


Abstract
The HpaR-mediated regulation of the hpa-meta operon (Pg promoter) of the 4-hydroxyphenylacetic acid catabolic pathway of Escherichia coli has been studied. The HpaR regulator was purified to homogeneity showing that it is able to bind selectively to 4-hydroxyphenylacetic, 3-hydroxyphenylacetic and 3,4-dihydroxyphenylacetic acids, which act as inducers of the system. The role of HpaR as a repressor and the requirement for cAMP receptor protein for maximal activity have been confirmed by in vitro transcription analyses. Two DNA operators, OPR1 and OPR2, have been identified in the intergenic region located between the hpa-meta operon and the hpaR gene. The OPR1 operator contains a perfect palindromic sequence overlapping the transcriptional +1 start site of the Pg promoter. The OPR2 operator shows a similar but imperfect palindromic sequence and is located far downstream of the +1 start site of the Pr promoter. The binding of HpaR to OPR2 displays a clear cooperativity with OPR1 binding. Based on the above observations and the results of permanganate footprinting experiments, a repression mechanism for HpaR is postulated. A 3-dimensional model of HpaR, generated by comparison with the crystal structures of the homologous regulators, MarR and MexR, suggests that HpaR is a dimer that contains a typical winged-helix DNA binding motif in each subunit.
INTRODUCTION
The hpa cluster of Escherichia coli W codes for a group of proteins involved in the catabolism of 4-hydroxyphenylacetic acid (4HPA) (1) (Fig. 1). The hpa catabolic genes are organized in two operons: the upper operon (hpaBC) encoding the two-component 4HPA monooxygenase, which transforms 4HPA to 3,4-dihydroxyphenylacetic acid (3,4HPA) (2,3), and the meta operon (hpaGEDFHI) encoding the enzymes that cleave the aromatic ring of 3,4HPA and allows its complete mineralization (1,4). The hpa pathway is regulated by two proteins named HpaA and HpaR (1). Although the role of the HpaA activator of the hpaBC operon has been analyzed in some detail (5), very few data are available on the function of HpaR, the putative regulator of the hpa-meta operon (6). It has been suggested that transcription of the homologous hpc-meta operon of E.coli C is repressed by the product of hpcR (4) and we have postulated that the homologous hpaR gene of E.coli W might play a similar role (1,6). Recently, we have demonstrated that the transcription from the Pg promoter of the hpa-meta operon is strictly controlled by a global regulatory system, which allows the expression of the hpa catabolic genes only in the absence of a preferred carbon source (6). This unusually strong catabolite repression control of the Pg promoter of the hpa-meta operon is mediated by the cAMP receptor protein (CRP). When E.coli W cells are grown on glucose plus 4HPA, Pg is only active in stationary phase and the activation mechanism requires the global regulator integration host factor (IHF) (6) (Fig. 1).
Figure 1.

Regulation of the hpa cluster and sequence of the hpaR-hpaG intergenic region. The organization of the catabolic (hpaBCDEFGHI) transport (hpaX) and the regulatory genes (hpaA and hpaR) of the hpa cluster, and their regulation by HpaA and HpaR and the global regulators CRP and IHF are represented. The open arrows indicate the directions of gene transcription. PR, PG, PX, PA and PBC are promoter regions. The white square indicates the active form of HpaR repressor; the black diamond indicates the inactive form of HpaA activator; the white circle represents the inducer. The lines ending in arrows or bars indicate positive or negative effects, respectively. The complete nucleotide sequence of the hpaR-hpaG intergenic region (PR-PG probe) is indicated. DNA regions located between triangles correspond to PG probe from –80 to +46 (filled triangles) and PR probe from –216 to –87 (empty triangles) numbered relative to the Pg transcription start, taken as +1. The –35 and –10 boxes of the Pg and Pr promoters, the ribosome binding sites (RBS), the transcription start site of Pg and Pr (+1) and the CRP and IHF sites are indicated. ATG start codons of hpaR and hpaG are shown in italic. – and + indicate transcriptional repression and activation, respectively. The OPR1 and OPR2 operators are indicated as dotted boxes.
In this work we have used different genetic and biochemical approaches to characterize the interaction of HpaR with its target DNA and its cognate inducer molecules. We have also demonstrated that hpaR controls both the expression of the hpa-meta operon (Pg promoter) and its own expression (Pr promoter) through an unusual repression mechanism, suggesting that although HpaR is related in amino acid sequence and structure to the MarR family of regulators (7,8), it is not related in its mode of action.
MATERIALS AND METHODS
Bacterial strains, plasmids and growth conditions
Bacteria were grown in LB medium (9) or M63 minimal medium (10) at 30°C supplemented with thiamine (1 µg/ml) and vitamin B12 (1 µg/ml) and 20 mM glycerol as carbon source as described previously (6). When required, 1 mM appropriate aromatic compound was added to the medium. The aromatic compounds used as putative inducers were purchased from Sigma. The abbreviations used are: 4HPA, 4-hydroxyphenylacetic acid; 3,4HPA, 3,4-dihydroxyphenylacetic acid; 2HPA, 2-hydroxyphenylacetic acid; 3HPA, 3-hydroxyphenylacetic acid; PP, phenylpropionic acid; PA, phenylacetic acid; 2,5HPA, homogentisic acid. When needed, antibiotics were added at the following concentrations: ampicillin, 100 µg/ml; tetracycline, 10 µg/ml; kanamycin, 50 µg/ml; rifampicin, 50 µg/ml. The E.coli strains and plasmids used in this work are listed in Table 1. The pUTminiTn5 derivatives are mini-Tn5 delivery plasmids used for the insertion of genes into the chromosome of different E.coli strains by the filter-mating technique using E.coli S17-1λpir as donor according to the method described previously (15).
Table 1. Bacterial strains and plasmids.
| Strain or plasmid | Revelant genotype or phenotype | Reference |
|---|---|---|
| Escherichia coli K-12 | ||
| S17-1λpir | Host for pUT-derived plasmids | (11) |
| MC4100 | F– araD319 Δ(argF-lac)U169 relA1flbB5301 deoC1 ptsF25 rbsR | (5) |
| AFMC | MC4100 Rifr | (12) |
| MCR14 | AFMC derivative, Pr::lacZ, Tcr | This work |
| MCR141 | MCR14 derivative, Pr::lacZ, hpa complete pathway, Kmr, Tcr | This work |
| MCR142 | MCR14 derivative, Pr::lacZ, Pr::hpaR, Kmr, Tcr | This work |
| Escherichia coli W | ||
| W14 | W derivative (Δpaa) | (13) |
| AF15 | W14 derivative (ΔlacZ), Rifr | (12) |
| WPG11 | W14 derivative (ΔlacZ, Pg::lacZ), Kmr | (6) |
| WPR13 | W14 derivative (ΔlacZ, Pr::lacZ), Kmr | This work |
| Plasmids | ||
| pUJ9 | Promoterless lacZ vector, Apr | (14) |
| pAJ40 | pUC18 derivative containing hpa pathway | (1) |
| pHCR2 | pACYC184 derivative with an EcoRI DNA fragment containing hpaR | (1) |
| pUTminiTn5-Km | MiniTn5 delivery plasmid, Kmr, Apr | (14) |
| pUTminiTn5-Tc | MiniTn5 delivery plasmid, Tcr, Apr | (14) |
| pUC18Not | Identical to pUC18 but with NotI sites flanking MCS | (15) |
| pUCR1 | pUC18Not derivative overexpressing the hpaR gene under Plac | This work |
| pJCD01 | pUC19 derivative, vector for in vitro transcription | (16) |
| pBF1 | pJCD01 derivative containing Pr-Pg promoters | This work |
| pBM2 | pUJ9 derivative, Pr::lacZ, Apr | This work |
| pPR13 | pUTminiTn5-Km derivative, Pr::lacZ, Kmr, Apr | This work |
| pPR14 | pUTminiTn5-Tc derivative, Pr::lacZ, Tcr, Apr | This work |
| pAJ402 | pUTminiTn5-Km derivative containing hpa pathway | (1) |
| pBA1 | pUCNot derivative, Pr::hpaR, Kmr, Apr | This work |
| pBA2 | pUTminiTn5-Km derivative, Pr::hpaR, Kmr, Apr | This work |
DNA manipulations and sequencing
Isolation of plasmid DNA, digestion with restriction enzymes, ligation with T4 DNA ligase and transformation were carried out as described elsewhere (9). DNA fragments were purified using the Geneclean Turbo Kit (BIO101 Inc.). Oligo nucleotides were synthesized in an Oligo-1000M nucleotide synthesizer (Beckman Instruments). Nucleotide sequences were determined directly from plasmids by using the dideoxy chain termination method (9). The manufacturer’s standard protocols for Taq DNA polymerase-initiated cycle sequencing reactions with fluorescently labeled dideoxynucleotide terminators (Applied Biosystems Inc.) were used.
