

Abstract
Heparanase activity is strongly implicated in structural remodeling of the extracellular matrix underlying tumor and endothelial cells that leads to cellular invasion. In addition, heparanase augments signaling cascades leading to enhanced phosphorylation of selected protein kinases and increased gene transcription associated with aggressive tumor progression. This function is apparently independent of heparan sulfate and enzyme activity and is mediated by a novel protein domain localized at the heparanase C-terminus (C-domain). Moreover, the functional repertoire of heparanase is expanded by its regulation of syndecan clustering, shedding, and mitogen binding. Recently, modified glycol-split heparin that inhibits heparanase activity was demonstrated to profoundly inhibit the progression of tumor xenografts produced by myeloma and carcinoma cells thus moving anti-heparanase therapy closer to reality.
Keywords: Heparanase, heparan sulfate, syndecan, structure-function, signaling, myeloma, glycol-split, heparin
Introduction
The activity responsible for heparan sulfate (HS) cleavage, commonly known as heparanase, is abundant in blood-borne and tumor-derived cells [1, 2]. Heparanase cleaves HS side chains presumably at sites of low sulfation, thus facilitating structural alterations of the extracellular matrix (ECM) and basement membrane underlying epithelial and endothelial cells. Importantly, heparanase activity correlates with the metastatic potential of cancer cells, a notion that is now well supported experimentally and clinically [3-7]. In addition to secreted proteoglycans assembled in the ECM, heparan sulfate proteoglycans (HSPGs) are highly abundant on the cell surface as membrane-spanning (i.e., syndecans) or glycosylphosphatidylinositol (GPI)-anchored proteoglycans (i.e., glypicans), which are also potentially subjected to cleavage by heparanase, releasing HS-bound growth factors. Recent evidence indicates, however, that heparanase function at the cell surface is not limited to HS cleavage or the release of sequestered growth factors. In fact, heparanase was noted to affect syndecan ecology in a manner that involves clustering, shedding, and mitogen binding. In addition, heparanase augmented signaling cascades leading to enhanced phosphorylation of selected protein kinases and promoted gene transcription associated with aggressive tumor progression [3]. This HS-independent function is appears to be mediated by an as yet unidentified cell surface heparanase binding protein/receptor.
In this review we discuss recent progress in the heparanase field, focusing on HS-dependent and -independent functions of heparanase at the cell surface and its clinical relevance. Aspects such as heparanase gene regulation, proteolytic processing, cellular localization, and the development of heparanase inhibitors have been the subject of several recent review articles [4, 8-10] and will not be discussed in detail here.
Internalization, clustering and activation of syndecans by heparanase
Syndecans are a family of four transmembrane proteins having covalently attached HS chains. Some syndecans contain both heparan sulfate and chondroitin sulfate. The HS chains mediate syndecan interactions with a large number of proteins, including plasma proteins (i.e., antithrombin), ECM proteins (i.e., fibronectin), and a multitude of biological mediators. By doing so, syndecans function as co-receptors, facilitating growth factors, morphogens, and integrin activity [11-13]. In addition to its co-receptor properties, clustering of syndecan-4 has been shown to directly initiate signaling cascades that result in PKCα and Rac1 activation which appears instrumental for cell adhesion and directional migration [14-17]. Syndecan clustering was mainly investigated, however, in the context of its interaction with matrix proteins such as fibronectin, while clustering by soluble proteins has not been demonstrated.
Formation and internalization of heparanase-syndecan complexes
Heparanase interacts with syndecans by virtue of their HS content and the typical high affinity that exists between an enzyme and its substrate. This high affinity interaction directs rapid and efficient uptake of heparanase [3, 18]. Heparanase uptake is considered a prerequisite for the delivery of latent heparanase to lysosomes and its subsequent proteolytic processing and activation by cathepsin L [18-23]. Efficient uptake of heparanase was evident also by GPI-deficient cells (i.e., lack cell surface glypicans), suggesting preferential involvement of syndecans in this process [18]. Notably, syndecan-1 and 4 are internalized by cells following addition of exogenous heparanase, and colocalize with heparanase in endocytic vesicles [18]. As a result, syndecans apparently disappear from the cell surface [18] (Fig. 1a, Hepa), altering the ability of cells to communicate with each other and with their immediate extracellular micro-environment. Decreased levels of cell surface syndecans, though likely temporary, follows syndecan clustering and emerges as the first layer of the cross-talk between heparanase and syndecans.
Figure 1. Heparanase stimulates syndecan-1 clustering, Rac1 activation, and cell spreading.
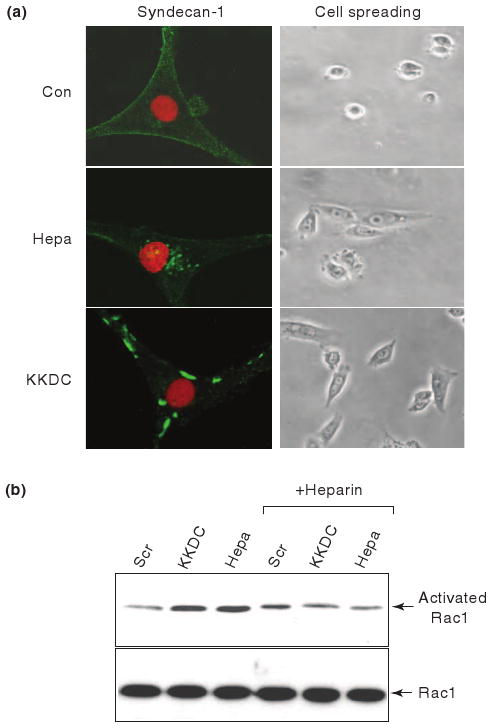
a. Syndecan-1 clustering and cell spreading. U87 glioma cells were serum starved for 24 h and were then incubated with control scrambled peptide (Con; upper panel, left), KKDC peptide (50 μM, lower panel, left) or heparanase (Hepa; 1 μg/ml; second panel, left) for 1 h. Cells were then gently washed, fixed with cold methanol and subjected to fluorescent staining with anti-syndecan-1 antibodies (green) merged with nuclear staining (red; left panels). Cell morphology was evaluated by light microscopy (right panels). b. Rac1 activation. U87 cells were kept in serum-free medium for 24 h and were then stimulated with the KKDC/Scr peptides (50 μM) or heparanase (Hepa; 1 μg/ml) for 30 min without or with heparin (50 μg/ml; + Heparin). Cell lysates were then subjected to pull-down with GST-PAK-agarose beads for detection of active Rac1 (upper panel) and subjected to immunoblotting with anti-Rac1 antibody (lower panel).
