

Abstract
Objective
To use human cartilage samples and a mouse model of osteoarthritis (OA) to determine whether extracellular superoxide dismutase (EC-SOD) is a constituent of cartilage and to evaluate whether there is a relationship between EC-SOD deficiency and OA.
Methods
Samples of human cartilage were obtained from femoral heads at the time of joint replacement surgery for OA or femoral neck fracture. Samples of mouse tibial cartilage obtained from STR/ort mice and CBA control mice were compared at 5, 15, and 35 weeks of age. EC-SOD was measured by enzyme-linked immunosorbent assay, Western blotting, and immunohistochemistry techniques. Real-time quantitative reverse transcription-polymerase chain reaction was used to measure messenger RNA for EC-SOD and for endothelial cell, neuronal, and inducible nitric oxide synthases. Nitrotyrosine formation was assayed by Western blotting in mouse cartilage and by fluorescence immunohistochemistry in human cartilage.
Results
Human articular cartilage contained large amounts of EC-SOD (mean ± SEM 18.8 ± 3.8 ng/gm wet weight of cartilage). Cartilage from patients with OA had an ~4-fold lower level of EC-SOD compared with cartilage from patients with hip fracture. Young STR/ort mice had decreased levels of EC-SOD in tibial cartilage before histologic evidence of disease occurred, as well as significantly more nitrotyrosine formation at all ages studied.
Conclusion
EC-SOD, the major scavenger of reactive oxygen species in extracellular spaces, is decreased in humans with OA and in an animal model of OA. Our findings suggest that inadequate control of reactive oxygen species plays a role in the pathophysiology of OA.
Osteoarthritis (OA) is a disabling disease of the joints that affects up to 50% of the population but has neither a clear cause nor an effective treatment (1). It causes stiffness, pain, and deformity, most often in the major weight-bearing joints, and results in a focal, progressive loss of articular cartilage, with formation of bony osteophytes around the joint margins. Advancing age, genetic factors, and mechanical damage all play a role in OA (2). There are no diagnostic tests to confirm the disease, and radiographic evidence of OA often occurs late in the disease course, after significant loss of cartilage tissue.
Known features of OA include inflammatory cytokine production, activation of matrix metalloproteinases (MMPs), and dysregulation of the extracellular matrix (ECM) such that the mechanical integrity of the tissue is impaired (3). Recent studies suggest that in cartilage, there are common signaling pathways for both inflammatory cytokines and mechanical forces and that these signaling pathways may be mediated in part by reactive oxygen species (ROS). Proinflammatory cytokines and the mechanical signaling pathways in cartilage both appear to affect the nuclear transcription factor NF-κB, such that mechanical signals of tissue loading can stimulate or inhibit inflammatory cytokines in chondrocytes (4). Damage from ROS and alteration of redox-sensitive signaling pathways may be common mechanisms in the mediation of the effects of inflammatory cytokines and mechanical forces in OA.
ROS can cleave collagen (5) and proteoglycans (6,7), activate MMPs (8), and modulate signaling pathways (9), as well as alter cellular synthetic activity (10) and chondrocyte apoptosis (11). Because ROS are evanescent and difficult to measure directly in vivo, the evidence for their ability to produce damage in cartilage and for their association with arthritis is indirect. However, a number of studies have confirmed that chondrocytes can produce ROS and that ROS can damage ECM proteins. Hiran et al (12) demonstrated that in isolated chondrocytes stimulated with ionomycin, the rate of production of ROS was 0.03-0.15 nmoles/minute/106 cells. Ramamurthy et al (13) showed that a 5 μM concentration of NaOCl activated procollagenase in an in vitro model of MMP activation. Burkhardt et al (8) showed that 1-9 nmoles/minute of superoxide was able to cause direct oxidant damage and to further increase tissue degradation by activating collagenase. Tiku et al (14) demonstrated that chondrocytes produce hydrogen peroxide and that the production is increased by cytokines. ROS vary in their capacity to cause injury, depending on the reactivity of the molecule as well as the concentrations of the reactants and the vulnerability of the substrates.
As important as the rate of production of ROS is the scavenging capacity of the system relative to the production of ROS. Together, they determine the redox balance in a system. Extensive mechanisms for scavenging ROS have evolved in species that utilize oxygen for energy production. These include superoxide dismutase, catalase, peroxidases, and noncatalytic antioxidants. Either excess production of ROS or inadequate scavenging leads to a net oxidized state, with the potential for oxidant damage to proteins, lipids, or DNA. This net oxidized state within a cell or tissue is referred to as oxidative stress and can lead to direct oxidant damage or altered cell signaling by causing oxidation of thiol residues. Oxidative stress within the cell, the system, or the tissue is identified by indirect markers of the oxidant damage, such as nitrotyrosine formation in proteins or isoprostanes in lipids (15).
Mechanical forces generate oxidant molecules in cartilage and can inhibit or replicate the effects of the proinflammatory cytokine interleukin-1β, depending on the type and intensity of the force (16,17). In articular cartilage, the increased production of superoxide is associated with both the inflammatory cytokines and injurious mechanical forces (18,19).
Nitric oxide (NO) has also been identified as a mechanotransduction signaling molecule in chondrocytes (20-25). NO alone is a molecule whose low reactivity enhances its signaling function, but production of NO in an oxidizing environment, as when superoxide is present, leads to the formation of reactive molecules such as peroxynitrite. Peroxynitrite is formed from the rapid reaction of superoxide with NO. Since both molecules are produced by chondrocytes, it appears that with damaging mechanical force or inflammatory cytokines, there may be a shift toward a more oxidized state with greater production of superoxide.
One of the primary scavengers of ROS is superoxide dismutase (SOD), which catalyzes the dismutation of superoxide to hydrogen peroxide and then to water. Local deficiency of SOD can lead to the formation of peroxynitrite and other oxidizing species, while adequate SOD can scavenge superoxide and permit NO to remain active as a signaling molecule. Three distinct SODs are found in the human body: SOD1, or CuZnSOD, which localizes primarily to the cytosol, SOD2, or MnSOD, which is found in the mitochondria, and SOD3, or extracellular SOD (EC-SOD). EC-SOD is a secreted tetrameric glycoprotein with a positively charged heparin-binding site (15). The active center contains copper and zinc and is homologous with SOD1, but the EC-SOD molecule is immunologically distinct, and the C-terminal region contains a positively charged heparin-binding region. It localizes to the ECM of tissues by binding to the negatively charged proteoglycans and collagen (26,27). In this location, it can protect the vulnerable proteins and macromolecules of the ECM from oxidant injury. This important antioxidant has been studied in the vascular, pulmonary, and neurologic systems, but not in cartilage or joints (15,28,29).
We postulated that EC-SOD may be one of the major antioxidants in the cartilage, and its deficiency would lead to oxidant damage in the matrix and clinical disease. To test this hypothesis, we studied human femoral head cartilage obtained from OA patients at the time of joint replacement surgery, representing the late stage, severe form of the disease. Because identifying and studying the early stages of OA in humans is difficult, an animal model of OA was also evaluated. Progressive OA of the cartilage of the tibial plateau spontaneously develops by 35 weeks of age in ~80% of mice of the STR/ort strain. This model has been well characterized and includes many of the biochemical and histologic features of the idiopathic disease in humans (30,31). We measured EC-SOD expression and sought signs of oxidant damage in the tibial cartilage of the STR/ort mouse in order to understand the possible role of EC-SOD in the early phases of OA.
MATERIALS AND METHODS
Human articular cartilage
Human cartilage was collected from patients undergoing total joint replacement because of degenerative arthritis (OA patients; n = 34) or patients undergoing bipolar joint replacement because of a displaced fracture of the femoral neck (controls; n = 17). Exclusion criteria included a history of rheumatoid arthritis, local joint infection, or gout. Control patients were excluded if there was radiographic evidence of occult arthritis or if visual inspection of the articular surfaces demonstrated lesions consistent with OA. Written informed consent was obtained from the study patients, and the study protocol was reviewed and approved by the institutional review boards of Exempla Hospital and the National Jewish Medical and Research Center.