Construction of strains harboring a translational Pr::lacZ fusion in the chromosome
To construct a translational fusion of the Pr promoter region of hpaR and the lacZ reporter gene, a 314 bp DNA fragment covering this promoter region was amplified by PCR using 10 ng of plasmid pAJ40 (Table 1) as template and the primers PAR3′ (5′-GCACGGATCCGTAATAGAAAAGGGGAC-3′; an engineered BamHI site is underlined) and PAR5′ (5′-CCGAATTCCTTTCATGGTACCACTCC-3′; the start codon is indicated in bold and an engineered EcoRI site is underlined). To create plasmid pBM2 (Table 1), the PCR amplified fragment was cut with EcoRI and BamHI endonucleases and ligated to the EcoRI and BamHI double-digested promoterless lacZ vector pUJ9 (Table 1). The correct fusion was verified by sequence analysis. Plasmids pPR13 and pPR14 were constructed by subcloning the NotI cassette of pBM2 into the mini-Tn5 delivery plasmids pUTminiTn5-Km and pUTminiTn5-Tc, respectively (Table 1), and they were used for insertion of the Pr::lacZ fusion into the chromosome of E.coli AF15 and AFMC (Table 1), respectively, by the filter-mating technique (15). The generated exconjugants containing the lacZ translational fusions inserted into their chromosomes were selected for the transposon marker, kanamycin, on rifampicin-containing LB medium to give the strain WPR13 and selected on tetracycline-containing LB medium for MCR14. In each case the final strain was selected from among three different exconjugants with similar expression levels and expression profiles of the reporter gene. The relevant genotypes of the resulting strains are indicated in Table 1.
Overexpression and purification of HpaR
The hpaR coding sequence was amplified by PCR using oligonucleotides HpaR5′ (5′-GGGAATTCTAAATGAAGG AGAAAGATAATGCACGACTC-3′; the start codon is indicated in bold and an engineered EcoRI site is underlined) and HpaR3′ (5′-GGGTACCAAGCTTAGATACTAAAAAGTT ATTC-3′; an engineered KpnI site is underlined) and plasmid pAJ40 (Table 1) as DNA template. The amplified DNA fragment was digested with EcoRI and KpnI and then inserted into vector pUC18Not (Table 1) under control of the Plac promoter. The resulting recombinant plasmid pUCR1 was transformed into E.coli MC4100 (Table 1). Escherichia coli MC4100 (pUCR1) cells overproduced a 17 kDa protein (17% of total protein) that corresponded to the expected size of HpaR (Fig. 2).
Figure 2.
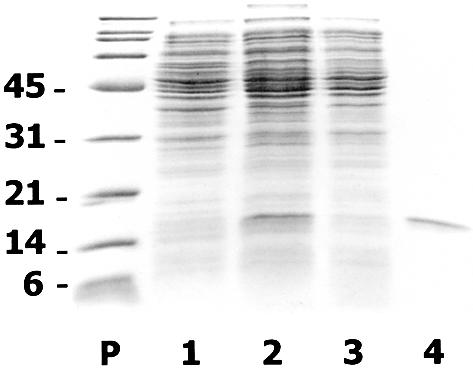
Overexpression and purification of HpaR protein. SDS–PAGE analysis of HpaR purification from E.coli MC4100 (pUCR1) cells. Lane P, the molecular mass markers in kDa; lane 1, soluble control extract from E.coli MC4100 (pUC18Not); lane 2, soluble crude extract from E.coli MC4100 (pUCR1); lane 3, supernatant after polyethylimine precipitation and dialysis of crude extract from E.coli MC4100 (pUCR1); lane 4, purified HpaR protein by the method based on precipitation of HpaR under low ionic strength conditions followed by phosphocellulose chromatography.
To purify HpaR, E.coli MC4100 (pUCR1) cells cultured overnight at 37°C in 500 ml of ampicillin-containing LB medium were harvested by centrifugation, washed with saline solution (0.9% NaCl) and, finally, resuspended in 25 ml of HpaR buffer (20 mM Tris–HCl pH 7.5, containing 10% glycerol, 2 mM β-mercaptoethanol, 1 mM EDTA and 100 mM KCl). All the steps were performed at 4°C. Cells were lysed by passage through a French press (Aminco Corp.) operated at a pressure of 20 000 p.s.i. The crude extract was spun in an ultracentrifuge at 150 000 g for 45 min and the supernatant, which contains the soluble HpaR, was collected. Nucleic acids present in the supernatant were removed by precipitation with 0.5% polyethyleneimine at pH 8.0. The solution was stirred gently over 15 min followed by centrifugation at 18 000 g for 15 min. Then, the DNA free supernatant was dialyzed for several hours against several changes of HpaR buffer (total volume 5 l). After dialysis the protein solution becomes cloudy due to the precipitation of a complex between HpaR and traces of remaining polyethyleneimine. The precipitate was recovered by centrifugation (18 000 g for 15 min) and solubilized in 30 ml of HpaR buffer supplemented with KCl to a concentration of 1 M. To eliminate the traces of polyethyleneimine, the solubilized material was loaded onto a phosphocellulose P11 (Whatman) column (10 ml), equilibrated with the same buffer. In these conditions polyethyleneimine binds to the column while HpaR does not. Thus, the eluted unbound fraction, containing a polyethyleneimine-free protein of 17 kDa that corresponded to HpaR, was dialyzed against HpaR buffer. The purity of HpaR was estimated by SDS–PAGE and Coomassie staining (17). Using this procedure we obtained 2 mg of purified HpaR at a concentration of 0.2 mg/ml (Fig. 2). Further attempts to concentrate HpaR were unfruitful due to its low solubility.
Phenyl-Sepharose chromatography
The DNA-free solution containing the HpaR protein was added to 8 ml of phenyl-Sepharose CL-4B (Amersham-Pharmacia Biotech AB) previously equilibrated with HpaR buffer plus 1 M KCl. Then, the matrix was washed with 24 ml of the same buffer without KCl to elute most of the retained proteins. After this washing step, HpaR remained adsorbed to the matrix by a specific interaction with the phenyl group. To determine the compounds that could compete with the phenyl group of the matrix releasing the HpaR protein, the washed matrix containing the HpaR protein was eluted with different solutions of 100 mM Tris–HCl pH 7.5 buffer containing one of the putative competing compounds (4HPA, 2HPA, 3HPA, 3,4HPA, PP, PA, 3-hydroxycinnamic acid or 2,5HPA) at a final concentration of 0.2 M. The corresponding eluted fractions were analyzed by SDS–PAGE and Coomassie staining.