Heparanase-mediated syndecan clustering augments Rac activation and cell adhesion
In-depth appraisal of heparanase-syndecan interaction and its significance was accomplished when the heparin-binding domains of heparanase were identified. Based on consensus sequences considered to mediate the interaction between polypeptides and heparin, three potential heparin-binding domains have been recognized [21]. Particular attention was given to the Lys158-Asp171 domain since a peptide corresponding to this sequence [KKFKNSTYSRSSVD(C); termed KKDC] interacted physically with heparin and HS with high affinity and inhibited heparanase enzymatic activity [21]. Notably, the KKDC peptide associates with the plasma membrane and induces clustering of syndecans [24] (Fig. 1, KKDC). The clustering of syndecan-1 induced by heparanase or the KKDC peptide was associated with improved spreading of primary and tumor-derived cells (Fig. 1). In fact, cells treated with the KKDC peptide spread better and formed more focal contacts compared with cells treated with heparanase, reflecting the generation of syndecan clusters [24]. Remarkably, syndecan clusters formed by the KKDC peptide were exceptionally large and failed to be internalized at the time points examined [24] (Fig. 1a, KKDC), while the heparanase-syndecan complexes are rapidly and efficiently internalized [18, 24] (Fig. 1a, Hepa).
Mechanistically, syndecan clustering by heparanase or the KKDC peptide enhanced cell spreading and was associated with PKC, Src, and Rac1 activation [24] (Fig. 1b), molecular determinants shown to be induced by syndecans [14, 16, 17, 25]. Thus, while heparanase has been reported to facilitate the adhesion, spreading and migration of several cell types in a manner that appeared independent of its enzymatic activity [26-29] and to induce Rac1 activation [29], studies applying the KKDC peptide combined these observations in a linear mode in which syndecan clustering by the heparin binding domains of heparanase initiates signaling cascades, resulting in enhanced cell spreading. This mode of action likely represents a non-enzymatic function of heparanase in its simplest term (Fig. 3a).
Figure 3. Enzymatic activity-independent function of heparanase at the cell surface.

Clustering of syndecan family members, and possibly glypicans, by the KKDC peptide dimer or the two heparin binding domains of heparanase facilitates cell-adhesion and cell-spreading (a). Enhanced cell adhesion and spreading is mediated by the recruitment and activation of PKCα, Rac1, and Src (a). In addition, heparanase is thought to interact with a heparanase-binding cell surface protein/receptor, leading to HS-independent Akt, p38, and Src activation. This results in enhanced transcription of genes such as vascular endothelial growth factor (VEGF-A, VEGF-C) [47, 61], tissue factor (TF) [62], and Cox2 [63], and further contributes to cell adhesion, spreading and motility. Src activation, in turn, phosphorylates a number of substrates including the EGFR, leading to increased cell proliferation and tumorigenesis [49] (b).
The heparanase molecule appears, nevertheless, versatile and can function in a manner that operates beyond merely interaction with HS. For example, heparanase appeared more efficient in stimulating the adhesion of floating ARH-77 leukemia1and U266 myeloma cells compared with the KKDC peptide [24]. This result indicates that syndecan activation by the KKDC peptide may not be sufficient for efficient cell adhesion, and that cooperation with additional adhesion molecules such as integrins is required [13]. In fact, β1-integrin activation was noted upon heparanase over expression or exogenous addition [28, 29] and does not seem to involve enzymatic aspects since mutated, inactive heparanase was as potent as wild type heparanase in facilitating cell adhesion [24]. Although the molecular mechanism underlying integrin activation by heparanase is yet to be defined, enhanced adhesion of cancer cells is thought to confer cell adhesion mediated drug resistance (CAM-DR). This aspect, mediated in part by β1-integrin, appears particularly relevant to multiple myeloma [30], where an emerging role of heparanase has been revealed [31, 32].
Heparanase and syndecan-1 in multiple myeloma: beyond shedding
In myeloma patients, heparanase enzyme activity was elevated in the bone marrow plasma of 86% of the patients examined [31]. A subsequent study confirmed this observation by gene array analysis showing that 92% of myeloma patients exhibited elevated heparanase expression [32]. Heparanase up-regulation in myeloma patients was associated with enhanced microvessel density and syndecan-1 expression [31]. While heparanase is pro-angiogenic in myeloma, which is a common feature shared with solid tumors [3], syndecan-1 regulation has emerged as highly relevant to multiple myeloma progression.
Syndecan-1 is particularly abundant in myeloma, and is the dominant and often the only HSPG present on the surface of myeloma cells [6]. Cell surface syndecan-1 promotes adhesion of myeloma cells and inhibits cell invasion in vitro [33]. In contrast, high levels of shed syndecan-1 can be found in the serum of some myeloma patients and are associated with poor prognosis [34]. Shed syndecan-1 becomes trapped within the bone marrow ECM where it likely acts to enhance the growth and metastasis of myeloma cells within the bone [33, 35]. This is supported by the finding that enhanced expression of soluble syndecan-1 by myeloma cells promotes tumor growth and metastasis in an animal model [33, 35]. Notably, heparanase up regulates both the expression and shedding of syndecan-1 from the surface of myeloma cells [32, 36]. In agreement with this notion, heparanase gene silencing was associated with decreased levels of shed syndecan-1 [32]. Importantly, both syndecan-1 up-regulation and shedding requires heparanase enzymatic activity, because over expression of mutated inactive heparanase failed to stimulate syndecan-1 expression or shedding [36]. Syndecan-1 shedding was similarly augmented by the addition of recombinant active heparanase to CAG myeloma cells, and even more dramatic shedding was observed following the addition of bacterial heparinase III (heparitinase) [36]. These findings indicate that cleavage of HS by heparanase or heparinase III may render syndecan-1 more susceptible to proteases mediating the shedding of syndecan-1. However, it appears that heparanase may play an even more direct role in regulating shedding of syndecan-1. It was recently demonstrated that enhanced expression of heparanase leads to increased MMP-9 levels (a syndecan-1 sheddase), while heparanase gene silencing resulted in reduced MMP-9 activity [37]. Up-regulation of MMP-9 expression has significant biological relevance because inhibition of MMP-9 reduces syndecan-1 shedding [37]. Moreover, not only MMP-9 but also urokinase-type plasminogen activator (uPA) and its receptor (uPAR), molecular determinants responsible for MMP-9 activation, are up-regulated by heparanase. These findings provide the first evidence for cooperation between heparanase and MMPs in regulating HSPGs on the cell surface and likely in the ECM. These proteases have multiple functions within the tumor microenvironment beyond simply enhancing syndecan-1 shedding and thus this finding has potential for far reaching clinical relevance. This and other results [38] suggest that heparanase acts as a master regulator of protease expression within the tumor microenvironment.
Similar to the induction of syndecan-1 shedding, MMP-9 induction required heparanase enzyme activity and is mediated by the ERK signaling pathway [37] (Fig. 2a). ERK phosphorylation has previously been reported to be induced following the addition of latent heparanase to primary T-cells [28], suggesting that this pathway prevails in hematopoietic lineage cells, while Akt and Src activation predominate in firmly attached cells (see below).
Figure 2. Remodeling of syndecan-1 by heparanase enzymatic activity modulates cell behavior and alters the tumor microenvironment.