Cartilage from the femoral heads of cadaver donors younger than age 40 (n = 7) was obtained from a tissue bank (AlloSource, Centennial, CO). Donors were excluded if there was a history of arthritis or if there was any visible abnormality of the articular cartilage.
The height, weight, age, and sex of the subjects were recorded, and the body mass index (BMI; weight [kg]/height [m2]) was calculated. Each cartilage sample was cut from the underlying subchondral bone, frozen in liquid nitrogen, and stored at -80°C. For the OA cartilage, the tissue was obtained from the residual, normal appearing cartilage generally present at the posterolateral and anterolateral margins of the femoral head. A representative portion of the sample was fixed overnight in 10% formalin and then embedded in paraffin for Safranin O staining and histologic analysis.
EC-SOD enzyme-linked immunosorbent assay (ELISA)
Antibody 97-5, a rabbit polyclonal antibody raised against alkylated purified human EC-SOD, was used as the detection antibody. Antibody 4G11G6, a mouse monoclonal antibody raised against human EC-SOD purified from aortas, was used as the capture antibody. Antibodies were provided by one of us (JC) and prepared in our laboratory according to the methods described elsewhere (27). Samples of human cartilage (~100 mg) were pulverized under liquid nitrogen and extracted in radioimmunoprecipitation assay lysis buffer consisting of 5 mM EDTA, 50 mM Tris, pH 8, 150 mM NaCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS), and 0.5% sodium deoxycholate, with protease inhibitors (Complete Mini tablets, EDTA free; Roche, Indianapolis, IN) added. The samples were sonicated and rocked overnight at 4°C then centrifuged at 1,000 revolutions per minute for 5 minutes, and the lysate was removed. Protein concentrations in the lysates were determined using the bicinchoninic acid method (BCA Protein Assay kit; Pierce, Rockford, IL) (32).
EC-SOD protein was quantified using a sandwich ELISA. MaxiSorp immunoplates (96 well; Nunc, Naperville, IL) were coated overnight at 4°C with capture antibody 4G11G6 at 1 μg/ml in 100 mM sodium phosphate, pH 8.4. Plates were washed with Tris buffered saline-0.1% Tween 20 after all incubations. Wells were blocked with 1% bovine serum albumin (fraction V; fatty acid free)-10% normal goat serum-phosphate buffered saline (PBS) at 200 μl/well. Samples, including cartilage lysates and recombinant human EC-SOD standard (31 pg/ml to 2 ng/ml), were diluted in 1% bovine serum albumin-0.05% Tween 20. Detection antibody 97-5 was added at a dilution of 1:10,000 in blocking solution. Secondary antibody, goat anti-rabbit biotin (ABC Elite Vectastain kit for rabbit IgG; Vector, Burlingame, CA), was used at a dilution of 1:200 in blocking solution. Avidin-biotin-peroxidase complex solution was incubated for 15 minutes at 37°C. Avidin-biotinylated enzyme complex substrate solution was added at 100 μl/well.
Absorbance was measured at 405 nm. The amount of EC-SOD per microgram of extracted total protein was calculated for all of the samples. A subset of 4 control samples and 4 OA samples was used to compare the amount of EC-SOD protein per wet weight of cartilage with the amount of ECSOD per microgram of extracted total protein.
RNA extraction and real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR)
After surgery, cartilage samples were collected into microfuge tubes and placed in either 500 μl of RNAlater (Ambion, Austin, TX) or liquid nitrogen. The snap-frozen samples were stored at -70°C. Samples preserved in RNAlater were incubated at 4°C to allow the preservative to soak into the tissue. Tissue pieces were then removed from the RNAlater, placed in new microfuge tubes, and stored at -70°C. Tissues were pulverized using liquid nitrogen in a Bio-Pulverizer (BioSpec Products, Bartlesville, OK). RNA was extracted with an RNeasy Midi Prep kit obtained from Qiagen. A mixture of buffer RLT and proteinase K (Qiagen, Chatsworth, CA) along with 2 mg/ml of collagenase II (Worthington, Lakewood, NJ) and 120 units of RNase Out RNase inhibitor (Invitrogen, San Diego, CA) was used to further degrade tissues (overnight at 55°C in a shaking water bath). After enzymatic degradation, samples were sonicated (Branson Sonifier; VWR, West Chester, PA) using 10-second pulses at 30% output for 30 seconds.
RNA was quantified by ultraviolet spectroscopy (model UV-2401 PC; Shimadzu, Kyoto, Japan). Five microliters of total RNA was reverse transcribed using the RetroScript kit (Ambion), and real-time PCR was conducted to examine the levels of human EC-SOD in cartilage. Levels of human EC-SOD were normalized against 18S RNA and expressed as a ratio of human EC-SOD to 18S. Universal 18S primers were obtained from Ambion. The following primer pairs were used: for EC-SOD, 5′-CTGGCTGGGTGCAGCTCTCTTTTC-3′ (forward) and 5′-GGCGGTGGATGCGAAACAG-3′ (reverse); for inducible nitric oxide synthase (iNOS), 5′-CAACAACAATGTGGAGAAAGCCCC-3′ (forward) and 5′-AATGGGGTTGCATCCAGCTTGAC-3′ (reverse); for neuronal NOS (nNOS), 5′-ACATGTTCGGTGTTCAGCAAATCCA-3′ (forward) and 5′-CCGTTGACCGCAAGAATGATGTCT-3′ (reverse), and for endothelial cell NOS (eNOS), 5′-CCCTCGTGTGAAGAACTGGGAGGT-3′ (forward) and 5′-CGTGGAAATACCAGGGAGCCCA-3′ (reverse).
Twenty-five-microliter reactions were set up containing 2 μl of sample complementary DNA (cDNA), 2.5 mM MgCl2, 25× SYBR Green I (Molecular Probes, Eugene, OR), and one Ready-To-Go PCR bead (Amersham, Piscataway, NJ). Thermal cycling conditions consisted of denaturation at 94°C, followed by 40 (18S) or 55 (human EC-SOD) cycles of amplification (denaturation at 94°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 30 seconds). Quantification was based on the fluorescence generated from the binding of SYBR Green I to the PCR products in the extension step. PCR reaction products were run on a 2% SeaKem LE agarose gel (Cambrex, Walkersville, MD) to confirm the human EC-SOD (219 bp) and 18S (315 bp) amplicons.
Immunohistochemistry
All incubations were performed at room temperature, followed by washing in PBS (3 times for 5 minutes each) except where noted otherwise. Paraffin sections were deparaffinized in CitriSolv (2 times for 7 minutes each), 100% ethanol (2 times for 5 minutes each), 95% ethanol (2 times for 5 minutes each), and 70% ethanol (2 times for 5 minutes each). Cryosections were fixed in 4% paraformaldehyde for 15 minutes and washed in PBS. Samples were blocked for endogenous H2O2 with 2% H2O2/methanol for 30 minutes, and then washed in water. Sections were treated with hyaluronidase (1 mg/ml in PBS) for 20 minutes at 37°C, then blocked in 5% normal goat serum/PBS for 30 minutes, followed by incubation with primary antibody 97-5 (1:100 dilution in 2% normal goat serum/PBS) for 1 hour. Prior to addition of secondary antibody, sections were again blocked in 5% normal goat serum/PBS for 10 minutes.
Biotinylated goat anti-rabbit IgG secondary antibody (ABC Elite Vectastain kit) was used at a dilution of 1:500 in 2% normal goat serum/PBS and incubated for 30 minutes. Avidin-biotin complex solution was premixed 30 minutes prior to use and incubated with the sections for 30 minutes. Sections were then developed using a diaminobenzidine peroxidase substrate kit (Vector) for 5 minutes and counterstained with 1% methyl green (Zymed, Burlingame, CA).