Gel retardation assays (EMSA)
The DNA fragments PR-PG, PR and PG of 314, 179 and 147 bp, respectively, used as probes were amplified by PCR using 10 ng of plasmid pAJ40 (Table 1) as template and the following primers: PG3′(5′-GATAGTGGGATCCATGGTA CCACTCCTCGGATTCGATC-3′) and PGDE (5′-CCGG AATTCTGTAAATAGTTTGTTAATTAG-3′) for the PG fragment; PG5′ (5′-AACGCAAGAATTCGTGAGTCGTGC ATTATCTTTCCCC-3′) and PRDE (5′-CCGGAATTC GATAAGAATATATTAAATATC-3′) for the PR fragment. The DNA fragments were labeled at the 5′-end with phage T4 DNA polynucletotide kinase and [γ-32P]ATP (3000 Ci/mmol) (Amersham Pharmacia Biotech). These fragments were purified on a glassfiber column (High Pure PCR purification kit; Roche). Complexes with the labeled promoter region were formed for 20 min at room temperature in 10 µl of buffer A (40 mM HEPES pH 8.0, 10 mM magnesium chloride, 100 mM potassium glutamate and 500 µg/ml bovine serum albumin) using purified HpaR protein. The mixture was loaded into a 7.5% native polyacrylamide gel. The gel was fixed and dried before being quantified using a phosphorimager (Molecular Dynamics).
KMnO4 and DNase I footprinting
Activators (100 nM CRP or IHF), prepared as described (6), repressor HpaR (100 nM) and RNA polymerase (RNAp) (100 nM), prepared as described (16), were allowed to form complexes with the radioactively labeled PR-PG fragment for 20 min at 37°C in 15 µl of buffer A. In one set of experiments 2.5 µl of DNase I solution (1 µg/ml in 10 mM Tris–HCl, 10 mM magnesium chloride, 10 mM calcium chloride, 125 mM potassium chloride) were added and incubated at 37°C for 20 s, or for 30 s when RNAp was present in the mixture. The reaction was stopped by addition of 200 µl of a solution containing 0.4 M sodium acetate, 2.5 mM EDTA and 50 µg/ml calf thymus DNA, and put on ice. In the other set, 2.5 µl of KMnO4 solution (40 mM) was added to the complexes for 30 s at 37°C. The reaction was stopped by adding 2.5 mM 2-mercaptoethanol (2 M). Then all the samples were phenol extracted and precipitated with ethanol. With the KMnO4 samples, the ethanol precipitates were resuspended in 100 µl of piperidine (1 M), heated at 90°C for 30 min and evaporated until dryness. Then 20 µl of water was added and evaporated (twice). KMnO4 and DNase I samples were resuspended in 5 µl of loading buffer (20 mM EDTA in 80% v/v formamide containing xylene cyanol blue and bromophenol blue) and loaded on a 7% denaturing polyacrylamide gel.
Run-off transcription assays
Single round transcription by E.coli RNAp was carried out under standard conditions (16), using buffer B (40 mM Tris–HCl pH 8.0, 10 mM MgCl2, 100 mM KCl, 200 µM cAMP and 500 µg/ml acetylated bovine serum albumin) and supercoiled DNA plasmid pBF1 (Table 1). To construct pBF1, the oligonucleotides PG3′ and PG5′ (see above) were used for PCR amplification of the hpaR-hpaG intergenic region using plasmid pAJ40 as DNA template. The final volume of the run-off reaction mixture was 9 µl, containing the plasmid DNA (5 nM) with CRP (100 nM) and HpaR (100 nM) or buffer. This mixture was incubated at room temperature for 20 min. Then, 3 µl of RNAp at 375 nM in buffer B was added and the mixture was incubated at 37°C for 5 min in a final volume of 12 µl. Elongation was started by the addition of 3 µl of a prewarmed mixture containing 1 mM ATP, 1 mM GTP, 1 mM CTP, 50 µM UTP, 1 µCi [α-32]UTP and 500 µg/ml heparin in buffer B to the template–polymerase mix and allowed to proceed for 5 min at 37°C. Reactions were stopped by the addition of 12 µl of loading buffer (see above) containing 1% SDS. After heating to 70°C, samples were subjected to electrophoresis on 7% sequencing gels. Run-off products were quantified using a phosphorimager (Molecular Dynamics).
Assay for β-galactosidase activity
Activities of the Pg and Pr promoters were monitored by assaying β-galactosidase accumulation in cells harboring either Pg::lacZ or Pr::lacZ fusions. Cells were grown in M63 minimal medium and, when indicated, Pg and Pr expression was induced with different aromatic compounds. β-Galactosidase activity was measured as described by Miller (10) and expressed in Miller units.
Primer extension
Primer extension analysis was used to determine the start site of the transcription of hpaR. The reverse transcriptase AMV primer extension system was used according to the protocol supplied by the manufacturer (Promega). The primer used to map the hpaR start site was R2+1 (5′-GCAACGCAAT GGTTAGTGAG-3′). Primer extension products were analyzed on a 6% polyacrylamide gel next to a sequence ladder generated with the same primer. Sequencing reactions were performed with Deaza G/A T7 Sequencing Mixes (Pharmacia). RNA was prepared with a RNeasy Mini Kit from E.coli MC4100 (pBM2) according to the protocol supplied by the manufacturer (Qiagen).
Homology modeling of HpaR
The 3-dimensional structure of HpaR was modeled using Swiss PDB Viewer 3.7 (18). The template used was MarR (8) as a ligand-bound model. Raw structures obtained from fitting were subjected to steepest descent energy minimization. The salicylate molecules were removed from the MarR-based structure and substituted by 4HPA molecules by manual docking using RASTOP 2.0.2. (http://www.geneinfinity.org/rastop/).
RESULTS
In vitro and in vivo analysis of the HpaR ligands
At the time we were testing different chromatographic supports to purify HpaR, we discovered that this protein could be strongly and specifically retained on phenyl-Sepharose due to its ability to interact with aromatic compounds. This property allowed us to determine in vitro the aromatic compounds that might act as ligands of HpaR. Figure 3 shows that HpaR can be eluted from phenyl-Sepharose by 4HPA, 3HPA and 3,4HPA. However, other structurally related aromatic compounds, such as PP, PA, 2HPA, 2,5HPA and 3-hydroxycinnamic acid, were unable to release HpaR from the phenyl-Sepharose support. These results suggested that the interaction of HpaR with the phenyl group of the matrix most likely takes place through the effector recognition site of the protein, rather than by a non-specific hydrophobic interaction. Moreover, they are in perfect agreement with the observation that repression of the hpc-meta operon of E.coli C caused by the homologous HpcR regulator can be released in vivo by 4HPA, 3HPA or 3,4HPA (4).
Figure 3.


Inducers of HpaR protein. (A) SDS–PAGE analysis of the phenyl-Sepharose chromatography of HpaR. Lane P, molecular mass markers shown in kDa; lanes 1–8, fractions eluted with 4HPA (lane 1), 2HPA (lane 2), 3HPA (lane 3), 3,4HPA (lane 4), PP (lane 5), PA (lane 6), 3-hydroxycinnamic acid (lane 7) and 2,5HPA (lane 8). (B) Effect of several aromatic compounds on the expression of Pg promoter. Cells of E.coli WPG11 (Pg::lacZ) were grown in glycerol-containing minimal medium in the absence (no aromatic) or in the presence of 1 mM different aromatics (4HPA, PA, 2HPA, 3HPA, PP, 2,5HPA, 3,4HPA and 3-hydroxycinnamic acid) until the cultures reached an OD600 of 0.8. β-Galactosidase activities were measured with permeabilized cells as described in Materials and Methods.