a. Heparanase-mediated syndecan-1 shedding. We suggest that active heparanase, secreted by tumor cells, cleaves HS side-chains of syndecan-1 and releases sequestered growth factors while still associated with small HS fragments. The HS-growth factor (i.e., HGF) complex can now interact with its high affinity receptor (i.e., c-Met) in an autocrine or paracrine manner, augmenting ERK phosphorylation. The resulting signaling induces MMP-9 expression and secretion. Similarly, the expression of uPA and uPAR is increased, resulting in MMP-9 activation. Active MMP-9 cleaves the syndecan-1 core protein, and syndecan-1 is shed from the cell surface, concentrates in the tumor microenvironment and facilitates myeloma progression. b. Heparanase mediated twist in syndecan-1 mitogenic function. Lacritin is a small epithelial glycoprotein which, through PKCα dephosphorylation and phospholipase D activation, regulates cell differentiation and proliferation. A role for heparanase in lacritin signaling was concluded by showing that heparanase gene silencing is associated with reduced lacritin-dependent cell proliferation, while exogenous addition of heparanase rescued cell proliferation. The emerging theme suggests a scenario by which heparanase activity at the cell surface cleaves the HS chains of syndecan-1, making the core protein accessible for lacritin binding and lacritin-induced cell proliferation [60].
The heparanase-syndecan axis is a target for therapy
Results from studies using several in vivo model systems support the notion that enzymatic activities responsible for syndecan-1 modification are valid targets for myeloma therapy. For example, enhanced expression of either of the extracellular endosulfatases HSulf-1 or HSulf-2, enzymes that remove 6-0 sulfate groups from HS, attenuated myeloma tumor growth [39]. Even a more dramatic inhibition of tumor growth was evident by the administration of heparinase III (heparitinase) to SCID mice inoculated with either CAG myeloma cells or cells isolated from the bone marrow of myeloma patients [40]. This effect is thought to be mediated by the release of heparinase III-generated HS fragments having anti-tumor activity [40].
Although heparinase III and human heparanase both degrade HS chains, their cleavage products are chemically distinct. While heparinase III extensively degrades HS, heparanase cleaves more selectively and generates fragments of HS that are 5-7 kDa in size, yielding strictly distinct outcomes in the context of tumor progression. While administration of heparinase III is associated with reduced tumor growth, heparanase activity is elevated in many hematological and solid tumors, and correlates with poor prognosis and shorter post-operative survival rate [3, 7, 9]. Accordingly, inhibition of heparanase activity is expected to suppress tumor progression. To examine this in myeloma, a chemically modified heparin, which is 100% N-acetylated and 25% glycol-split was tested. This flexible molecule is a potent inhibitor of heparanase enzymatic activity, lacks anti-coagulant activity typical of heparin and does not displace ECM-bound bFGF or potentiate its mitogenic activity [41, 42]. The modified heparin profoundly inhibits the progression of tumor xenografts produced by CAG myeloma cells inoculated subcutaneously, in a dose responsive manner [40]. Importantly, mice treated with this compound for 28 days exhibited no signs of adverse side effects, lending optimism that this and related compounds will prove safe in human clinical trials. These studies support the notion that heparanase activity not only facilitates tumor metastasis, but also the progression of primary tumors and that heparanase inhibitors are capable of inhibiting tumor growth with no or only minimal side effects.
HS-independent signaling by heparanase
The heparanase-syndecan axis described above governs enzymatic activity-dependent (i.e., shedding) and -independent (i.e., clustering, internalization) modifications of syndecans by heparanase, thus substantiating and expanding the significance of this pathway. In addition, evidence accumulated in recent years indicates that heparanase also elicits HS-independent signaling. Signaling is considered to be HS-independent if it occurs in HS-deficient cells (i.e., CHO 745) or in the presence of heparin, as has been demonstrated for enhanced Akt phosphorylation by heparanase [26]. In fact, laminaran sulfate or heparin, two both potent inhibitors of heparanase enzymatic activity, when added together with heparanase augmented Akt phosphorylation [26], thus critically implying that heparanase enzymatic activity is not required for Akt activation. In endothelial cells, heparanase added exogenously [26] facilitated cell migration, invasion, and the formation of tube-like structures in a PI 3-kinase-dependent manner [26]. In several cases, where tumor xenograft development was examined, heparanase over-expression resulted in tumors bigger in volume and weight [29, 43, 44] coupled with Akt induction [29, 44]. Importantly, heparanase gene silencing was associated with reduced Akt phosphorylation levels [45], further substantiating a role for endogenous heparanase in Akt modulation.
Signaling via the heparanase-Src axis
Signaling by inactive heparanase is not restricted to Akt activation. Activation of ERK was noted following addition of latent heparanase to primary T-cells [28] and p38 phosphorylation was noted to be augmented in cells over expressing heparanase or following its exogenous addition [46, 47]. Heparanase over expression was also associated with VEGF up-regulation, coinciding with induction of p38 phosphorylation [47]. Similar to Akt, heparanase gene silencing was associated with reduced p38 phosphorylation and VEGF expression levels [47], lending further support for the intimate involvement of endogenous heparanase in modulating signaling pathways and gene transcription [38]. Interestingly, however, inhibitors of p38 had no effect on VEGF expression, suggesting modulation by signaling pathways other than p38. Utilizing a screen for small molecule inhibitors of signal transduction, Zetser et al. have identified Src as the underlying signaling mechanism responsible for VEGF gene induction by heparanase [47]. Notably, increased Src phosphorylation was associated with equivalent elevation of Src kinase activity. This is best exemplified by enhanced p120 catenin phosphorylation levels, a well known Src substrate [48], in heparanase transfected cells [47]. Although the consequences of p120 catenin phosphorylation are unclear, this observation led Cohen-Kaplan et al. to suspect that additional Src substrates are induced by heparanase [49].
Given the large repertoire of Src activities and substrates, focus was directed to those most closely associated with tumor progression, such as the EGF receptor (EGFR). EGFR phosphorylation was markedly increased in cells over expressing heparanase or following its exogenous addition, while heparanase gene silencing was accompanied by reduced EGFR and Src phosphorylation levels [49]. Notably, enhanced EGFR phosphorylation by heparanase was restricted to selected tyrosine residues (i.e., 845, 1173) thought to be direct targets of Src rather than a result of receptor autophosphorylation [50]. Indeed, enhanced EGFR phosphorylation on tyrosine residues 845 and 1173 by heparanase was abrogated in cells treated with Src inhibitors or anti-Src siRNA [49]. The functional significance of EGFR modulation by heparanase emerged by monitoring cell proliferation. Thus, heparanase gene silencing was accompanied by a decrease in cell proliferation, while heparanase over expression resulted in enhanced cell proliferation and formation of larger colonies in soft agar, in a Src- and EGFR-dependent manner [49].