The specificity of the EC-SOD antibodies was assessed by absorbing the EC-SOD antibody with purified recombinant EC-SOD and demonstrating an absence of staining in the cartilage sections.
Grading of immunohistochemistry features
Staining of the cells and the matrix were graded separately by one of us (ER), who was blinded to the source of the tissues. Staining of cells was graded on a scale of 1-3, where 1 = dark brown staining of ≥50% of the cells, 2 = dark brown staining of 10-49% of the cells, and 3 = light or dark brown staining of <10% of the cells. Staining of the matrix was graded on a scale of 0-3, where 0 = no brown staining (only purple staining), 1 = light brown staining of the matrix around chondrocytes, with at least 75% of the area between cells unstained, 2 = light brown staining that was confluent in some areas, with up to 50% of the matrix unstained, and 3 = brown staining of the matrix that was confluent over most of the section. Scores were summed to produce a final grade that ranged from 1 to 6, where 6 = EC-SOD largely secreted into the matrix, with little stimulation of cellular production and 1 = little or no evidence of matrix-based EC-SOD staining, but large stimulation of cellular production.
Safranin O staining
Tissue samples were fixed overnight in 10% formalin and then embedded in paraffin. Each section was stained with hematoxylin for 10 minutes, followed by fast green for 4 minutes, with a rinse in 1% glacial acetic acid. Safranin O stain was applied for 5 minutes, and the sections were dehydrated and mounted. An unselected subset of 13 control and 19 OA samples were graded blindly using the Mankin grading system for OA cartilage, assessing tissue structure, cells, and proteoglycan content, as marked by the Safranin O stain (33). No assessment was made of the tidemark integrity, since all samples were cut above the tidemark.
Nitrotyrosine fluorescence immunohistochemistry
We studied 1 sample of OA cartilage and 1 sample of control cartilage for the formation of 3-nitrotyrosine in human tissues. Fluorescence images were obtained of 0.4μ-thick sections that had been prepared by ultracryotomy from 0.2-mm slices of cartilage and fixed by immersion in phosphate buffered (pH 7.4) 2% formaldehyde plus 0.2% glutaraldehyde. Rabbit polyclonal antinitrotyrosine was kindly provided by Dr. Harry Ischiropoulos (Stokes Research Institute, Philadelphia, PA). Secondary antibody was commercial Cy3-labeled goat anti-rabbit IgG.
Extraction of mouse articular cartilage
Tibial plateau cartilage was obtained from male STR/ort mice and CBA control mice (ages 5, 15, and 25 weeks). Cleavage was obtained at the junction of the calcified cartilage zone and the underlying bone. The cartilage was extracted overnight at 4°C, with agitation, in 500 mM NaCl, 25 mM Tris, pH 7.5, with a protease inhibitor cocktail, without EDTA. The cartilage extracts were used for subsequent Western analysis of EC-SOD. The cartilage residue was stored at -20°C and used in a hydroxyproline analysis to measure the collagen content of the tissue.
Polyacrylamide gel electrophoresis (PAGE) of mouse proteins
Samples were solubilized in reducing SDS-PAGE loading buffer containing a cocktail of protease inhibitors (Roche Diagnostics, Lewes, UK). Samples were boiled for 5 minutes and resolved on 10% SDS-PAGE gels, and proteins were transferred to a polyvinylidene difluoride membrane filter (Immobilon-P; Millipore, Watford, UK). Immunodetection was performed essentially as described by Towbin et al (34).
EC-SOD protein was detected with a polyclonal rabbit anti-mouse EC-SOD antibody (1:2,000 dilution) and goat anti-rabbit peroxidase-conjugated secondary antibody (1: 10,000 dilution). Purified mouse lung EC-SOD was used as a reference standard. Nitrotyrosinylated proteins were detected using a polyclonal rabbit anti-nitrotyrosine primary antibody (1:1,500 dilution; Upstate Biotechnology, Lake Placid, NY) and goat anti-pig antibody (1:10,000 dilution). A mixture of 3 nitrosylated proteins was used as a reference standard (Upstate Biotechnology). Enhanced chemiluminescence detection was used in both cases (Amersham, Little Chalfont, UK).
The Western blots and agarose gels were scanned with an Epson scanner (Epson, Hertfordshire, UK) into Adobe software (Adobe Systems, San Jose, CA). To minimize error in the scanning procedure or resulting from variations in bandwidth or thickness, the procedure was repeated 3 times, and the mean value was used. The nitrotyrosinylated proteins were quantified by densitometry, by comparing the total relative density of proteins on Western blots that reacted with the antinitrotyrosine antibody on each lane (normalized against hydroxyproline).
Hydroxyproline assay of mouse cartilage
The hydroxyproline assay was performed essentially as described by Prockop and Udenfriend (35), with some modifications. After extraction of the mouse tibial cartilage in 500 mM NaCl and 25 mM Tris, pH 7.5, the cartilage residue was digested overnight in 6M HCl at 110°C. The dried hydrolysate was then resuspended in 400 mM citric acid, 100 mM sodium acetate buffer, pH 6. Then, 6 mM chloramine T was added to oxidize the hydroxyproline, followed by the addition of perchloric acid and 100 mM Ehrlich's reagent (dimethylaminobenzaldehyde) at 60°C for 30-45 minutes, until the color was adequately developed for detection at 570 nm.
Semiquantitative RT-PCR analysis of mouse cartilage
For RNA analyses, mouse tibial cartilage samples were dissected as described above. Cartilage was powdered under liquid nitrogen, then extracted in TRIzol. The expression of EC-SOD was determined by RT-PCR. Aliquots of total RNA were reverse transcribed into cDNA and was amplified using forward and reverse primers (5′-TTCTTGTTCTACGGCTTGCTA-3′ and 5′-GAACTCATGCACGTGGATGGC-3′). Specific oligonucleotides were also used to amplify transcripts of mouse GAPDH, a reference control gene.
Statistical analysis
Linear regression modeling using EC-SOD protein levels as the outcome variable in studies of human tissues was used to test for effects of age, total protein concentration, sex, and BMI on the relationship between disease status and EC-SOD. Messenger RNA (mRNA) for EC-SOD, iNOS, and nNOS from the 2 groups of human tissues were compared using nonparametric tests. Histologic scores in human tissues were also compared using nonparametric tests. Comparisons between the control and STR/ort mice for EC-SOD protein, EC-SOD mRNA, and nitrotyrosine were performed using Student's t-test with Bonferroni correction for multiple comparisons. All reported values are the mean ± SEM.
RESULTS
Histologic features of human articular cartilage
Cartilage was obtained from patients undergoing hip joint replacement because of OA. Samples were taken from areas that retained largely normal architecture and demonstrated only mild surface irregularities. Because OA tends to occur focally and then progress throughout the cartilage, it was usually possible to identify suitable areas for sampling, even in joints in which advanced arthritis had led to joint replacement. Safranin O staining for proteoglycans was used to grade the tissue according to the Mankin classification (33). The mean Mankin grade was 6.5 in the OA group and 1.1 in the normal control group (femoral neck fracture). The major factors contributing to the differences between groups were the degree of cellularity and the loss of proteoglycan from the tissue. The baseline characteristics and EC-SOD levels in the OA and control subjects are shown in Figure 1A.
Figure 1.
