

Abstract
Adipose LPL (lipoprotein lipase) plays an important role in regulating plasma triacylglycerols and lipid metabolism. We have previously demonstrated that PKCα (protein kinase Cα) depletion inhibits LPL translation in 3T3-F442A adipocytes. Using in vitro translation experiments, the minimum essential region on the 3′UTR (3′-untranslated region) of LPL mRNA required for the inhibition of translation was identified as the proximal 39 nt. These results were confirmed by RNase protection analysis using cytoplasmic proteins isolated from the adipocytes treated with PKCα antisense oligomers and the LPL 3′UTR transcript (LPL 3′UTR nt: 1512–1640). The protein components involved in this RNA-binding interaction from PKCα depletion were passed through an affinity column containing a sequence of the LPL 3′UTR and, after Western blotting, the RNA-binding proteins were identified as the catalytic and the regulatory subunits of PKA (protein kinase A), Cα and RIIβ, and AKAP (A-kinase-anchoring protein) 121. This RNA inhibitory complex consisted of the same RNA-binding proteins that have been identified previously as mediators of LPL translational inhibition by PKA activation, suggesting that PKCα depletion inhibits LPL translation through PKA activation. In additional experiments, PKC depletion by prolonged PMA treatment or PKCα antisense oligomers resulted in an increase in PKA activity in 3T3-F442A adipocytes, comparable with PKA activation with adrenaline (epinephrine) treatment. These results demonstrate that LPL translational inhibition occurs through an RNA-binding complex involving PKA subunits and AKAP121, and this complex can be activated either through traditional PKA activation methods or through the depletion of PKCα.
Keywords: cell signalling, kinase cross-talk, post-transcriptional regulation, protein kinase
Introduction
LPL (lipoprotein lipase) plays a central role in lipid metabolism by regulating the hydrolysis of triacylglycerol (triglyceride)-rich lipoproteins in plasma (chylomicrons and very low density lipoproteins), and generates fatty acids for storage or energy utilization [1]. LPL is synthesized mostly by adipose tissue and muscle and is regulated in a tissue-specific manner in response to nutritional and hormonal stimuli [2–4]. The regulation of LPL is complex and occurs at the level of transcription, translation or post-translational processing, depending on the cell and the stimulus [5]. Insulin and catecholamines are reciprocal physiological regulators of LPL. LPL activity is increased in the hyperinsulinaemic environment of the fed state and inhibited during fasting, when insulin is low and catecholamine-mediated stimulation of lipolysis predominates [6].
The regulation of LPL by catecholamines is complex. Studies using rat adipocytes and 3T3 adipocyte cell lines indicate that there is a 3–5-fold inhibition of LPL synthesis following adrenaline (epinephrine) treatment, with no corresponding change in LPL mRNA [7,8]. This inhibition of LPL translation by adrenaline is mediated by an RNA-binding protein complex, containing the catalytic subunit of PKA (protein kinase A) and AKAP (A-kinase-anchoring protein) 121/149, which interact with the 3′UTR (3′-untranslated region) of the LPL mRNA [9,10]. The activation of PKA by cAMP binding results in the formation of this complex and LPL translation inhibition, along with the activation of hormone sensitive lipase-mediated lipolysis.
LPL is also translationally repressed following depletion of cellular PKC (protein kinase C), either through prolonged treatment with phorbol esters or through the use of antisense oligonucleotides to PKCα [11]. This PKCα-mediated inhibition of LPL translation also involved the 3′UTR of the LPL mRNA, although the precise nature of the RNA-binding protein is not known. As depletion of PKCα and activation of PKA both inhibited LPL translation, we speculated whether these two pathways share common mechanisms with regard to LPL translational regulation. Indeed, previous studies have demonstrated an inverse relationship between PKC and PKA in a number of different situations, such as in mitogenic pathways, and in response to Raf1 phosphorylation [12–14]. Therefore, in the present study we examined the inhibition of translation of LPL resulting from PKCα depletion. We demonstrate that PKCα depletion results in an increase in intracellular cAMP and the activation of PKA, resulting in the formation of an RNA-binding complex that inhibits LPL translation through binding to the 3′UTR of LPL mRNA.
Experimental
Cell culture and differentiation
3T3-F442A adipocytes were obtained from Dr Howard Green (Harvard Medical School, Boston, MA, U.S.A.). Cells were maintained in DMEM (Dulbecco's modified Eagle's medium) (Gibco BRL), supplemented with 10% calf serum. For experiments, cells were grown to confluence and stimulated to differentiate in DMEM containing 10% fetal bovine serum and 100 nM insulin for 14 days. Cells were treated with 10−5 M adrenaline for 2 h, for PKA activation studies, or with 100 nM phorbol ester (PMA) for 16 h to deplete PKC isoforms.