The clinical relevance of the heparanase-Src-EGFR pathway has been elucidated for head and neck carcinoma. Notably, heparanase expression in head and neck carcinomas correlated with phospho-EGFR immunostaining [49]. Even more significant was the correlation between heparanase cellular localization and phospho-EGFR levels. Thus, while cytoplasmic heparanase was associated with elevated EGFR phosphorylation, nuclear localization of heparanase was associated with reduced phospho-EGFR levels [49], in agreement with a favorable outcome of patients exhibiting nuclear heparanase [51]. These studies provide a more realistic view of heparanase function in the course of tumor progression. Hence, while heparanase enzymatic activity has traditionally been implicated in tumor metastasis, the current view points to a multifaceted protein engaged in multiple aspects of tumor progression. While enzymatic activity-dependent and -independent signaling through HS appears straightforward in its rational, HS-independent signaling by heparanase requires a mediator, possibly in the form of cell surface receptor(s) (Fig. 3b).
Is there a cell surface receptor for heparanase?
Apart of HSPGs, several cell surface proteins have been shown to bind heparanase and mediate its uptake. These include mannose 6-phosphate receptor (MPR), cation-independent mannose 6-phosphate receptor (CD222), and low density lipoprotein receptor-related protein (LRP) [22, 52] which potentially can mediate heparanase signaling. Akt induction by heparanase was noted, however, in MPR-, and LRP-deficient cells [45]. Similarly, Akt phosphorylation elicited by heparanase was not affected by recombinant receptor-associated protein (RAP) [45], an antagonist of the LRP receptor family [53], altogether suggesting that LRP, and/or MPR do not account for Akt induction by heparanase. Instead, enhanced Akt phosphorylation by heparanase was markedly inhibited in cells treated with lovastatin or methyl-β-cyclodextrin, drugs which disrupt the integrity of lipid rafts [45]. Indeed, low, yet detectable amounts of heparanase were noted to be localized to lipid rafts, resulting in Akt activation at the raft microdomain [45]. This would imply that while the bulk of heparanase is associated with non-raft HSPGs, a small fraction is associated with raft-resident molecule(s) which mediate heparanase signaling to Akt.
The existence of cell surface heparanase receptor is supported by binding experiments. Applying iodinated heparanase to HeLa cells revealed the presence of two distinct types of binding sites exhibiting low-affinity (Kd=3 mM), high abundant (βmax = 1×108), and high affinity (Kd = 2 nM), low abundant (βmax = 1.7×104) characteristics [54]. Binding studies performed with wild type CHO-KI cells and their HS-deficient CHO-745 counterpart cells have demonstrated that heparanase binding to the high affinity binding sites is almost identical in both cell types. In contrast, the number of low affinity binding sites was significantly reduced in CHO-745 vs. CHO-KI cells, and a similar decrease was noted in CHO-KI cells treated with heparinase III [54]. These studies reinforce the notion that while HSPGs serve as low affinity, high abundant binding sites, heparanase also associates with high affinity, low abundant cell surface receptor(s). A first indication for the protein nature of this receptor and its molecular weight emerged from cross-linking experiments, revealing two distinct complexes representing 130 and 170 kDa proteins associated with heparanase [55] (Fig. 4e).
Figure 4. Three dimensional model and function of the heparanase C-domain.
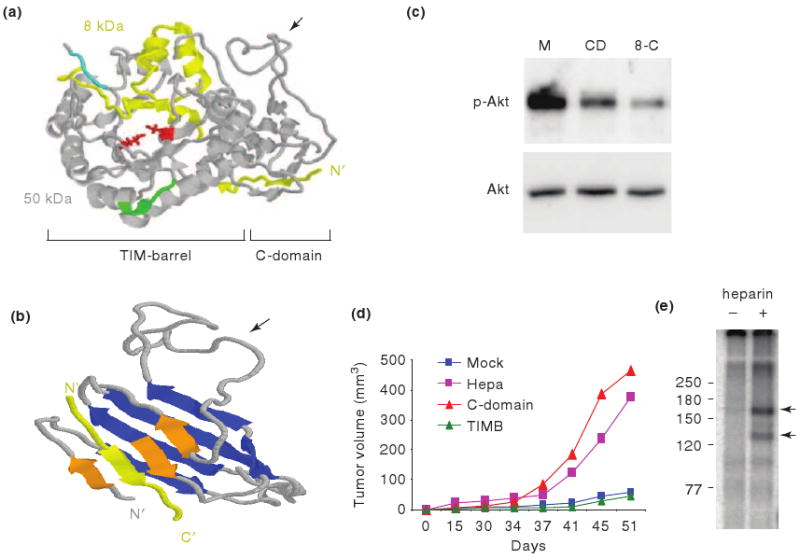
a. The model, including the 8 kDa (yellow) and 50 kDa (gray) protein subunits, amino acids critical for heparanase catalysis (Glu225 and Glu343, red), and heparin binding regions (Lys158-Asp171 and Gln270-Lys280; cyan and green, respectively) is shown. b. A more detailed structure of the C-domain is shown. The model illustrates eight β-strands, one of which is contributed by the 8 kDa subunit (yellow), arranged in two sheets (blue and orange) which are connected by an unstructured, flexible loop (arrow). c. Akt induction. FaDu pharynx carcinoma cells were stably transfected with control (Mock), C-domain (CD), or 8-C gene constructs and cell lysates were subjected to immunoblotting applying phospho-Akt (p-Akt) and Akt antibodies. d. Shown are tumor volumes of control (Mock), heparanase (Hepa), C-domain, and TIM-barrel (TIMB) transfected U87 cells inoculated subcutaneously at the flank of Balb/C nude mice. e. 125I-labeled 8-C protein was added to HeLa cells in the absence (-) or presence (+) of heparin (10 μg/ml). Crosslinking of the 8-C protein to its putative high affinity receptors resulted in the formation of two major protein complexes exhibiting molecular weights of ∼130 and 170 kDa (arrows). Molecular weight markers (kDa) are shown in the left.
Structure-function aspects of heparanase signaling: emerging role for the protein C-terminus
In an attempt to shed more light on heparanase structure, Fux et al. utilized a prediction server (www.robetta.org) to better delineate the three dimensional structure of active heparanase [55]. A similar approach was first adopted by Hulett et al. and led the authors to suggest that heparanase structure resembles glycosyl hydrolases [56]. On this ground, glutamic acids 225 and 343 were recognized critical for heparanase catalysis, acting as proton donor and nucleophil, mechanism typical of glycosyl hydrolases [56]. Furthermore, the alternating β-strands and α-helices, among other considerations, led to the prediction of heparanase bearing a TIM-barrel structure, common to glycosyl hydrolase families within clan GH-A [56]. A similar structure was subsequently concluded by others [20, 57]. While Hulett et al. introduced the full length sequence of heparanase as a template [56], Abboud-Jarrous introduced the linker segment and the 50 kDa subunit sequences [20], both representing inactive forms of the enzyme. In contrast, Fux et al. predicted the structure of enzymatically active, single chain, heparanase enzyme, in which the linker segment was replaced by three glycine-serine repeats (GS3), resulting in a constitutively active enzyme [57]. The structure obtained clearly illustrates a TIM-barrel fold (Fig. 4a), in agreement with previous predictions [20, 56]. In addition, conserved glutamic acid residues critical for heparanase catalysis (Glu225 and Glu343), as well as heparin/HS binding regions (Lys158-Asp171 and Gln270-Lys280; cyan and green, respectively; Fig. 4a) [21] were situated in close proximity to the micro-pocket active site, corroborating the relevance of the model. Notably, the structure also delineates a C-terminus fold positioned next to the TIM-barrel structure (Fig. 4a, b) [55]. The C-terminus domain (C-domain) appears to be comprised of 8 β strands arranged in two sheets, as well as a flexible, unstructured loop that lies in-between (Fig. 4b, arrow). The two sheets appear packed against each other, stabilized by hydrophobic interactions between the upper and lower β sheets. Notably, one of the C-domain β strands seems to be contributed by the 8 kDa protein subunit, suggesting that the protein N-terminus plays a structural role in the establishment of the TIM-barrel [56] and C-domain folds.