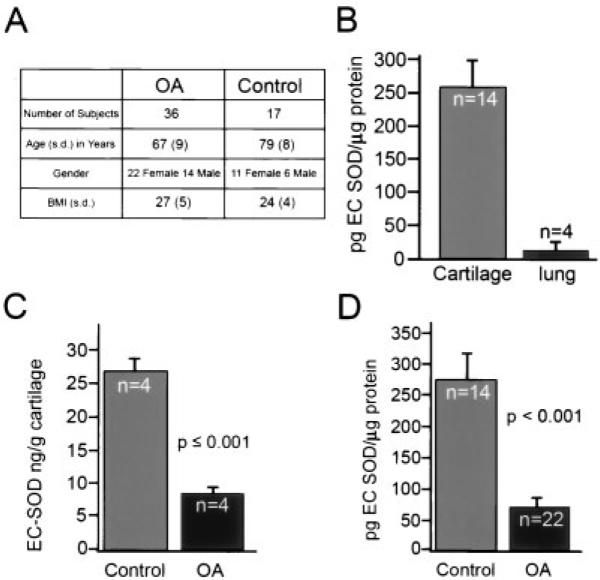
Extracellular superoxide dismutase (EC-SOD) in normal and osteoarthritic (OA) human cartilage and in human lung tissues. A, Baseline data in the OA and femoral neck fracture (normal control) patients. There was a preponderance of women in both groups. The control group was slightly older than the OA group and had a trend toward a higher body mass index (BMI). Linear regression modeling showed that only disease state (OA or normal control) was a significant predictor of the EC-SOD level. B, Levels of EC-SOD in lung tissue from cadaver donors and in normal articular cartilage from patients with femoral neck fracture, as determined by enzyme-linked immunosorbent assay (ELISA) using a polyclonal antibody against recombinant human EC-SOD. Cartilage, with its extensive extracellular matrix, has large amounts of EC-SOD compared with lung tissue. C, Levels of EC-SOD per gram of cartilage wet weight in a subset of OA and normal control cartilage, as determined by ELISA. There is an ~4-fold decrease in EC-SOD content in OA cartilage compared with control cartilage. D, Levels of EC-SOD protein in a random subset of cartilage samples from OA patients and normal controls, as determined by ELISA. Again, there is an ~4-fold decrease in EC-SOD protein in OA cartilage compared with control cartilage. Except where indicated otherwise, values are the mean and SEM.
EC-SOD levels in human articular cartilage and lung tissues
EC-SOD levels were extremely high in human cartilage samples. The mean ± SEM concentration in the 4 OA and 4 control samples together was 18.8 ± 3.8 ng/gm wet weight of cartilage. The concentration in the 4 OA samples was 7.6 ± 0.65 ng/gm of cartilage, and the concentration in the 4 control samples was 26.8 ± 0.65 ng/gm of cartilage (Figure 1C). In a previous study (28), EC-SOD was found to be most highly expressed in arterial walls and to have higher expression in the lung than in other major organs. Figure 1B shows the concentrations of EC-SOD in cartilage as compared with lung tissue. We found that cartilage had a 10-fold higher concentration of EC-SOD than lung tissue.
EC-SOD levels in human OA articular cartilage
We compared levels of EC-SOD protein in cartilage from patients with femoral neck fractures with those in cartilage from patients with OA. Because there were differences between the 2 groups in terms of age, BMI, and amount of protein extracted from the tissue, we fit a general linear univariate model with EC-SOD protein concentration as the outcome variable and disease status, age, sex, protein concentration, BMI, and their interactions as predictors. Using a planned, backwards, stepwise testing procedure, age, sex, protein concentration, BMI, and their interactions were eliminated from the model. In the final model, disease status (OA versus control) was a strong predictor of EC-SOD levels (F = 20.633, P < 0.001), with significantly decreased EC-SOD in the cartilage of OA patients. Cartilage from young cadaver donors who had no evidence of arthritis had a mean ± SEM EC-SOD level of 141 ± 70 pg/μg of extracted protein. This level was increased to 278.2 ± 36.5 pg/μg of extracted protein in older control subjects with femoral neck fracture (Figure 1D). The OA patients had a mean level of EC-SOD of 67.2 ± 28.7 pg/μg of extracted protein (Figure 1D), which was substantially lower than that in either the young or the older controls.
Immunolocalization of EC-SOD in human articular cartilage
A subset of cartilage samples was used to investigate the immunolocalization of EC-SOD (Figure 2). Seven OA samples and 8 control samples (2 from patients with femoral neck fractures and 6 from young cadaver donors) were compared using a grading scale to rank the degree of both matrix staining and intracellular immunostaining for EC-SOD. The OA samples showed a clear pattern of decreased EC-SOD in the matrix, but increased intracellular staining (mean score 3.0 in the OA group and 4.8 in the control group; z = -2.4, P = 0.016, by Kruskal-Wallis nonparametric test).
Figure 2.

Immunolocalization of extracellular superoxide dismutase (EC-SOD) in normal and osteoarthritic (OA) human cartilage. Immunohistochemistry of EC-SOD in human cartilage samples was performed using polyclonal antibody to human EC-SOD and peroxidase staining. Negative controls were prepared with EC-SOD-absorbed antiserum. A, Cartilage section from a 37-year-old male cadaver donor, showing significant EC-SOD staining in the matrix surrounding the chondrocytes, with variable light staining intracellularly. B, Negative control for the section shown in A. C, Cartilage from a 58-year-old woman with OA, showing increased cellularity, degeneration of the superficial cartilage layer, and pronounced lack of matrix staining. Note the increased intracellular staining for EC-SOD compared with the nonarthritic chondrocytes. D, Negative control for the section shown in C. (Original magnification × 100.)
There was a striking difference in the distribution of EC-SOD. The control samples (Figures 2A and B), demonstrated a smooth distribution of staining in the ECM, with some variable intracellular staining. In the OA samples (Figures 2C and D), there was a dramatic decrease in the staining of the matrix, whereas the chondrocytes showed increased staining. The intracellular appearance of EC-SOD in the OA samples was consistent with the pattern of increased mRNA seen in this group, which suggests that there is up-regulation of EC-SOD in response to oxidative stress.
Levels of mRNA for EC-SOD and NOS in human articular cartilage
Cartilage samples were evaluated for levels of mRNA for EC-SOD, iNOS, nNOS, and eNOS, using real-time quantitative RT-PCR (Figure 3). The OA patients showed a strong trend toward increased EC-SOD message compared with controls (human ECSOD:18S mRNA 685 ± 200 in OA samples and 97.6 ± 36 in controls; z = -2.11, P = 0.03, by Wilcoxon's rank sum test) (Figure 3A). The levels of mRNA for both iNOS and nNOS were also increased in the OA samples compared with the controls (Figures 3B and C). We were not able to detect eNOS mRNA in either the OA or control samples (data not shown). The percentage change in the levels of mRNA in OA samples compared with control samples were 25.3% for EC-SOD, 17.3% for iNOS, and 20.4% for nNOS.
Figure 3.