To determine in vivo the range of HpaR inducers on the Pg promoter of E.coli W, we used the reporter strain WPG11 (Pg::lacZ, hpa+) (Table 1). WPG11 was cultured for 2 h in M63 minimal medium containing 20 mM glycerol and different aromatic acids (1 mM) as inducers. Figure 3B shows that the β-galactosidase activity was only increased in the presence of 4HPA, 3HPA and 3,4HPA, corroborating the hypothesis that these compounds are the prevalent and primary inducers of the HpaR repressor. It is worth noting that since WPG11 is a hpa+ strain, we could not at this stage rule out the possibility that the effect of 3HPA and 4HPA might be due to their transformation into 3,4HPA or into other metabolites of the pathway. However, the observation that these compounds specifically interact with HpaR in vitro as well as the in vivo experiments performed on the homologous HpcR regulator (4) support the idea that these compounds could be true HpaR effectors (see also in vitro transcription experiments below).
In vitro HpaR binding to the hpaG-hpaR intergenic region
The ability of HpaR to bind to the hpaR-hpaG intergenic region containing the Pg and Pr promoters (Fig. 1) was tested in vitro by gel retardation assays (EMSA) (Fig. 4) using three different DNA fragments as probes. While the PR-PG (Pg and Pr promoters) fragment covers the entire DNA region located between the hpaR genes and the hpa-meta operon, the PR (Pr promoter) and PG (Pg promoter) fragments contain the 179 bp region upstream of hpaR and the 147 bp region upstream of the hpa-meta operon, respectively (Fig. 1). Remarkably, whereas two HpaR–DNA complexes were detected with the PR-PG probe (complexes I and II), only one was visible with each of the PG and PR probes (Fig. 4). To localize the HpaR binding sites more precisely in the hpaR-hpaG intergenic region, DNase I footprinting experiments were performed using the PR-PG fragment as probe. These experiments revealed that HpaR protects two operators, named OPR1 (centered at position +2) and OPR2 (centered at position –199, relative to the transcription start site +1 of the Pg promoter) (Figs 1 and 5). OPR1 and OPR2 both consist of a 27 bp region containing two inverted half-sites of 9 bp separated by 4 bp (Fig. 5). These results are in agreement with those presented above (Fig. 4) showing that one HpaR binding site was located in the PG fragment (corresponding to OPR1) and the other within the PR fragment (corresponding to OPR2). In addition, EMSA experiments indicated that the OPR1 site had an affinity about 10-fold higher than the OPR2 site on the PR-PG fragment (Fig. 4A). Interestingly, when the two operator sites are located on separate fragments, the difference in their affinities appeared much greater, reaching a factor of 1000-fold, indicating that binding of HpaR to OPR2 on the PR-PG fragment is clearly cooperative with the binding to OPR1 (Fig 4). Other experiments performed in the presence of 4HPA on the PR-PG fragment revealed that the affinity of the Pg operator was only slightly decreased by the presence of this compound (less than 2-fold) (data not shown).
Figure 4.

HpaR binding to the hpaR-hpaG intergenic region. EMSAs were performed as indicated in Materials and Methods. The DNA probes used were (A) PR-PG and (B) PG and PR. Increasing concentrations of purified HpaR were used. (A) Lane 1, 0 nM; lane 2, 3 nM; lane 3, 10 nM; lane 4, 30 nM; lane 5, 50 nM; lane 6, 75 nM; lane 7, 150 nM; lane 8, 300 nM. (B) Lanes 1 and 9, 0 nM; lane 2, 3 nM; lane 3, 10 nM; lane 4, 30 nM; lane 5, 50 nM; lane 6, 75 nM; lane 7, 100 nM; lane 8, 200 nM; lane 10, 10 nM, lane 11, 30 nM; lane 12, 50 nM; lane 13, 75 nM; lane 14, 100 nM; lane 15, 200 nM; lane 16, 1000 nM.
Figure 5.


DNase I footprinting analysis of the interaction of HpaR with the Pr-Pg promoter region. The reaction mixture was treated as described in Materials and Methods using as probe the 5′-end-labeled non-coding strand of the Pr-Pg region of the hpaGEDFHI operon showing (A) the OPR1 and (B) the OPR2 protected regions. HpaR concentrations were as follows. (A) Lane 1, 0 nM; lane 2, 10 nM; lane 3, 30 nM; lane 4, 100 nM; lane 5, 200 nM. (B) Lane 1, 0 nM; lane 2, 30 nM; lane 3, 100 nM; lane 4, 200 nM; lane 5, 400 nM; lane 6, 1000 nM. An A+ G sequencing ladder is indicated with an asterisk. (C) OPR1 and OPR2 operators. Operators are indicated and conserved nucleotides are shown in bold. Arrows represent the inverted repeat sequences.
In vitro transcription from the Pg promoter
Although we have demonstrated that 4HPA interacts with HpaR in vitro and induces the hpa-meta operon in vivo, it was only able to very slightly reduce the HpaR–DNA binding affinity in vitro according to the footprinting analysis (see above). Thus, to ascertain the role of 4HPA in the induction of HpaR-repressed Pg promoter, we developed an in vitro transcription assay using plasmid pBF1 as DNA template. Plasmid pBF1 contains the hpaR-hpaG intergenic region flanked by transcriptional terminators (Table 1). Since, as we showed previously, the Pg promoter is CRP dependent (6), it was not active when RNAp was the unique protein added to the reaction mixture and, thus, only the RNA1 control transcript is detected in the assay (Fig. 6). However, when CRP was supplied to the previous reaction mixture, the Pg activity was stimulated and a 139 nt transcript generated from Pg was detected (Fig. 6). As expected, the addition of HpaR clearly decreased the Pg transcription rate, demonstrating that this protein was able to work in vitro as a repressor. Moreover, the addition of 4HPA (Fig. 6), but not that of PA or 2HPA (data not shown), restored the transcription activity of the promoter. These results demonstrate, for the first time, repression by HpaR in vitro. But more importantly, it should be noted that in these in vitro experiments 4HPA cannot be transformed into other intermediates of the hpa catabolic pathway, which could occur in the in vivo experiments shown above and, therefore, these results confirm unequivocally that 4HPA is a true inducer of the hpa-meta operon.
Figure 6.
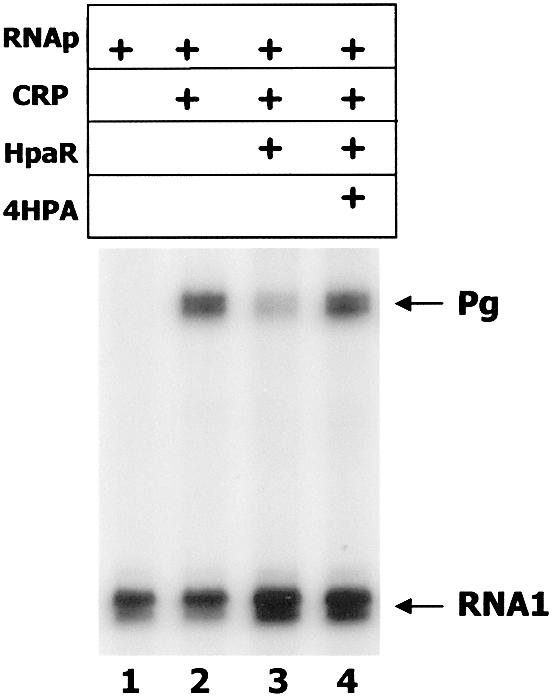
In vitro transcription assay of the Pg promoter. Run-off transcription from Pg. Single round in vitro transcription was carried out by using the plasmid pBF1 as template. RNAp, CRP and HpaR were added at final concentrations of 100 nM each and the final concentration of the inducer 4HPA was 10 mM. The Pg-derived mRNA (139 nt) and the vector-derived RNA1 are indicated by arrows.