Fux et al. [55] thus, hypothesized that the seemingly distinct protein domains observed in the three dimensional model, namely the TIM-barrel and C-domain regions, mediate enzymatic and non-enzymatic functions of heparanase, respectively. Interestingly, cells transfected with the TIM-barrel construct (amino acids 36-417) failed to display heparanase enzymatic activity, suggesting that the C-domain is absolutely required for the establishment of an active heparanase enzyme, possibly by stabilizing the TIM-barrel fold [55]. Deletion and site directed mutagenesis approaches further indicated that the C-domain plays a decisive role in heparanase enzymatic activity and secretion [55, 58, 59]. Notably, Akt phosphorylation was stimulated by cells over expressing the C-domain (amino acids 413-543), while the TIM-barrel protein variant yielded no Akt activation compared with control, mock transfected cells [55] (Fig. 4c). These findings clearly indicate that the non-enzymatic signaling function of heparanase leading to activation of Akt is mediated by the C-domain. Because the C-domain gene construct lacks the 8 kDa segment which, according to the predicted model, contributes one beta strand to the C-domain structure (Fig. 4b), the resulting protein may exhibit suboptimal Akt activation.
Indeed, Akt phosphorylation was markedly enhanced by cells transfected with a mini gene comprising a segment of the 8 kDa subunit, predicted by the model to contribute a beta strand (Gln36-Ser55) to the C-domain structure, linked to the C-domain sequence (8-C, Fig. 4c). These findings further support the predicted three dimensional model (Fig. 4a), indicating that the C-domain is indeed a valid functional domain responsible for Akt phosphorylation. The cellular consequences of C-domain over expressionare best revealed by monitoring tumor xenograft development. Notably, tumor xenografts produced by C-domain-transfected glioma cells appeared comparable to those produced by cells transfected with the full length heparanase, while progression of tumors produced by TIM-barrel-transfected cells appeared comparable with control mock transfected cells (Fig. 4d) [55]. These results show, that in some tumor systems (i.e., glioma) heparanase facilitates primary tumor progression regardless of its enzymatic activity, while in others (i.e., myeloma) heparanase enzymatic activity dominates [40].
Conclusions and perspective
Ten years following its cloning, the functional repertoire of heparanase is only starting to be revealed. From activity mainly implicated in cell invasion associated with tumor metastasis, heparanase has turned into a multifaceted protein that appears to participate in essentially all major aspects of tumor progression. Accumulating evidence support an important role of heparanase in the aggressive behavior of myeloma. This and the apparent efficacy of glycol-split heparin as a heparanase inhibitor urge the continued evaluation of such heparin derived compounds in clinical trials, given their lack of anticoagulant activity and apparent lack of other side effects. It is conceivable, however, that heparanase activity is critical for myeloma progression, while enzymatic activity-independent functions of heparanase may direct the progression of solid tumors such as glioma [55]. In this regard, identification and characterization of a cell surface receptor for heparanase remains a most exciting challenge in the field. Thus, new strategies are needed for the development of heparanase inhibitors directed against its non-enzymatic functions. These, combined with inhibitors of heparanase enzymatic activity, are expected to neutralize heparanase functions and untangle its significance in homeostasis and pathological disorders such as cancer, inflammation, and diabetic nephropathy.
Perspective
Heparanase/C-domain crystal structure
Attempts to inhibit heparanase enzymatic activity were initiated in the early days of heparanase research, in parallel with the emerging clinical relevance of this activity. Following heparanase cloning and the availability of recombinant enzyme and high-throughput screening methods, a variety of inhibitory molecules have been developed, including neutralizing antibodies, peptides, small molecules, modified non-anticoagulant species of heparin, as well as several other polyanionic molecules, such as laminaran sulfate, suramin and PI-88 [64, 65]. PI-88 has been shown to have activity on melanoma patients (phase I), and some efficacy in patients with advanced melanoma (phase II) [66, 67]. It seems, therefore, that better characterization of heparanase structure is required for the development of highly specific and efficacious inhibitors. Resolving the heparanase/C-domain crystal structure is expected to markedly advance heparanase-based therapy.
Heparanase receptor
Targeted therapy directed against heparanase enzymatic activity provides one aspect of inhibition strategy. Identification and characterization of cell surface molecule(s) that functions as a high affinity receptor for heparanase and mediates its signaling activity will significantly advance our basic understating of heparanase function in health and disease. Moreover, the receptor will serve as a target for therapy which, together with heparin-derived inhibitors will neutralize enzymatic and non-enzymatic functions of heparanase. Thus, identification and characterization of a cell surface receptor for heparanase is perhaps the most exciting challenge in the field.
Substrate binding specificity
The ability of the KKDC peptide to interact with heparin and to cluster syndecans is unique, since peptides derived from two additional heparin binding domains (Gln270-Lys280 and Lys411-Arg432) of heparanase exhibited only a very weak interaction with heparin and failed to inhibit heparanase enzymatic activity. Thus, the mere presence of heparin-binding consensus sequences appears insufficient to ensure biologically active peptide. What turns a heparin binding sequence to a functional domain is not entirely clear. Similarly, the observed preferred interaction of heparanase with syndecans over glypicans (i.e., efficient uptake of heparanase by glypican-deficient cells) is unclear. Given that HS side chains are expected to share high levels of resemblance between syndecans and glypicans, interaction preference is likely dictated by the core protein. If indeed the core protein turns to be a critical determinant for heparanase interaction, this would imply that only certain types of HSPGs are susceptible to cleavage by heparanase, contrasting the more simplistic view by which all HS species are equally good substrates for heparanase.