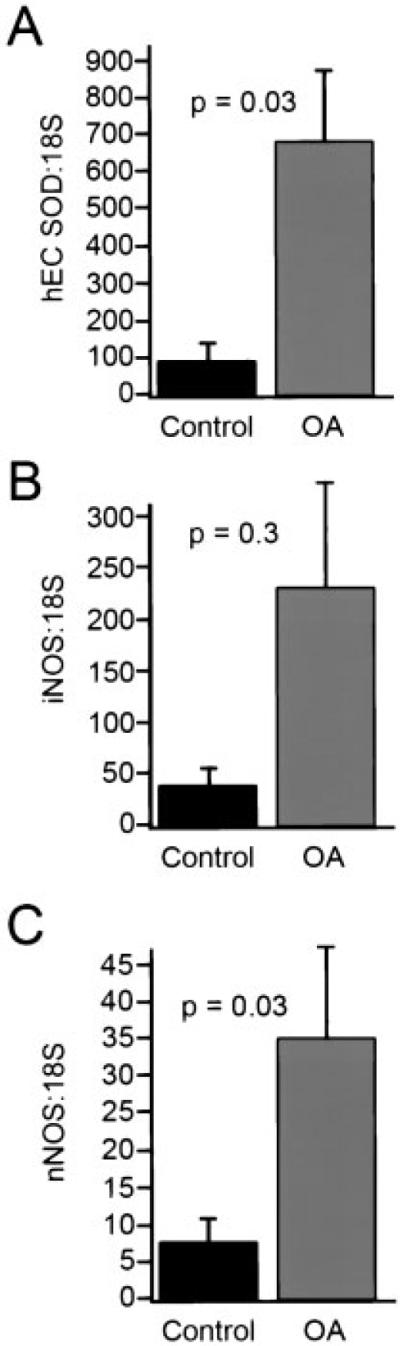
Messenger RNA for human extracellular superoxide dismutase (EC-SOD), inducible nitric oxide synthase (iNOS), neuronal NOS (nNOS), and endothelial cell NOS (eNOS) in human articular cartilage. RNA was extracted from OA (n = 36) and control (n = 16) cartilage, and quantitative real-time reverse transcription-polymerase chain reaction was used to measure levels of mRNA for human EC-SOD (hEC SOD) (A), iNOS (B), nNOS (C), and eNOS (data not shown). Values were normalized against 18S and expressed as a ratio to 18S. We did not detect eNOS in any of the cartilage samples. Levels of mRNA for EC-SOD (z = -2.11, P = 0.03) and nNOS (z = -2.2, P = 0.03) were significantly increased in OA patients compared with controls, whereas levels of mRNA for iNOS were increased 4.6-fold, but the increase was not statistically significant (P = 0.3), by Wilcoxon's rank sum test. Values are the mean and SEM.
Findings in mouse cartilage
Levels of EC-SOD protein and mRNA and levels of nitrotyrosine residues in tibial plateau cartilage from CBA mice, which do not develop OA, were compared with those in cartilage from STR/ort mice, which develop OA almost universally. Comparisons were made at ages 5 weeks (before the development of histopathologic OA lesions), 15 weeks, and 25 weeks. Cartilage samples were extracted and analyzed for EC-SOD protein (Figure 4A), mRNA for EC-SOD (Figure 4B), and nitrotyrosinylated proteins (Figures 4C and D).
Figure 4.

Levels of extracellular superoxide dismutase (EC-SOD) and nitrotyrosine formation in the STR/ort mouse model of osteoarthritis (OA). Tibial plateau cartilage samples from STR/ort and control CBA mice ages 5, 15, and 25 weeks were compared for levels of EC-SOD protein and mRNA by Western blotting and reverse transcription-polymerase chain reaction techniques (RT-PCR), as well as for evidence of nitrotyrosine formation. A, EC-SOD protein expression (mean and SEM). At 5 weeks of age, there is a dramatic decrease in EC-SOD protein in cartilage from STR/ort mice compared with CBA mice, which do not develop arthritis. At this age, there is no histologic evidence of arthritis in STR/ort mice. * = t = 8.48, P < 5 × 10-5. B, EC-SOD mRNA levels (mean and SEM), as measured by RT-PCR. At 5 weeks of age, there is a decrease in EC-SOD mRNA in cartilage from STR/ort mice compared with control mice, suggesting either a failure of transcription or a rapid turnover of the pool. * = t = 10.54, P < 0.001. C, Levels of extractable nitrotyrosinylated proteins (mean and SEM). There is a small, but significant, decrease in STR/ort mice compared with control mice at ages 5 weeks (* = t = 3.93, P < 0.001), with progressive increases over time (* = t = 5.4, P < 0.0009 at 15 weeks and * = t = 6.27, P < 0.0004 at 25 weeks). The progressive increase in nitrotyrosinylated proteins despite the apparent correction of EC-SOD protein and mRNA levels suggests ongoing oxidant stress in the tissue following the initial insult. The initial EC-SOD deficiency with oxidative damage at 5 weeks may compromise the function of key proteins or signaling pathways in chondrocytes or in the matrix, leading to more oxidative damage and arthritis. D, Representative Western blot of nitrotyrosinylated proteins in cartilage from 2 CBA mice and 2 STR/ort mice ages 25 weeks. At least 3 proteins with molecular weights of ~75, 66, and 60 kd, indicating nitrotyrosine formation, were dramatically increased in STR/ort cartilage compared with control cartilage.
An interesting pattern emerged, with cartilage from 5-week-old STR/ort mice showing a significant deficiency of EC-SOD protein as compared with cartilage from 5-week-old CBA mice (mean ± SEM 7.43 ± 2.2/μg of hydroxyproline in STR/ort mice versus 37.9 ± 3.7/μg of hydroxyproline in CBA mice; P < 0.001). At 15 and 25 weeks of age, the STR/ort mice no longer demonstrated a clear deficiency in EC-SOD, although there was evidence of ongoing oxidative stress. Western blotting of extractable proteins showed significantly increased nitrotyrosine residues in samples from STR/ort mice compared with control CBA mice at all ages tested (5, 15, and 25 weeks). Levels of mRNA for EC-SOD were significantly decreased at 5 weeks of age, but by 15 weeks of age, had increased to levels similar to those in the CBA controls.
Nitrotyrosine formation in human articular cartilage
We also studied the formation of 3-nitrotyrosine in articular cartilage from 1 OA patient and 1 control patient with femoral neck fracture. As shown in Figure 5, there was a distinct speckled pattern of nitrotyrosine staining in the pericellular ECM of the OA cartilage sample, as well as intracellular fluorescence. While the normal cartilage sample also showed intracellular fluorescence, there was no staining of the ECM.
Figure 5.

Nitrotyrosine in the extracellular matrix (ECM) of human cartilage samples, as determined by fluorescence immunohistochemistry. A, Osteoarthritic cartilage immunostained for 3-nitrotyrosine shows a distinct speckled pattern of labeling in the pericellular ECM as well as intracellular fluorescence. B, Control cartilage shows intracellular staining for 3-nitrotyrosine, but no staining of the ECM. (Original magnification × 100.)
DISCUSSION
EC-SOD has not previously been studied in articular cartilage. We found that it was present in large amounts in normal human cartilage and was decreased ~4-fold in OA cartilage. Extracellular SOD is the major scavenger of superoxide in the extracellular spaces and fluids. It is highly expressed in blood vessels, especially arteries, and vascular smooth muscle, as well as in lung tissue. Both tissues have shown important correlations between EC-SOD deficiency and diseases (28,36). Deficiency of EC-SOD increases bleomycin-induced pulmonary fibrosis (37) and increases sensitivity to hyperoxic lung injury (38). Two recent studies of the vascular system associated EC-SOD deficiency with cardiovascular disease (29,39). Our findings that EC-SOD is present in 10-fold higher concentrations in articular cartilage as compared with lung tissue and that it is markedly decreased in OA cartilage as compared with normal cartilage suggest that EC-SOD plays a key role in modulating ROS in cartilage.
Because cartilage is avascular and hypoxic, the concept that ROS may play a part in tissue regulation is counterintuitive. Oxygen concentrations in cartilage have been shown to range from 5% at the surface to 1% in the deeper zones (40). Chondrocytes do produce ROS when grown in culture at 21% oxygen and possess an NADPH oxidase-like complex (12), but the effect on ROS production has not been studied under conditions of low oxygen pressure. However, hypoxia has been shown in other systems to induce a paradoxical increase in oxygen radicals, especially superoxide, apparently due to a shift to 1-electron transfers in the electron transport chain (41,42). Identifying large amounts of EC-SOD, an important ROS scavenger, in cartilage tissue suggests that ROS scavenging is necessary for homeostasis and that ROS are produced in significant amounts.
Collagen is particularly vulnerable to oxidant damage, which impairs its mechanical properties (5). Proteoglycans in the ECM or extracellular fluids can be cleaved by ROS (6). Damage to these structural proteins from excess oxidants would leave the cartilage at risk of mechanical failure under normal loads and would change the water-holding capacity of the tissue. Decreased tensile strength is seen in OA cartilage in association with damage to collagen fibrils (43). Interestingly, this damage to the collagen fibrils has been shown to originate around chondrocytes (44). Since chondrocytes are the source of ROS, whether generated by mechanical forces or cytokines, the fact that the initial collagen damage is found in pericellular areas is consistent with a local pericellular deficiency of oxidant scavenging.