Influence of HpaR on open complex formation
To ascertain whether the repression mechanism mediated by HpaR involves inhibition of the formation of the open complex required for transcription initiation, we performed a potassium permanganate footprinting assay using the PR-PG fragment labeled at its Pg 5′-end. Figure 7 shows that RNAp alone induced only a weak reactivity of the thymine residues from +2 to –13, confirming the fact that the Pg promoter is inefficient in the absence of its cognate activators, CRP and IHF (6). Indeed, addition of CRP alone or a combination of CRP and IHF (data not shown) significantly increased open complex formation at the Pg promoter. The presence of HpaR abolished the permanganate reactivity of the thymine residues characteristic of the Pg promoter, in agreement with its inhibitory effect on Pg transcription. Although HpaR alone did not induce any pattern of reactivity to permanganate or open complex formation (Fig. 7), a new pattern of reactivity to permanganate was detected from position –22 to –32, between the –10 and –35 promoter boxes of the Pg promoter, just upstream of the HpaR binding site, when HpaR, CRP and RNAp were present in the same reaction mixture. This suggests that HpaR does not prevent RNAp from binding to the Pg promoter region but effectively blocks promoter escape by displacing RNAp from its usual location (Fig. 7).
Figure 7.
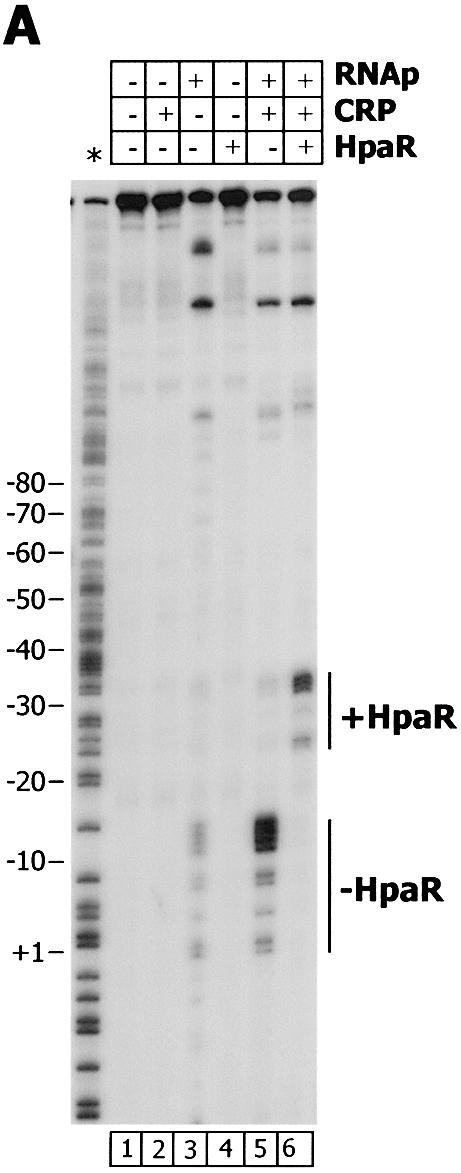

KMnO4 footprinting of the Pg promoter. (A) The reaction mixture was treated as described in Materials and Methods using as probe the 5′-end-labeled non-coding strand of the Pr-Pg region. RNAp, CRP and HpaR were added at final concentrations of 100 nM. The positions sensitive to KMnO4 are indicated. An A+ G sequencing ladder is indicated with an asterisk. (B) Nucleotide sequence of the Pg region showing the –35 and –10 promoter boxes and +1 site. The positions sensitive to KMnO4 corresponding to the open complex (empty boxes) and to the HpaR-dependent DNA bubble (hatched boxes) are indicated.
Regulation of the Pr promoter
To check whether expression of the divergently transcribed Pr promoter, which drives expression of the hpaR gene (Fig. 1), was also regulated by HpaR, we have engineered a reporter Pr::lacZ translational fusion within plasmid pBM2 (Table 1). Firstly, the transcriptional start site of hpaR was determined in plasmid pBM2 by primer extension (Fig. 8). This experiment revealed that the transcription of hpaR starts at a G residue located at position –153 relative to the transcriptional start site of the Pg promoter (Figs 1 and 8). A putative –10 box (TAAAAT) located 9 bp upstream of the Pr +1 site and a putative –35 box (TTAATA) with a spacer of 18 bp were identified. As expected, these Pr promoter boxes are similar to the typical boxes of σ70-dependent promoters.
Figure 8.
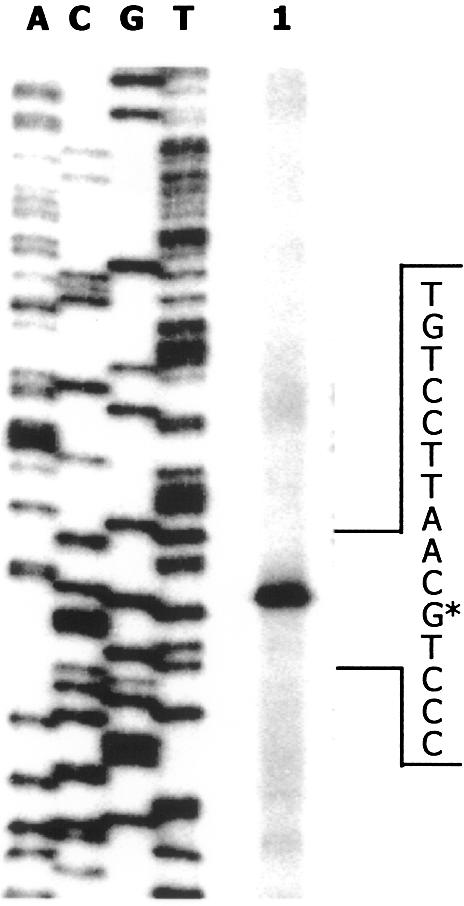
Mapping of the hpaR transcriptional start site by primer extension. Total RNA was isolated from LB grown E.coli MC4100 (pBM2) cells (lane 1). A sequence ladder generated with the same primer (R2+1) is shown (lanes A, C, G and T). The position of the primer extension product is indicated by the asterisk.
The Pr::lacZ translational fusion was subcloned in a mini-Tn5 vector resulting in plasmids pPR13 and pPR14 (Table 1). These plasmids were used to deliver, by transposition, the Pr::lacZ fusion into the chromosomes of E.coli W AF15 (hpa+, ΔlacZ) and E.coli K12 AFMC (hpa–, ΔlacZ), generating the reporter E.coli strains WPR13 (hpa+, Pr::lacZ) and MCR14 (hpa–, Pr::lacZ), respectively. β-Galactosidase assays performed with permeabilized E.coli WPR13 and E.coli MCR14 cells cultured in the presence or absence of 1 mM 4HPA revealed a significant effect of 4HPA on Pr expression in the WPR13 strain (2.6-fold increase in β-galactosidase activity), whereas Pr expression in the MCR14 strain was constitutive (Table 2). These results showed that the Pr promoter was regulated by a regulator of the hpa cluster, but since we have demonstrated that this cluster contains two regulators, HpaR and HpaA, that respond to 4HPA (5), they did not allow us to determine if Pr expression was regulated by HpaR or HpaA.
Table 2. Repression of Pr promoter by HpaR.
| Strain | β-galactosidase activity (Miller units) | |
|---|---|---|
| –4HPA | +4HPA | |
| MCR14 (–HpaR, –HpaA) | 1544 | 1495 |
| MCR141 (+HpaR, +HpaA) | 87 | 119 |
| MCR142 (+HpaR, –HpaA) | 72 | 109 |
| WPR13 (+HpaR, +HpaA) | 510 | 1350 |
To establish the role of HpaR in Pr expression, the E.coli strains S17-1λpir (pAJ402) and S17-1λpir (pBA2) (Fig. 9 and Table 1) were used to transfer the hpaR gene into the chromosome of E.coli MCR14, generating the exconjugants MCR141 and MCR142, respectively. Table 2 shows that in E.coli MCR141 and E.coli MCR142 Pr transcription decreased 18-fold in the presence of the hpaR gene, strongly suggesting that HpaR represses the Pr promoter. The presence of 4HPA produced only a slight increase in β-galactosidase activity in both strains. This result could be attributed to more efficient synthesis of the HpaR protein in the heterologous K12 strain than in the wild-type W strain.