Heparanase function in HS turnover in the lysosome
Latent heparanase secretion is directed by an N-terminal signal peptide (Met1-Ala35). The secreted protein interacts rapidly and efficiently with cell surface HSPGs, low density lipoprotein receptor-related protein (LRP), and mannose 6-phosphate receptor (MPR) [18, 22, 54], followed by internalization and processing, collectively defined as heparanase uptake [3, 18, 54]. Following uptake, heparanase was noted to reside primarily within endocytic vesicles identified as late endosomes and lysosomes [68, 69]. This observation led to the identification of late endosomes/lysosomes as the heparanase processing organelle [23, 70], and cathepsin family members, mainly cathepsin L, as the heparanase activating protease [19]. In spite of its localization to a highly active protein degradation environment such as the lysosome, heparanase exhibits a half life of about 30 hours [18], relatively long compared with a t1/2 of 2-6 hours, and 25 minutes of transmembrane and GPI-anchored HSPGs, respectively. Residence and accumulation of heparanase in late endosomes and lysosomes may indicate that the enzyme functions in physiological turnover of cellular HSPGs. This possibility relates to earlier observations [71] where endoglycosidase degradation of HS chains was shown to occur both in endosomes and lysosomes. This, and related aspects such as the role of heparanase in regulating levels of nuclear syndecan-1 [72] await in-depth examination.
Acknowledgments
This work was supported by grants from the Israel Science Foundation (grant 549/06); National Cancer Institute, NIH (grant RO1-CA106456); the Israel Cancer Research Fund (ICRF); and the Rappaport Family Institute Fund to I. Vlodavsky and NIH CA058819 and CA135075 to R. Sanderson. I. Vlodavsky is a Research Professor of the ICRF. We thank Prof. B. Casu (‘Ronzoni’ Institute, Milan, Italy), Dr. A. Naggi (‘Ronzoni’) and Drs. C. Pisano and S. Penco (Sigma-Tau, Rome, Italy) for kindly providing glycol-split species of heparin and for their continuous help and collaboration. We gratefully acknowledge the contribution, motivation and assistance of the research teams in the Rappaport Faculty of Medicine and the Hadassah Medical Center. Due to space limitation, we apologize for not citing several relevant articles.
Glossary Box
- Akt
A serine/threonine kinase that functions down stream to phosphoinositol 3- kinase (PI 3-K). Akt (also known as PKB) is involved in cellular survival pathways by inhibiting apoptotic processes. Since it can block apoptosis, and thereby promote cell survival, Akt1 has been implicated as a major factor in various types of cancer.
- EGFR
EGFR (epidermal growth factor receptor) exists on the cell surface and is activated by binding of its specific ligands, including epidermal growth factor and transforming growth factor α (TGFα). Upon activation by its growth factor ligands, EGFR undergoes transition from an inactive monomeric form to an active homodimer. In addition, EGFR may pair with another member of the ErbB receptor family, such as ErbB2/Her2/neu, to create an activated heterodimer. EGFR dimerization stimulates its intrinsic intracellular protein-tyrosine kinase activity. As a result, autophosphorylation of several tyrosine (Y) residues in the cytoplasmic domain of EGFR occurs. This autophosphorylation elicits downstream activation and signaling by several other proteins that associate with the phosphorylated tyrosines. These downstream signaling proteins initiate several signal transduction cascades, primarily the MAPK, Akt and STAT pathways, leading to DNA synthesis and cell proliferation.
- Extracellular matrix (ECM)
In a given organ, the cells normally occupy only a certain portion of the volume. A substantial part is filled with a network of macromolecules defined as extracellular matrix (ECM). The ECM is composed of a variety of proteins and polysaccharides secreted by the cells. Protein components are adhesive molecules (i.e., laminin, fibronectin, vitronectin), structural molecules (i.e., collagen, elastin), and heparan sulfate proteoglycans (HSPGs, see below).
- Glycosaminoglycan (GAG) and heparan sulfate (HS)
Glycosaminoglycans (GAGs) are polymers of disaccharide units. In heparan sulfate (HS), uronic acid (either glucuronic acid or iduronic acid) and glucosamine (either N-acetyl or N-sulfamino) repeats compose its basic structure. These GAG chains are long, linear carbohydrate polymers that are negatively charged under physiological conditions, due to the presence of sulfate and uronic acid groups. Despite the seemingly simple single repeating structural motif, these sugar polymers show a great deal of structural diversity generated by complex pattern of deacetylation, sulfation, and epimerization.
- Heparin
Heparin is a highly sulfated analogue of HS. Heparin is a potent inhibitor of heparanase enzymatic activity, being a relatively poor heparanase substrate. Heparin is widely used as an anticoagulant, thus urging the development of more specific, non-anticoagulant heparin-derived heparanase inhibitors.
- Glycol-split heparin
Chemical modification (oxidation of nonsulfated uronic acid residues) of heparin that results in flexible joints along the sulfated polysaccharide chain, thereby strengthening its binding to heparanase.
- Proteoglycan
Proteoglycans consist of a core protein with one or more covalently attached glycosaminoglycan (GAG) side chain(s). The major cell membrane HSPGs are the transmembrane syndecans and the glycosylphosphatidylinositol (GPI) anchored glypicans. In the ECM, especially basement membranes, perlecan, agrin and collagen XVIII core proteins are the main HS-bearing species.
- Heparanase
Heparanase is an endo-β-glucuronidase that cleaves HS side chains presumably at sites of low sulfation, releasing saccharide products with appreciable size (4-7 kDa) that can still associate with protein ligands and modulate their biological potency. Heparanase cleaves HS utilizing hydrolase mechanism as opposed to bacterial heparinase III (heparitinase) that cleaves HS more extensively by a β-eliminase mechanism. The heparanase mRNA encodes a 61.2 kDa latent pro-enzyme that is post translationally cleaved into 8 and 50 kDa subunits that non-covalently associate to form the active heparanase heterodimer.
- Myeloma
Myeloma is part of a broad group of diseases called hematological malignancies. Multiple myeloma is a plasma cell malignancy characterized by complex heterogeneous cytogenetic abnormalities. The bone marrow microenviornment promotes myeloma cell growth and resistance to conventional therapies.
- Rac1
Rac1 is a small (∼21 kDa) signaling protein and is a member of the Rho family of GTPases. Members of this superfamily appear to regulate a diverse array of cellular events, including the control of cell growth, cytoskeletal reorganization, and the activation of protein kinases.
- Src
Src is a family of proto-oncogenic tyrosine kinases. pp60src is a cytoplasmic protein with tyrosine kinase activity that associates with the cytoplasmic face of the plasma membrane. Src kinase plays a key role in multiple signaling pathways regulating diverse cell functions from proliferation and survival to invasion and angiogenesis. Src has been implicated in the pathogenesis of a number of cancers, and has been found to be over expressed in breast, prostate, colorectal, pancreatic and non-small-cell lung tumors. The discovery of Src family proteins has been instrumental to the understanding of cancer as a disease where normal cellular signaling has gone awry.