A superoxide molecule produced in the extracellular space by a membrane oxidase would potentially remain in the ECM until it is either scavenged or reacts with one of the molecules of the ECM, such as collagen or aggrecan. The diffusion path, or distance that the molecule can travel before reacting, has not been studied for superoxide in the ECM, but in the vascular system, the diffusion path was found to be ~40μ (45). Superoxide is highly reactive with hemoglobin, and thus, the diffusion path is likely to be greater in avascular cartilage. The distance that a reactive molecule can travel in a tissue is related to its reduction potential and charge. Because of its high reactivity (reduction potential 2.31V), the hydroxyl radical would have a much shorter diffusion path than superoxide (reduction potential 0.94V). Since superoxide is negatively charged, it does not diffuse through plasma membranes.
The local pericellular loss of EC-SOD that we found in the OA cartilage, as shown in Figure 3, could account in part for the pericellular collagen damage and the mechanical deficiency of the OA cartilage. Our observation that EC-SOD is decreased in OA cartilage may represent an important etiologic factor, representing either an initial trigger for the disease due to a congenital deficiency in EC-SOD or an accelerator of the process after some of the proteoglycans and their EC-SOD-binding sites are lost from the tissue.
Superoxide and nitric oxide react rapidly in biologic systems to produce toxic species, including peroxynitrite and the hydroxyl radical. With adequate local extracellular SOD, superoxide is removed from intercellular spaces and NO signaling can occur (46). Because superoxide is formed in response to both inflammatory cytokines and damaging mechanical forces, the capacity to scavenge excess superoxide may represent an important control point for maintaining protection of the ECM structural molecules. Although low levels of hydrogen peroxide have been shown to stimulate synthesis and may help to repair matrix after mechanical injury (10), with inadequate EC-SOD, the balance may shift to an oxidized/inflammatory state with formation of the more toxic radicals.
Del Carlo and Loeser (47) have shown that NO alone is not sufficient to cause chondrocyte apoptosis, but that it requires the addition of more ROS. This finding correlates well with the dualistic effects of NO shown in other biologic systems where NO alone is a physiologic signaling molecule and the damaging effects require an oxidizing environment (48). Excess ROS can result in damage to the structural proteins, collagen, and proteoglycans if no mechanism is present to scavenge the reactive molecules, particularly if the proinflammatory cytokines activate NF-κB, inducing iNOS expression and producing NO in association with superoxide (49).
We found that EC-SOD mRNA was increased in OA cartilage in concert with the local deficiency of EC-SOD protein. We also found mRNA for both iNOS and nNOS were increased proportionately to the ECSOD mRNA in the OA cartilage (Figure 4). In dermal fibroblasts, EC-SOD has been shown to be up-regulated by the inflammatory cytokines tumor necrosis factor α and interferon-γ (50). EC-SOD was also shown to be temporally coregulated with iNOS in response to NF-κB activation (51). Although the factors that regulate ECSOD expression in chondrocytes have not been defined, our findings are consistent with a coordinated response.
Our subject population represents the severe end of the OA disease spectrum, given that the disease had progressed to necessitate joint replacement. Since tissue samples were obtained late in the disease process, the findings may not represent the biochemical situation at disease onset. Although alterations in the redox state of the chondrocytes may constitute a part of the initiation or propagation of OA, the changes in EC-SOD in the tissue may also represent secondary effects of proteoglycan loss and lack of binding sites in the matrix. We therefore turned to an animal model of spontaneous OA to define whether there were changes in the oxidative state of cartilage early in the disease.
The STR/ort mouse model allowed us to study alterations in EC-SOD during the prelesional period, which is not possible to study in human OA. We found that the STR/ort mouse is deficient in EC-SOD weeks before developing histologic lesions, which suggests that inadequate scavenging of ROS occurs before the matrix is disrupted. The presumed increase in ROS due to EC-SOD deficiency is associated with a progressive increase in nitrotyrosine residues in the cartilage. The response of increased EC-SOD in the tibial cartilage was not adequate to prevent continued oxidative stress in the tissue, as demonstrated by the increasing nitrotyrosine formation at 15 and 25 weeks of age.
The fact that the EC-SOD level in the STR/ort mice increased to the level in the control CBA mice does not mean that it retained adequate scavenging capacity for tissue that had already sustained oxidative damage and had increased production of ROS. Studies in other systems have shown that when oxidative stress occurs, the response of the antioxidant enzymes is to rise to supranormal levels in order to control the excess ROS being generated. This supranormal increase in antioxidants did not occur in the STR/ort mice, and the state of oxidative stress was not corrected. Demonstrating deficiency of EC-SOD in the young mice in association with progressive increases in nitrotyrosine over the time course of the disease represents an important corroboration of the postulated role of ROS and EC-SOD deficiency in the initiation of OA.
We believe that the mouse cartilage, with lower levels of EC-SOD found in 5-week-old animals before the development of OA lesions, reflect an initiating event that causes oxidative stress/damage to key proteins in the murine joint. We postulate that this continuing oxidative damage, despite the reactive increase in ECSOD levels in the cartilage, acts in an unknown manner to trigger the progressive loss of cartilage. The reactive increase in EC-SOD in the STR/ort mouse cartilage is inadequate to manage the ROS that are being generated, even though the chondrocytes are able to respond in part to the oxidative stress.
In the cartilage samples from the OA patients, we are looking at the very late stages of the disease. Proteoglycan has been lost, although the tissue architecture is retained, and adjacent tissue has undergone complete destruction. Further study of human tissues is needed to define the disease process, but we would postulate that after a period of compensation for increased oxidative stress, there is a late stage in which the continued production of EC-SOD is inadequate and that this, coupled with losses of binding sites, leads to a late phase of low EC-SOD in the tissue.
EC-SOD is secreted with a positively charged binding site and binds to collagen and proteoglycans in the ECM. The high levels of EC-SOD that we found in normal cartilage may play several critical roles in the tissue, such as to protect the vulnerable structural proteins in the ECM from direct oxidant damage, to limit the formation of peroxynitrite and prevent its deleterious effects on proteins and the cytoskeleton, to permit the unopposed signaling of NO in the tissue, and to modulate the local redox state and signaling pathways in the pericellular matrix and chondrocytes.
Articular cartilage is a mechanically sensitive tissue in which alterations in mechanical loading affect the synthetic and degradative pathways, and chondrocytes are able to respond to harmful or beneficial forces with different cellular biochemical messages (52). OA chondrocytes exposed to shear stress up-regulate iNOS in association with an inhibition of collagen and aggrecan synthesis, and both NO and superoxide are produced in cartilage tissue exposed to prolonged compression (16,53). Both the mechanical messages and cytokine messages in cartilage appear to be mediated by NO and ROS. Since EC-SOD scavenges superoxide, it can modulate the signaling events on the cell surface between NO alone and within an oxidizing environment. Unopposed signaling of NO may reduce inflammation by inhibiting NF-κB (54). Use of SOD mimetics has been shown to reduce the severity of inflammation in collagen-induced arthritis (55).
OA is a multifactorial disease in which genetic influences and environmental factors affect the occurrence of the disease and the age at onset. Increased body weight, excessive shearing forces from a malaligned or unstable joint, or unusual loading from occupational activities might be transduced as increased reactive molecules in the cartilage, but the variation in scavenging capacity within the joint would account for the individual differences in the incidence of arthritis or the age at onset. The individual capacity to buffer ROS can be related to genetic factors, possibly affected by hormones, or decreased by other disease processes that generate excess ROS (e.g., iron overload). By demonstrating that cartilage contains large amounts of ECSOD, we propose that its role is to protect the extracellular structural proteins from ROS that is routinely generated in the tissue. EC-SOD deficiency may be involved in the disease process as a result of early matrix changes and loss of binding sites in the tissue or as a result of an inadequate production of the enzyme. In showing an association between OA and a decrease in a major extracellular ROS scavenger in cartilage, we suggest that damage from ROS plays a key role in either the initiation or the propagation of OA.
