (1999) Conserved amino acids in the ligand-binding and tau(i) domains of the peroxisome proliferator-activated receptor alpha are necessary for heterodimerization with RXR. Mol. Cell. Endocrinol., 147, 37–47. [DOI] [PubMed] [Google Scholar]
- 40.Chen S., Costa,C.H., Nakamura,K., Ribeiro,R.C. and Gardner,D.G. (1999) Vitamin D-dependent suppression of human atrial natriuretic peptide gene promoter activity requires heterodimer assembly. J. Biol. Chem., 274, 11260–11266. [DOI] [PubMed] [Google Scholar]
- 41.Ribeiro R.C., Feng,W., Wagner,R.L., Costa,C.H., Pereira,A.C., Apriletti,J.W., Fletterick,R.J. and Baxter,J.D. (2001) Definition of the surface in the thyroid hormone receptor ligand binding domain for association as homodimers and heterodimers with retinoid X receptor. J. Biol. Chem., 276, 14987–14995. [DOI] [PubMed] [Google Scholar]
- 42.Torres-Padilla M.E., Sladek,F.M. and Weiss,M.C. (2002) Developmentally regulated N-terminal variants of the nuclear receptor HNF4alpha mediate multiple interactions through coactivator and corepressor/HDAC complexes. J. Biol. Chem., 277, 44677–44687. [DOI] [PubMed] [Google Scholar]
- 43.Gay F., Anglade,I., Gong,Z. and Salbert,G. (2000) The LIM/homeodomain protein islet-1 modulates estrogen receptor functions. Mol. Endocrinol., 14, 1627–1648. [DOI] [PubMed] [Google Scholar]
- 44.Shyr C.R., Hu,Y.C., Kim,E. and Chang,C. (2002) Modulation of estrogen receptor-mediated transactivation by orphan receptor TR4 in MCF-7 cells. J. Biol. Chem., 277, 14622–14628. [DOI] [PubMed] [Google Scholar]
- 45.Hong Y.H., Varanasi,U.S., Yang,W. and Leff,T. (2003) AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem., 278, 27495–27501. [DOI] [PubMed] [Google Scholar]
- 46.Valentine J.E., Kalkhoven,E., White,R., Hoare,S. and Parker,M.G. (2000) Mutations in the estrogen receptor ligand binding domain discriminate between hormone-dependent transactivation and transrepression. J. Biol. Chem., 275, 25322–25329. [DOI] [PubMed] [Google Scholar]
- 47.Bourguet W., Vivat,V., Wurtz,J.M., Chambon,P., Gronemeyer,H. and Moras,D. (2000) Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol. Cell, 5, 289–298. [DOI] [PubMed] [Google Scholar]
- 48.Weatherman R.V., Fletterick,R.J. and Scanlan,T.S. (1999) Nuclear-receptor ligands and ligand-binding domains. Annu. Rev. Biochem., 68, 559–581. [DOI] [PubMed] [Google Scholar]