- Shedding
Protein shedding refers to transmembrane proteins which can undergo proteolytic cleavage at the cell surface to release soluble ectodomain; a process often referred to as “ectodomain shedding”. Many transmembrane molecules in diverse organisms are subjected to shedding, including cytokines and receptors, adhesion molecules and other factors. Ectodomain shedding has most often been linked to multiple zinc-binding metalloproteases of the matrix metalloproteinase (MMP) and a disintegrin and metalloproteinase (ADAM) families.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parish CR, et al. Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta. 2001;1471:M99–108. doi: 10.1016/s0304-419x(01)00017-8. [DOI] [PubMed] [Google Scholar]
- 2.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–347. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ilan N, et al. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 4.McKenzie EA. Heparanase: a target for drug discovery in cancer and inflammation. Br J Pharmacol. 2007;151:1–14. doi: 10.1038/sj.bjp.0707182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miao HQ, et al. Development of heparanase inhibitors for anti-cancer therapy. Curr Med Chem. 2006;13:2101–2111. doi: 10.2174/092986706777935230. [DOI] [PubMed] [Google Scholar]
- 6.Sanderson RD, et al. Heparan sulfate proteoglycans and heparanase--partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004;23:341–352. doi: 10.1016/j.matbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Vlodavsky I, et al. The impact of heparanese and heparin on cancer metastasis and angiogenesis. Pathophysiol Haemost Thromb. 2006;35:116–127. doi: 10.1159/000093553. [DOI] [PubMed] [Google Scholar]
- 8.Ferro V, et al. PI-88 and novel heparan sulfate mimetics inhibit angiogenesis. Seminars in thrombosis and hemostasis. 2007;33:557–568. doi: 10.1055/s-2007-982088. [DOI] [PubMed] [Google Scholar]
- 9.Vlodavsky I, et al. Heparanase: structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr Pharm Des. 2007;13:2057–2073. doi: 10.2174/138161207781039742. [DOI] [PubMed] [Google Scholar]
- 10.Vreys V, David G. Mammalian heparanase: what is the message? J Cell Mol Med. 2007;11:427–452. doi: 10.1111/j.1582-4934.2007.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beauvais DM, Rapraeger AC. Syndecans in tumor cell adhesion and signaling. Reprod Biol Endocrinol. 2004;2:3. doi: 10.1186/1477-7827-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couchman JR. Syndecans: proteoglycan regulators of cell-surface microdomains? Nat Rev Mol Cell Biol. 2003;4:926–937. doi: 10.1038/nrm1257. [DOI] [PubMed] [Google Scholar]
- 13.Saoncella S, et al. Syndecan-4 signals cooperatively with integrins in a Rho-dependent manner in the assembly of focal adhesions and actin stress fibers. Proc Natl Acad Sci U S A. 1999;96:2805–2810. doi: 10.1073/pnas.96.6.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bass MD, et al. Syndecan-4-dependent Rac1 regulation determines directional migration in response to the extracellular matrix. J Cell Biol. 2007;177:527–538. doi: 10.1083/jcb.200610076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dovas A, et al. PKCbeta-dependent activation of RhoA by syndecan-4 during focal adhesion formation. J Cell Sci. 2006;119:2837–2846. doi: 10.1242/jcs.03020. [DOI] [PubMed] [Google Scholar]
- 16.Tkachenko E, et al. Syndecan-4 clustering induces cell migration in a PDZ-dependent manner. Circ Res. 2006;98:1398–1404. doi: 10.1161/01.RES.0000225283.71490.5a. [DOI] [PubMed] [Google Scholar]
- 17.Tkachenko E, et al. Syndecans: new kids on the signaling block. Circ Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 18.Gingis-Velitski S, et al. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J Biol Chem. 2004;279:44084–44092. doi: 10.1074/jbc.M402131200. [DOI] [PubMed] [Google Scholar]
- 19.Abboud-Jarrous G, et al. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J Biol Chem. 2008;283:18167–18176. doi: 10.1074/jbc.M801327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abboud-Jarrous G, et al. Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. J Biol Chem. 2005;280:13568–13575. doi: 10.1074/jbc.M413370200. [DOI] [PubMed] [Google Scholar]
- 21.Levy-Adam F, et al. Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. J Biol Chem. 2005;280:20457–20466. doi: 10.1074/jbc.M414546200. [DOI] [PubMed] [Google Scholar]
- 22.Vreys V, et al. Cellular uptake of mammalian heparanase precursor involves low density lipoprotein receptor-related proteins, mannose 6-phosphate receptors, and heparan sulfate proteoglycans. J Biol Chem. 2005;280:33141–33148. doi: 10.1074/jbc.M503007200. [DOI] [PubMed] [Google Scholar]
- 23.Zetser A, et al. Processing and activation of latent heparanase occurs in lysosomes. J Cell Sci. 2004;117:2249–2258. doi: 10.1242/jcs.01068. [DOI] [PubMed] [Google Scholar]
- 24.Levy-Adam F, et al. Heparanase facilitates cell adhesion and spreading by clustering of cell surface heparan sulfate proteoglycans. PLoS ONE. 2008;3:e2319. doi: 10.1371/journal.pone.0002319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woods A, Couchman JR. Syndecan-4 and focal adhesion function. Curr Opin Cell Biol. 2001;13:578–583. doi: 10.1016/s0955-0674(00)00254-4. [DOI] [PubMed] [Google Scholar]
- 26.Gingis-Velitski S, et al. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem. 2004;279:23536–23541. doi: 10.1074/jbc.M400554200. [DOI] [PubMed] [Google Scholar]
- 27.Goldshmidt O, et al. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003;17:1015–1025. doi: 10.1096/fj.02-0773com. [DOI] [PubMed] [Google Scholar]
- 28.Sotnikov I, et al. Enzymatically quiescent heparanase augments T cell interactions with VCAM-1 and extracellular matrix components under versatile dynamic contexts. J Immunol. 2004;172:5185–5193. doi: 10.4049/jimmunol.172.9.5185. [DOI] [PubMed] [Google Scholar]
- 29.Zetser A, et al. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–7741. [PubMed] [Google Scholar]
- 30.Meads MB, et al. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–2526. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 31.Kelly T, et al. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003;63:8749–8756. [PubMed] [Google Scholar]
- 32.Mahtouk K, et al. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood. 2007;109:4914–4923. doi: 10.1182/blood-2006-08-043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanderson RD, Yang Y. Syndecan-1: a dynamic regulator of the myeloma microenvironment. Clinical & Experimental Metastasis. 2008;25:149–159. doi: 10.1007/s10585-007-9125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidel C, et al. Serum syndecan-1: a new independent prognostic marker in multiple myeloma. Blood. 2000;95:388–392. [PubMed] [Google Scholar]