We propose that an altered redox state in articular cartilage can lead to some of the major findings that are associated with OA and offer a means of integrating the role of mechanical factors and the varying predisposition of individuals to the disease. In concert with the direct oxidant damage to the structural proteins, the additional effects of abnormal redox signaling may lead to a failure of the repair processes or to a decreased responsiveness to growth factors (56). Our findings of profoundly decreased levels of EC-SOD in OA cartilage, both in humans and in an animal model of spontaneous OA, support our thesis and suggest that this is an important area for further research into new treatment options.
ACKNOWLEDGMENTS
The authors thank the surgeons of the orthopedic Department of Colorado Permanente (Denver, CO), their patients for the donation of surgical tissue samples, and the staff of Exempla St. Joseph's Hospital (Denver, CO), as well as AlloSource (Centennial, CO) for the donation of additional cartilage samples. We also thank Dr. Henry Mankin for information on grading the cartilage samples.
REFERENCES
- 1.Lawrence RC, Helmick CG, Arnett FC, Deyo RA, Felson DT, Giannini EH, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778–99. doi: 10.1002/1529-0131(199805)41:5<778::AID-ART4>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 2.Anderson JJ, Felson DT. Factors associated with osteoarthritis of the knee in the first national Health and Nutrition Examination Survey (HANES I): evidence for an association with overweight, race, and physical demands of work. Am J Epidemiol. 1988;128:179–89. doi: 10.1093/oxfordjournals.aje.a114939. [DOI] [PubMed] [Google Scholar]
- 3.Pelletier JP, Martel-Pelletier J, Abramson SB. Osteoarthritis, an inflammatory disease: potential implication for the selection of new therapeutic targets [review] Arthritis Rheum. 2001;44:1237–47. doi: 10.1002/1529-0131(200106)44:6<1237::AID-ART214>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal S, Deschner J, Long P, Verma A, Hofman C, Evans CH, et al. Role of NF-κB transcription factors in antiinflammatory and proinflammatory actions of mechanical signals. Arthritis Rheum. 2004;50:3541–8. doi: 10.1002/art.20601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monboisse JC, Borel JP. Oxidative damage to collagen. EXS. 1992;62:323–7. doi: 10.1007/978-3-0348-7460-1_32. [DOI] [PubMed] [Google Scholar]
- 6.McCord JM. Free radicals and inflammation: protection of syno-vial fluid by superoxide dismutase. Science. 1974;185:529–31. doi: 10.1126/science.185.4150.529. [DOI] [PubMed] [Google Scholar]
- 7.Klamfeldt A, Marklund S. Enhanced breakdown in vitro of bovine articular cartilage proteoglycans by conditional synovial medium: the effect of superoxide dismutase and catalase. Scand J Rheumatol. 1987;16:41–5. [PubMed] [Google Scholar]
- 8.Burkhardt H, Schwingel M, Menninger H, Macartney HW, Tschesche H. Oxygen radicals as effectors of cartilage destruction: direct degradative effect on matrix components and indirect action via activation of latent collagenase from polymorphonuclear leukocytes. Arthritis Rheum. 1986;29:379–87. doi: 10.1002/art.1780290311. [DOI] [PubMed] [Google Scholar]
- 9.Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL, Schwartz GD, et al. Redox regulation of cell signalling. Nature. 1996;381:380–1. doi: 10.1038/381380a0. [DOI] [PubMed] [Google Scholar]
- 10.Burdon RH. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic Biol Med. 1995;18:775–94. doi: 10.1016/0891-5849(94)00198-s. [DOI] [PubMed] [Google Scholar]
- 11.Blanco FJ, Ochs RL, Schwarz H, Lotz M. Chondrocyte apoptosis induced by nitric oxide. Am J Pathol. 1995;146:75–85. [PMC free article] [PubMed] [Google Scholar]
- 12.Hiran TS, Moulton PJ, Hancock JT. Detection of superoxide and NADPH oxidase in porcine articular chondrocytes. Free Radic Biol Med. 1997;23:736–43. doi: 10.1016/s0891-5849(97)00054-3. [DOI] [PubMed] [Google Scholar]
- 13.Ramamurthy NS, Vernillo AT, Greenwald RA, Lee HM, Sorsa T, Golub LM, et al. Reactive oxygen species activate and tetracyclines inhibit rat osteoblast collagenase. J Bone Miner Res. 1993;8:1247–53. doi: 10.1002/jbmr.5650081013. [DOI] [PubMed] [Google Scholar]
- 14.Tiku ML, Liesch JB, Robertson FM. Production of hydrogen peroxide by rabbit articular chondrocytes: enhancement by cytokines. J Immunol. 1990;145:690–6. [PubMed] [Google Scholar]
- 15.Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med. 2003;35:236–56. doi: 10.1016/s0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- 16.Miyagi I, Kikuchi H, Hamanishi C, Tanaka S. Auto-destruction of the articular cartilage and free radical mediators. J Lab Clin Med. 1998;131:146–50. doi: 10.1016/s0022-2143(98)90156-1. [DOI] [PubMed] [Google Scholar]
- 17.Gassner R, Buckley MJ, Georgescu H, Studer R, Stefanovic-Racic M, Piesco NP, et al. Cyclic tensile stress exerts antiinflammatory actions on chondrocytes by inhibiting inducible nitric oxide synthase. J Immunol. 1999;163:2187–92. [PMC free article] [PubMed] [Google Scholar]
- 18.Lo YY, Cruz TF. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J Biol Chem. 1995;270:11727–30. doi: 10.1074/jbc.270.20.11727. [DOI] [PubMed] [Google Scholar]
- 19.Kurz B, Lemke A, Kehn M, Domm C, Patwari P, Frank EH, et al. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis Rheum. 2004;50:123–30. doi: 10.1002/art.11438. [DOI] [PubMed] [Google Scholar]
- 20.Fermor B, Weinberg JB, Pisetsky DS, Misukonis MA, Banes AJ, Guilak F. The effects of static and intermittent compression on nitric oxide production in articular cartilage explants. J Orthop Res. 2001;19:729–37. doi: 10.1016/S0736-0266(00)00049-8. [DOI] [PubMed] [Google Scholar]
- 21.Lee MS, Trindade MC, Ikenoue T, Schurman DJ, Goodman SB, Smith RL. Intermittent hydrostatic pressure inhibits shear stressinduced nitric oxide release in human osteoarthritic chondrocytes in vitro. J Rheumatol. 2003;30:326–8. [PubMed] [Google Scholar]
- 22.Ikenoue T, Trindade MC, Lee MS, Lin EY, Schurman DJ, Goodman SB, et al. Mechanoregulation of human articular chondrocyte aggrecan and type II collagen expression by intermittent hydrostatic pressure in vitro. J Orthop Res. 2003;21:110–6. doi: 10.1016/S0736-0266(02)00091-8. [DOI] [PubMed] [Google Scholar]
- 23.Lee DA, Frean SP, Lees P, Bader DL. Dynamic mechanical compression influences nitric oxide production by articular chondrocytes seeded in agarose. Biochem Biophys Res Commun. 1998;251:580–5. doi: 10.1006/bbrc.1998.9520. [DOI] [PubMed] [Google Scholar]