Figure 9.


Regulation of the Pr promoter. (A) Schematic construction of E.coli MCR142 (Pr::hpaR, Pr::lacZ). (B) Effect of several aromatic compounds on expression of the Pr promoter. Cells of E.coli WPR13 (Pr::lacZ) were grown in glycerol-containing minimal medium in the absence (no aromatic) or in the presence of 1 mM different aromatics (4HPA, PA, 2HPA, 3HPA, PP, 2,5HPA, 3,4HPA and 3-hydroxycinnamic acid) until the cultures reached an OD600 of 0.8. β-Galactosidase activities were measured with permeabilized cells as described in Materials and Methods.
Finally, the strain WPR13 was also used to determine in vivo the range of HpaR inducers which act on its own Pr promoter. As expected, β-galactosidase activity was only increased in the presence of 4HPA, 3HPA and 3,4HPA, confirming that these compounds are also inducers of the HpaR repressor when it regulates its own expression (Fig. 9).
Homology modeling of ligand-bound HpaR
Taking into account the sequence homology of HpaR with the MarR regulator (19% identity, 45% similarity; Fig. 10A), we have modeled the 3-dimensional structure of ligand-bound HpaR using the crystal coordinates of this regulator (8) (Fig. 10B and C). MarR is a dimer that contains a typical winged-helix DNA binding motif in each monomer. The two recognition helices are supposed to bind two adjacent major grooves of DNA, whereas the wings might well be positioned to make minor groove or phosphate backbone contacts to the distal parts of the inverted repeat. However, up to now, no structure of a DNA–protein complex is available for a protein of the MarR family. Based on the MarR crystal, HpaR might bind two molecules of 4HPA per monomer, one of them between the DNA recognition helix and the ‘wing’ structure (corresponding to the SAL-A site in MarR) and the other between the recognition helix and the α2 helix (corresponding to the SAL-B site in MarR) (Fig. 10B and C). In both cases, the ligands would be solvent exposed. A comparison of the modeled 4HPA binding sites in HpaR with those of salicylate in MarR reveals that their overall configuration is retained in HpaR, which is not surprising considering the similarity of the salicylate and 4HPA structures. Moreover, the non-conservative changes between the two sequences can be explained in terms of the accommodation of the hydroxyl group located in a different position in the aromatic ring of the ligand. This, and the fact that no notable steric clashes are produced in the model, support the reliability of the latter.
Figure 10.


Model of ligand-bound HpaR structure. (A) Amino acid sequence alignment of the HpaR and MarR proteins. Residues corresponding to the SAL-A type binding site are shown in bold. Residues corresponding to the SAL-B type binding site are represented in lower case. The putative HpaR DNA recognition helix is boxed. Alignment was done with the BESTFIT program. (B) Detail of the ‘SAL-A’ type 4HPA binding site. The ligand is depicted in spacefill representation and interacting residues are shown in thick wireframe format. Note that 4HPA could also bind in an alternative orientation, rotated 180° around the z-axis. (C) Detail of the ‘SAL-B’ type 4HPA binding site. The scheme representation is as indicated for (B).
DISCUSSION
Based on sequence comparison analysis, the HpaR protein of the hpa catabolic cluster from E.coli W had been proposed to be a transcriptional regulator belonging to the MarR family, but until now, there was no experimental evidence concerning its function and properties (5,6). The activity of the MarR-like proteins found in bacteria and archaea are modulated in response to environmental signals and they play important roles in global health problems caused by bacterial pathogens since they control genes implicated in antibiotic resistance and hemolysis (7,19). Some members of this family are involved in specific responses to aromatic compounds, e.g. MarR (7,20), CinR (21), HpcR (4,5), BadR (22) and EmrR (23). Most of the regulators belonging to this family are transcriptional repressors with the exception of BadR (22), CbaR (24), SlyA (25) and NhhD (26), which function as activators. Moreover, MexR can act as either a transcriptional repressor or activator (27). Recently, the crystal structures of MarR and MexR have been described (8,28) and these findings have increased the interest in these regulators since they have paved the way for structural studies on other members of the family.
Remarkably, although many positive regulators have been shown to control the aromatic catabolic pathways in bacteria, only a few repressors have been described in these pathways (19). The known repressors include the CymR regulator in the catabolism of p-cimene in Pseudomonas putida F1 (29), the AphS repressor of phenol catabolism in Comamonas testosteroni TA441 (30,31) and the PaaX and PaaN regulators of the PA catabolic pathway from E.coli (12) and P.putida U (32), respectively. In this context it appeared very interesting to perform the first rigorous analysis of the HpaR repressor. In this work, we have demonstrated that HpaR negatively regulates not only expression of the hpa-meta operon but also its own expression. The fact that HpaR can regulate its own expression agrees with observations for other MarR-like proteins (23,33) and may represent a common feature of this family of regulators (21). In addition, we have illustrated experimentally that HpaR binds specifically to both the Pg and Pr promoter regions. The intrinsic binding affinity for the Pg operator (OPR1) was 100-fold higher than that for the Pr operator (OPR2) when the operators were carried on different fragments (Fig. 4B). Although HpaR bound cooperatively to both operators on the native PR-PG template, OPR1 remains the first operator to be occupied by the HpaR repressor. These data indicate that as soon as the inducers are depleted from the medium, HpaR binds to OPR1 to shut off expression of the catabolic enzymes before HpaR starts to inhibit its own expression by binding to OPR2. DNase I footprinting (Fig. 5) revealed that OPR1 comprises a 27 bp region containing a palindromic sequence of 9 bp on each side separated by 4 bp (Fig. 5). A similar structural design has been described for the operators of other promoters controlled by MarR-like regulators (21,33). The DNA binding site of MarR consists of a 21 bp sequence organized into an inverted repeat (33) and CinR protects a DNA region of 16 bp showing similar characteristics (21). All these findings are consistent with the dimeric structure proposed for MarR-like regulators where each subunit binds to one of the two inverted half-sites of the operator (8), and suggest an architecture conserved through evolution.
The fact that OPR1 is centered at position +2 of Pg, i.e. overlapping the transcriptional start site, suggests that HpaR should repress transcription from Pg by a mechanism based on steric hindrance, by inhibiting binding of RNAp to the promoter. However, in the presence of both RNAp and HpaR a new pattern of thymine residues reactive to permanganate appears ∼20 bp upstream of the cognate transcription bubble. This result suggests that RNAp and the repressor are bound simultaneously to the Pg promoter forming a ternary complex that inhibits formation of the open complex around the transcriptional start site. Taking into account the results of the in vitro transcription assay (Fig. 6), HpaR might repress transcription from Pg by generating an upstream displacement of RNAp from its functional promoter binding site and hence preventing transcription initiation by a road-block mechanism. Although this sort of repression is not very frequent, there are, in the literature, some examples of regulators that remain bound to the promoter in the presence of RNAp, e.g. protein ω of the broad host range plasmid pSM19035 (34), the factor for inversion stimulation of E.coli (Fis) at the bgl promoter (35), TyrR in the aroP2 promoter of E.coli (36), Spo0A at the abrB promoter of Bacillus subtillis (37) and Arc at the Pant promoter of phage P22 (38). Therefore, HpaR could be considered as a new representative of this type of regulator.