- 35.Yang Y, et al. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood. 2002;100:610–617. doi: 10.1182/blood.v100.2.610. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, et al. Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–13333. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- 37.Purushothaman A, et al. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zcharia E, et al. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS ONE. 2009;4:e5181. doi: 10.1371/journal.pone.0005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai Y, et al. HSulf-1 and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo. J Biol Chem. 2005;280:40066–40073. doi: 10.1074/jbc.M508136200. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, et al. The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood. 2007;110:2041–2048. doi: 10.1182/blood-2007-04-082495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casu B, et al. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol Haemost Thromb. 2008;36:195–203. doi: 10.1159/000175157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naggi A, et al. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J Biol Chem. 2005;280:12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 43.Cohen I, et al. Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int J Cancer. 2006;118:1609–1617. doi: 10.1002/ijc.21552. [DOI] [PubMed] [Google Scholar]
- 44.Doviner V, et al. Spatial and temporal heparanase expression in colon mucosa throughout the adenoma-carcinoma sequence. Mod Pathol. 2006;19:878–888. doi: 10.1038/modpathol.3800603. [DOI] [PubMed] [Google Scholar]
- 45.Ben-Zaken O, et al. Heparanase induces Akt phosphorylation via a lipid raft receptor. Biochem Biophys Res Commun. 2007;361:829–834. doi: 10.1016/j.bbrc.2007.06.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nadir Y, et al. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J Thromb Haemost. 2006;4:2443–2451. doi: 10.1111/j.1538-7836.2006.02212.x. [DOI] [PubMed] [Google Scholar]
- 47.Zetser A, et al. Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006;66:1455–1463. doi: 10.1158/0008-5472.CAN-05-1811. [DOI] [PubMed] [Google Scholar]
- 48.Reynolds AB. p120-catenin: Past and present. Biochim Biophys Acta. 2007;1773:2–7. doi: 10.1016/j.bbamcr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen-Kaplan V, et al. Heparanase augments epidermal growth factor receptor phosphorylation: correlation with head and neck tumor progression. Cancer Res. 2008;68:10077–10085. doi: 10.1158/0008-5472.CAN-08-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haskell MD, et al. c-Src tyrosine phosphorylation of epidermal growth factor receptor, P190 RhoGAP, and focal adhesion kinase regulates diverse cellular processes. Chemical Reviews. 2001;101:2425–2440. doi: 10.1021/cr0002341. [DOI] [PubMed] [Google Scholar]
- 51.Doweck I, et al. Heparanase localization and expression by head and neck cancer: correlation with tumor progression and patient survival. Neoplasia. 2006;8:1055–1061. doi: 10.1593/neo.06577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wood RJ, Hulett MD. Cell surface-expressed cation-independent mannose 6-phosphate receptor (CD222) binds enzymatically active heparanase independently of mannose 6-phosphate to promote extracellular matrix degradation. J Biol Chem. 2008;283:4165–4176. doi: 10.1074/jbc.M708723200. [DOI] [PubMed] [Google Scholar]
- 53.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ben-Zaken O, et al. Low and high affinity receptors mediate cellular uptake of heparanase. Int J Biochem Cell Biol. 2008;40:530–542. doi: 10.1016/j.biocel.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fux L, et al. Structure-function approach identifies a COOH-terminal domain that mediates heparanase signaling. Cancer Res. 2009;69:1758–1767. doi: 10.1158/0008-5472.CAN-08-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hulett MD, et al. Identification of active-site residues of the pro-metastatic endoglycosidase heparanase. Biochemistry. 2000;39:15659–15667. doi: 10.1021/bi002080p. [DOI] [PubMed] [Google Scholar]
- 57.Nardella C, et al. Mechanism of activation of human heparanase investigated by protein engineering. Biochemistry. 2004;43:1862–1873. doi: 10.1021/bi030203a. [DOI] [PubMed] [Google Scholar]
- 58.Lai NS, et al. Requirement of the conserved, hydrophobic C-terminus region for the activation of heparanase. Exp Cell Res. 2008;314:2834–2845. doi: 10.1016/j.yexcr.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 59.Simizu S, et al. Involvement of disulfide bond formation in the activation of heparanase. Cancer Res. 2007;67:7841–7849. doi: 10.1158/0008-5472.CAN-07-1053. [DOI] [PubMed] [Google Scholar]
- 60.Ma P, et al. Heparanase deglycanation of syndecan-1 is required for binding of the epithelial-restricted prosecretory mitogen lacritin. J Cell Biol. 2006;174:1097–1106. doi: 10.1083/jcb.200511134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cohen-Kaplan V, et al. Heparanase induces VEGF C and facilitates tumor lymphangiogenesis. Int J Cancer. 2008;123:2566–2573. doi: 10.1002/ijc.23898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nadir Y, et al. Heparanase induces tissue factor pathway inhibitor expression and extracellular accumulation in endothelial and tumor cells. Thromb Haemost. 2008;99:133–141. [PubMed] [Google Scholar]
- 63.Okawa T, et al. Heparanase is involved in angiogenesis in esophageal cancer through induction of cyclooxygenase-2. Clin Cancer Res. 2005;11:7995–8005. doi: 10.1158/1078-0432.CCR-05-1103. [DOI] [PubMed] [Google Scholar]
- 64.Ferro V, et al. The development of inhibitors of heparanase, a key enzyme involved in tumour metastasis, angiogenesis and inflammation. Mini Rev Med Chem. 2004;4:693–702. doi: 10.2174/1389557043403729. [DOI] [PubMed] [Google Scholar]
- 65.Simizu S, et al. Heparanase as a molecular target of cancer chemotherapy. Cancer Sci. 2004;95:553–558. doi: 10.1111/j.1349-7006.2004.tb02485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lewis KD, et al. A phase II study of the heparanase inhibitor PI-88 in patients with advanced melanoma. Invest New Drugs. 2008;26:89–94. doi: 10.1007/s10637-007-9080-5. [DOI] [PubMed] [Google Scholar]
- 67.Basche M, et al. A phase I biological and pharmacologic study of the heparanase inhibitor PI-88 in patients with advanced solid tumors. Clin Cancer Res. 2006;12:5471–5480. doi: 10.1158/1078-0432.CCR-05-2423. [DOI] [PubMed] [Google Scholar]
- 68.Goldshmidt O, et al. Human heparanase is localized within lysosomes in a stable form. Exp Cell Res. 2002;281:50–62. doi: 10.1006/excr.2002.5651. [DOI] [PubMed] [Google Scholar]
- 69.Nadav L, et al. Activation, processing and trafficking of extracellular heparanase by primary human fibroblasts. J Cell Sci. 2002;115:2179–2187. doi: 10.1242/jcs.115.10.2179. [DOI] [PubMed] [Google Scholar]
- 70.Cohen E, et al. Heparanase processing by lysosomal/endosomal protein preparation. FEBS Lett. 2005;579:2334–2338. doi: 10.1016/j.febslet.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 71.Yanagishita M, Hascall VC. Cell surface heparan sulfate proteoglycans. J Biol Chem. 1992;267:9451–9454. [PubMed] [Google Scholar]
- 72.Chen L, Sanderson RD. Heparanase regulates levels of syndecan-1 in the nucleus. PLoS ONE. 2009;4:e4947. doi: 10.1371/journal.pone.0004947. [DOI] [PMC free article] [PubMed] [Google Scholar]