- 24.Millward-Sadler SJ, Wright MO, Lee H, Nishida K, Caldwell H, Nuki G, et al. Integrin-regulated secretion of interleukin 4: a novel pathway of mechanotransduction in human articular chondrocytes. J Cell Biol. 1999;145:183–9. doi: 10.1083/jcb.145.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Z, Buckley MJ, Evans CH, Agarwal S. Cyclic tensile strain acts as an antagonist of IL-1β actions in chondrocytes. J Immunol. 2000;165:453–60. doi: 10.4049/jimmunol.165.1.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen SV, Oury TD, Ostergaard L, Valnickova Z, Wegrzyn J, Thogersen IB, et al. Extracellular superoxide dismutase (EC-SOD) binds to type I collagen and protects against oxidative fragmentation. J Biol Chem. 2004;279:13705–10. doi: 10.1074/jbc.M310217200. [DOI] [PubMed] [Google Scholar]
- 27.Oury TD, Chang LY, Marklund SL, Day BJ, Crapo JD. Immunocytochemical localization of extracellular superoxide dismutase in human lung. Lab Invest. 1994;70:889–98. [PubMed] [Google Scholar]
- 28.Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167:1600–19. doi: 10.1164/rccm.200212-1479SO. [DOI] [PubMed] [Google Scholar]
- 29.Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, et al. Genetically reduced antioxidative protection and increased ischemic heart disease risk: the Copenhagen City Heart Study. Circulation. 2004;109:59–65. doi: 10.1161/01.CIR.0000105720.28086.6C. [DOI] [PubMed] [Google Scholar]
- 30.Mason RM, Chambers MG, Flannelly J, Gaffen JD, Dudhia J, Bayliss MT. The STR/Ort mouse and its use as a model of osteoarthritis. Osteoarthritis Cartilage. 2001;9:85–91. doi: 10.1053/joca.2000.0363. [DOI] [PubMed] [Google Scholar]
- 31.Chambers MG, Bayliss MT, Mason RM. Chondrocyte cytokine and growth factor expression in murine osteoarthritis. Osteoarthritis Cartilage. 1997;5:301–8. doi: 10.1016/s1063-4584(97)80034-9. [DOI] [PubMed] [Google Scholar]
- 32.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 33.Reimann I, Mankin HJ, Trahan C. Quantitative histologic analyses of articular cartilage and subchondral bone from osteoarthritic and normal human hips. Acta Orthop Scand. 1977;48:63–73. doi: 10.3109/17453677708985113. [DOI] [PubMed] [Google Scholar]
- 34.Towbin H, Staehelin T, Gordon J. Immunoblotting in the clinical laboratory. J Clin Chem Clin Biochem. 1989;27:495–501. [PubMed] [Google Scholar]
- 35.Prockop J, Udenfriend S. A specific method for the analysis of hydroxyproline in tissues and urine. Anal Biochem. 1960;1:228–39. doi: 10.1016/0003-2697(60)90050-6. [DOI] [PubMed] [Google Scholar]
- 36.Fukai T, Folz RJ, Landmesser U, Harrison DG. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res. 2002;55:239–49. doi: 10.1016/s0008-6363(02)00328-0. [DOI] [PubMed] [Google Scholar]
- 37.Bowler RP, Nicks M, Warnick K, Crapo JD. Role of extracellular superoxide dismutase in bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2002;282:L719–26. doi: 10.1152/ajplung.00058.2001. [DOI] [PubMed] [Google Scholar]
- 38.Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad SciUSA. 1995;92:6264–8. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang XL, Adachi T, Sim AS, Wilcken DE. Plasma extracellular superoxide dismutase levels in an Australian population with coronary artery disease. Arterioscler Thromb Vasc Biol. 1998;18:1915–21. doi: 10.1161/01.atv.18.12.1915. [DOI] [PubMed] [Google Scholar]
- 40.Silver IA. Measurement of pH and ionic composition of pericellular sites. Philos Trans R Soc Lond B Biol Sci. 1975;271:261–72. doi: 10.1098/rstb.1975.0050. [DOI] [PubMed] [Google Scholar]
- 41.Sjostrom K, Crapo JD. Structural and biochemical adaptive changes in rat lungs after exposure to hypoxia. Lab Invest. 1983;48:68–79. [PubMed] [Google Scholar]
- 42.Fridovich I. Hypoxia and oxygen toxicity. Adv Neurol. 1979;26:255–9. [PubMed] [Google Scholar]
- 43.Kempson GE, Muir H, Pollard C, Tuke M. The tensile properties of the cartilage of human femoral condyles related to the content of collagen and glycosaminoglycans. Biochim Biophys Acta. 1973;297:456–72. doi: 10.1016/0304-4165(73)90093-7. [DOI] [PubMed] [Google Scholar]
- 44.Hollander AP, Pidoux I, Reiner A, Rorabeck C, Bourne R, Poole AR. Damage to type II collagen in aging and osteoarthritis starts at the articular surface, originates around chondrocytes, and extends into the cartilage with progressive degeneration. J Clin Invest. 1995;96:2859–69. doi: 10.1172/JCI118357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saran M, Bors W. Signalling by O2- and NO: how far can either radical, or any specific reaction product, transmit a message under in vivo conditions? Chem Biol Interact. 1994;90:35–45. doi: 10.1016/0009-2797(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 46.Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. 2003. Circ Res. 93:622–9. doi: 10.1161/01.RES.0000092140.81594.A8. [DOI] [PubMed] [Google Scholar]
- 47.Del Carlo M, Jr, Loeser RF. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002;46:394–403. doi: 10.1002/art.10056. [DOI] [PubMed] [Google Scholar]
- 48.Colasanti M, Suzuki H. The dual personality of NO. Trends Pharmacol Sci. 2000;21:249–52. doi: 10.1016/s0165-6147(00)01499-1. [DOI] [PubMed] [Google Scholar]
- 49.Jouzeau JY, Pacquelet S, Boileau C, Nedelec E, Presle N, Netter P, et al. Nitric oxide (NO) and cartilage metabolism: NO effects are modulated by superoxide in response to IL-1. Biorheology. 2002;39:201–14. [PubMed] [Google Scholar]
- 50.Marklund SL. Regulation by cytokines of extracellular superoxide dismutase and other superoxide dismutase isoenzymes in fibro-blasts. J Biol Chem. 1992;267:6696–701. [PubMed] [Google Scholar]
- 51.Brady TC, Chang LY, Day BJ, Crapo JD. Extracellular superoxide dismutase is upregulated with inducible nitric oxide synthase after NF-κB activation. Am J Physiol. 1997;273:L1002–6. doi: 10.1152/ajplung.1997.273.5.L1002. [DOI] [PubMed] [Google Scholar]
- 52.Lippiello L, Kaye C, Neumata T, Mankin HJ. In vitro metabolic response of articular cartilage segments to low levels of hydrostatic pressure. Connect Tissue Res. 1985;13:99–107. doi: 10.3109/03008208509152388. [DOI] [PubMed] [Google Scholar]
- 53.Lee MS, Trindade MC, Ikenoue T, Schurman DJ, Goodman SB, Smith RL. Effects of shear stress on nitric oxide and matrix protein gene expression in human osteoarthritic chondrocytes in vitro. J Orthop Res. 2002;20:556–61. doi: 10.1016/S0736-0266(01)00149-8. [DOI] [PubMed] [Google Scholar]
- 54.Park SK, Lin HL, Murphy S. Nitric oxide regulates nitric oxide synthase-2 gene expression by inhibiting NF-κB binding to DNA. Biochem J. 1997;322:609–13. doi: 10.1042/bj3220609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salvemini D, Mazzon E, Dugo L, Serraino I, de Sarro A, Caputi AP, et al. Amelioration of joint disease in a rat model of collagen-induced arthritis by M40403, a superoxide dismutase mimetic. Arthritis Rheum. 2001;44:2909–21. doi: 10.1002/1529-0131(200112)44:12<2909::aid-art479>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 56.Loeser RF, Carlson CS, del Carlo M, Cole A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: correlation of oxidative damage with the presence of interleukin-1β and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002;46:2349–57. doi: 10.1002/art.10496. [DOI] [PubMed] [Google Scholar]