Footprinting analyses have demonstrated that a second HpaR operator, OPR2, is centered at position +47 relative to the Pr transcriptional start site (Fig. 5). The location of OPR2 with respect to the +1 site of Pr suggests that the repression effect might be produced by inhibiting the transcription elongation process. OPR2 comprises a 27 bp region with a similar organization to OPR1. One of the half-sites of the operator is identical to that of OPR1 but the other consists of an imperfect repeat with four nucleotide changes (Fig. 5). These differences could explain the lower binding affinity of HpaR to OPR2 when compared to OPR1. On the other hand, we have observed that the binding to OPR2 is clearly cooperative with the binding to OPR1. Since HpaR binding experiments were performed in the absence of other DNA-binding proteins, such as CRP or IHF, this cooperative effect indicates that the binding of HpaR to OPR1 stabilizes its binding to OPR2 by protein–protein interactions. Preliminary experiments performed by analytical ultracentrifugation suggest that HpaR is a dimer in the absence of effector (data not shown). Nevertheless, we cannot eliminate the hypothesis of dimerization (or oligomerization) of the HpaR dimers to explain the striking cooperativity observed in HpaR binding to OPR1 and OPR2. Two dimers of HpaR bound at OPR1 and OPR2 could interact together leading to the formation of an oligomer which could generate a repression loop. The distance between the two operators is 200 bp, which corresponds to 19 turns of B-DNA helix (assuming a pitch of 10.5 bp per turn). Hence, the two dimers of HpaR should be positioned on the same face of the DNA helix. It is also likely that CRP and IHF help to stabilize the repression loop by bending DNA, whereas 4HPA might disturb the interactions between two HpaR dimers and HpaR–DNA complexes. A similar mechanism has been proposed in the case of the gal promoter region where both the presence of histone-like unstable nucleoid protein and the supercoiled state of the template are required to close the repression loop formed by dimerization of the gal repressor dimers bound to each operator (39,40). Validation of this hypothetical mechanism for HpaR repression requires further research.
The in vivo experiments performed with E.coli WPR13 (Pr::lacZ, hpa+) and E.coli WPG11 (Pg::lacZ, hpa+) have demonstrated that activation of the Pg and Pr promoters requires the presence of 4HPA, 3HPA or 3,4HPA as inducer. These results correlate with the observations obtained by phenyl-Sepharose chromatography where HpaR appears to be adsorbed to the matrix by a pseudo-affinity mechanism. The finding that only the compounds 4HPA, 3HPA and 3,4HPA were able to desorb the protein suggests that most likely the interaction of HpaR with this matrix takes place through the effector binding site of HpaR. However, it was surprising that although these results clearly demonstrate that the inducers interact directly with the protein, the presence of inducer even at high concentration (10 mM) (Fig. 4) and in different ionic strength conditions had little effect on HpaR binding. It decreased HpaR affinity for OPR1 by only 2-fold in DNase I footprinting assays and had hardly any effect on HpaR binding to DNA in EMSAs (data not shown). In this respect it has been previously documented that under in vitro conditions some repressors are not released from the DNA in the presence of the effector (39). It could be argued that a correct response of the HpaR–DNA interaction to the presence of the effector requires the presence of RNAp located in close contact with the OPR1–HpaR–4HPA complex in order to destabilize the repressor complex by protein–protein interactions. Moreover, the supercoiled structure of DNA might be necessary to observe derepression. The latter argument correlates with our observation that 4HPA was able to induce derepression in vitro in the run-off transcription experiments when the template was a supercoiled plasmid (Fig. 6B) and not a linear fragment (data not shown).
Analysis of the 3-dimensional structures of MexR and MarR regulators suggests that despite their similarities in sequence and structure their regulatory mechanisms are different (28). MexR recognizes an inverted repeat separated by 5 bp, which facilitates binding of the two subunits to the same face of the DNA, i.e. a single dimeric molecule can interact with the operator. It has been proposed that MexR regulates its DNA binding activity by shifting the distance between the DNA binding helices of each monomer as a result of a conformational change induced by interaction of the effector with the C-terminal region of MexR (28). The case of MarR protein is more puzzling since the two inverted repeat half-sites on the DNA are separated by only 2 bp, a distance unlikely to be able to accommodate the two recognition helices in two adjacent major grooves of DNA. Moreover, in the liganded protein complex, two arginine side chains known to be involved in specific DNA binding (41) are predicted to point away from the bases in the major groove. MarR binding to the operator site as a single dimer would only take place after a considerable rearrangement in both the spacing and the orientation of the recognition helices that could require the binding of a new effector. However, it still cannot be excluded that two dimers of the MarR protein bind on different faces of the DNA to each half-site. The recent structure of SlyA, a member of the MarR family in the unligated state at 1.6 Å resolution, does not shed more light on the mode of DNA binding of these proteins (41).
The above considerations indicate that the members of the MarR family of regulators may be diverse both in the types of effector molecules recognized and in the ways their DNA binding activity is regulated, and it appeared interesting to analyze the case of HpaR. Taking into account that the two inverted repeats in the HpaR operator are separated by 4 bp, HpaR repression could likely be explained by the binding of a single dimer to both repeats of the operator. The fact that HpaR might bind to DNA both in the absence and in the presence of 4HPA suggests that small changes in the quaternary structure of the repressor should be the key event for regulation, rather than binding itself. According to our 3-dimensional model (data not shown), binding of 4HPA, especially in the ‘SAL-B’ type site, may substantially affect the dimerization interface between the two monomers, leading to a different positioning of the recognition helices.
The analysis of HpaR presented here represents the most detailed characterization of a regulatory protein of an aromatic catabolic pathway from E.coli and provides clear evidence that the complex hpa regulatory system will provide new insights in the field of the regulation of secondary metabolic pathways of this model microorganism.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank E. Díaz, M. Carmona and J. Plumbrigge for helpful discussions and critical reading of the manuscript. We are indebted to F. Boccard for the kind gift of purified IHF protein. We gratefully acknowledge the help of E. Aporta with oligonucleotide synthesis, A. Díaz, G. Porra and S. Carbajo with sequencing and the technical assistance of E. Cano, M. Carrasco, E. Calvo and F. Morante. This work was supported by the Comisión Interministerial de Ciencia y Tecnología (grants ABM97-603-C02-02 and BMC2000-0125-C04-02) and by the Programme de Recherche Fondamentale en Microbiologie, Maladies Infectieuses et Parasitaires.
REFERENCES
- 1.Prieto M.A., Díaz,E. and García,J.L. (1996) Molecular characterization of the 4-hydroxyphenylacetate catabolic pathway of Escherichia coli W: engineering a mobile aromatic degradative cluster. J. Bacteriol., 178, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prieto M.A. and García,J.L. (1994) Molecular characterization of 4-hydroxyphenylacetate 3-hydroxylase of Escherichia coli. A two-protein component enzyme. J. Biol. Chem., 269, 22823–22829. [PubMed] [Google Scholar]
- 3.Galán B., Díaz,E., Prieto,M.A. and García,J.L. (2000) Functional analysis of the small component of the 4-hydroxyphenylacetate 3-monooxygenase of Escherichia coli W: a prototype of a new flavin:NAD(P)H reductase subfamily. J. Bacteriol., 182, 627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roper D.I., Fawcett,T. and Cooper,R.A. (1993) The Escherichia coli C homoprotocatechuate degradative operon: hpc gene order, direction of transcription and control of expression. Mol. Gen. Genet., 237, 241–250. [DOI] [PubMed] [Google Scholar]
- 5.Prieto M.A. and García,J.L. (1997) Identification of a novel positive regulator of the 4-hydroxyphenylacetate catabolic pathway of Escherichia coli.Biochem. Biophys. Res. Commun., 232, 759–765. [DOI] [PubMed] [Google Scholar]
- 6.Galán B., Kolb,A., García,J.L. and Prieto,M.A. (2001) Superimposed levels of regulation of the 4-hydroxyphenylacetate catabolic pathway in Escherichia coli. J. Biol. Chem., 276, 37060–37068. [DOI] [PubMed] [Google Scholar]
- 7.Sulavik M.C., Gambino,L.F. and Miller,P.F. (1995) The MarR repressor of the multiple antibiotic resistance (mar) operon in Escherichia coli: prototypic member of bacterial regulatory proteins involved in sensing phenolic compounds. Mol. Med., 1, 436–446. [PMC free article] [PubMed] [Google Scholar]
- 8.Alekshun M.N., Levy,S.B., Mealy,T.R., Seaton,B.A. and Head,J.F. (2001) The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3Å resolution. Nature Struct. Biol., 8, 710–714. [DOI] [PubMed] [Google Scholar]
- 9.Sambrook J. and Russell,D.W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 10.Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 11.De Lorenzo V. and Timmis,K.N. (1994) Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol., 235, 386–405. [DOI] [PubMed] [Google Scholar]
- 12.Ferrández A., García,J.L. and Díaz,E. (2000) Transcriptional regulation of the divergent paa catabolic operons for phenylacetic acid degradation in Escherichia coli. J. Biol. Chem., 275, 12214–12222. [DOI] [PubMed] [Google Scholar]
- 13.Ferrández A., Prieto,M.A., García,J.L. and Díaz,E. (1997) Molecular characterization of PadA, a phenylacetaldehyde dehydrogenase from Escherichia coli.FEBS Lett., 406, 23–27. [DOI] [PubMed] [Google Scholar]
- 14.De Lorenzo V., Herrero,M., Jakubzik,U. and Timmis,K.N. (1990) Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing and chromosomal insertion of clones DNA in Gram negative eubacteria. J. Bacteriol., 172, 6568–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrero M., de Lorenzo,V. and Timmis,K.N. (1990) Transposon vector containing non-antibiotic selection markers for cloning and stable chromosomal insertion of foreign DNA in gram-negative bacteria. J. Bacteriol., 172, 6557–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marschall C., Labrousse,V., Kreimer,M., Weichart,D., Kolb,A. and Hengge-Aronis,R. (1998) Molecular analysis of the regulation of csiD, a carbon starvation-inducible gene in Escherichia coli that is exclusively dependent on σS and requires activation by cAMP-CRP. J. Mol. Biol., 276, 339–353. [DOI] [PubMed] [Google Scholar]
- 17.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature, 277, 680–685. [DOI] [PubMed] [Google Scholar]
- 18.Guex N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 19.Díaz E. and Prieto,M.A. (2000) Bacterial promoters triggering biodegradation of aromatic pollutants. Curr. Opin. Biotechnol., 11, 467–475. [DOI] [PubMed] [Google Scholar]
- 20.Cohen S.P., Hächler,H. and Levy,S.B. (1993) Genetic and functional analysis of the multiple antibiotic resistance (mar) locus in Escherichia coli. J. Bacteriol., 175, 1484–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalrymple B.P. and Swadling,Y. (1997) Expression of Butyrivibrio fibrisolvens E14 gene (cinB) encoding an enzyme with cinnamoyl ester hydrolase activity is negatively regulated by the product of an adjacent gene (cinR). Microbiology, 143, 1203–1210. [DOI] [PubMed] [Google Scholar]
- 22.Egland P.G. and Hardwood,C. (1999) BadR, a new MarR family member, regulates anaerobic benzoate degradation by Rhodopseudomonas palustris in concert with AadR, an FNR family member. J. Bacteriol., 181, 2102–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.del Castillo I., González-Pastor,J.E., San Millán,J.L. and Moreno,F. (1991) Nucleotide sequence of the Escherichia coli regulatory gene mprA and construction and characterization of mprA-deficient mutants. J. Bacteriol., 173, 3924–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Providenti M.A. and Wyndham,R.C. (2001) Identification and functional characterization of CbaR, a MarR-like modulator of the cbaABC-encoded chlorobenzoate catabolism pathway. Appl. Environ. Microbiol., 67, 3530–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oscarsson J., Mizunoe,B., Uhlin,E. and Haydon,D.J. (1996) Induction of haemolytic activity in Escherichia coli by the slyA gene product. Mol. Microbiol., 20, 191–199. [DOI] [PubMed] [Google Scholar]
- 26.Komeda H., Kobayashi,M. and Shimizu,S. (1996) Characterization of the gene cluster of high-molecular-mass nitrile hydratase (H-Nhase) induced by its reaction products in Rhodococcus rhodocrous. Proc. Natl Acad. Sci. USA, 93, 4267–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poole K., Tetro,K., Zhao,Q., Neshat,S., Heinrichs,D.E. and Bianco,N. (1996) Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob. Agents Chemother., 40, 2021–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim D., Poole,K. and Strynadka,N.C. (2002) Crystal structure of the MexR repressor of the mexRAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Biol. Chem., 277, 29253–29259. [DOI] [PubMed] [Google Scholar]
- 29.Eaton R.W. (1997) p-Cymene catabolic pathway in Pseudomonas putida F1: cloning and characterization of DNA encoding conversion of p-cymene to p-cumate. J. Bacteriol., 179, 3171–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arai H., Akahira,S., Ohishi,T. and Kudo,T. (1999) Adaptation of Comamonas testosteroni TA441 to utilization of phenol by spontaneous mutation of the gene for a trans-acting factor. Mol. Microbiol., 33, 1132–1140. [DOI] [PubMed] [Google Scholar]
- 31.Arai H., Ohishi,T., Chang,M.Y. and Kudo,T. (2000) Arrangement and regulation of the genes for meta-pathway enzymes required for degradation of phenol in Comamonas testosteroni TA441. Microbiology, 146, 1707–1715. [DOI] [PubMed] [Google Scholar]
- 32.Garcia B., Olivera,E.R., Miñambres,B., Carnicero,D., Muniz,C., Naharro,G. and Luengo,J.M. (2000) Phenylacetyl-coenzyme A is the true inducer of the phenylacetic acid catabolism pathway in Pseudomonas putida U. Appl. Environ. Microbiol., 10, 4575–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin R.G. and Rosner,J.L. (1995) Binding of purified multiple antibiotic-resistance repressor protein (MarR) to mar operator sequences. Proc. Natl Acad. Sci. USA, 92, 5456–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de la Hoz A., Ayora,S., Sitkiewicz,I., Fernández,S., Pankiewvic,R., Alonso,J.C. and Ceglowski,P. (2000) Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc. Natl Acad. Sci. USA, 97, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caramel A. and Schnetz,K. (2000) Antagonistic control of the Escherichia coli bgl promoter by FIS and CAP in vitro. Mol. Microbiol., 36, 85–92. [DOI] [PubMed] [Google Scholar]
- 36.Yang J., Wang,P. and Pittard,A.J. (1999) Mechanism of repression of the aroP2 promoter by the TyrR protein of Escherichia coli. J. Bacteriol., 181, 6411–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greene E.A. and Spiegelman,G.B. (1996) The Spo0A protein of Bacillus subtilis inhibits transcription of the abrB gene without preventing binding of the polymerase to the promoter. J. Biol. Chem., 271, 11455–11461. [DOI] [PubMed] [Google Scholar]
- 38.Smith Y.L. and Sauer,R.T. (1996) Dual regulation of open-complex formation and promoter clearance by Arc explains a novel repressor to activator switch. Proc. Natl Acad. Sci. USA, 93, 8868–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rojo F. (2001) Mechanisms of transcriptional repression. Curr. Opin. Microbiol., 4, 145–151. [DOI] [PubMed] [Google Scholar]
- 40.Semsey S., Geanacopoulos,M., Lewis,D.E. and Adhya,S. (2002) Operator-bound GalR dimers close DNA loops by direct interaction: tetramerization and inducer binding. EMBO J., 21, 4349–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu R.Y., Zhang,R.G., Zagnitko,O., Dementieva,I., Maltzev,N., Watson,J.D., Laskowski,R., Gornicki,P. and Joachimiak,A. (2003) Crystal structure of Enterococcus faecalis SlyA-like transcriptional factor. J. Biol. Chem., 278, 20240–20244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.