Transfection of adipocytes with oligonucleotides
Oligonucleotides were synthesized with phospho-orothioate-containing 2-methoxyethyl modification at positions 1–5 and 15–20 (Isis Pharmaceuticals, Carlsbad, CA, U.S.A.). The sequences were as follows: Isis 14012 antisense PKCα, 5′-CAGCCATGGTTCCCCCCAAC; and Isis 17250 scrambled oligomer, 5′-CCAGTCACTCGCACCATCGC. All oligomers were synthesized using an Applied Biosystems 380B automated DNA synthesizer as described previously [15]. Differentiated 3T3-F442A adipocytes were transfected using serum-free DMEM containing 15 μg/ml Lipofectin® (Invitrogen) and 0.5 μM PKCα antisense oligomer for 16 h and incubated in differentiation medium for 72 h. Control cultures were treated similarly but with Lipofectin® and scrambled oligomer or Lipofectin® alone in the presence of DMEM.
Quantification of PKCα and LPL mRNA using real-time PCR
Total RNA was extracted using an RNeasy Lipid Tissue Mini kit from Qiagen. The quantity and quality of the isolated RNA was determined by UV spectrophotometry and formaldehyde-agarose gel electrophoresis respectively. Total RNA (0.5 μg) was reverse-transcribed using oligo-dT primers with TaqMan Reverse transcription kit (Applied Biosystems). Reverse-transcribed RNA was amplified with SYBR Green PCR Master Mix (Applied Biosystems) plus 0.3 μM of gene-specific upstream and downstream primers during 50 cycles on a Rotor-Gene 3000 Real-time Thermal Cycler (Corbett Research, Sydney, Australia). Each cycle consisted of denaturation at 94 °C for 20 s, annealing at 58 °C for 20 s and extension at 72 °C for 20 s. Amplified 18S expression was used as standard control to normalize the differences in individual samples. The primer sequences for mouse PKCα, LPL and 18S RNA are described below; primers were designed spanning an intron. All data were expressed in relation to 18S RNA and standard curves were generated using pooled cDNA from the samples being assayed. Therefore, the results represent arbitrary units which accurately compare samples within each assay. Primers were synthesized by Integrated DNA Technologies Inc. PKCα forward, 5′-CCCATTCCAGAAGGAGATGA; reverse, 5′-CGTTGACGTATTCCATGACG. LPL forward, 5′-ACTCGCTCTCAGATGCCCTA; reverse, 5′-TTGTGTTGCTTGCCATTCCT. 18S forward, 5′-TTCGAACGTCTGC CCTATCAA; reverse, 5′-ATGGTAGGCACGGCGACTA.
Preparation of cytoplasmic extracts
Approx. 100 dishes of 100 mm diameter containing 3T3-F442A adipocytes, representing ∼ 109 cells differentiated with insulin for 14 days, were transfected with Lipofectin® in the presence or absence of 0.5 μM PKCα antisense oligomers. A cytoplasmic extract was prepared as described previously [8]. Briefly, adipocytes were homogenized in lysis buffer (50 mM Tris/HCl, pH 7.4, 250 mM sucrose, 35 mM KCl, 10 mM MgCl2, 0.5 mM EDTA and 7 mM 2-mercaptoethanol) with 2 mM PMSF and protease inhibitor cocktail (Sigma). The post-nuclear extract was used to prepare a high-speed supernatant fraction, S-100; this cytosolic fraction was precipitated with ammonium sulfate to 60% saturation for 30 min on ice. Precipitated proteins were collected, dissolved and dialysed against Buffer A (20 mM Tris/HCl, pH 7.4, 20 mM KCl, 10% glycerol, 7 mM 2-mercaptoethanol, 0.1 mM EDTA and 2 mM PMSF).
LPL constructs used in in vitro translation experiments
Construct A contains 175 nt of the 5′UTR and 1600 nt of the 3′UTR, creating a 3.2 kb cDNA clone that extends up to the first polyadenylation signal [16]. Construct B is the originally cloned 2435 kb human LPL35 cDNA, and contains 175 nt of the 5′UTR and 822 nt of the 3′UTR [17]. Construct C was made by digesting construct B with EcoR1 at nt 1638. Construct D lacks the 5′UTR and contains the complete coding sequence and 44 nt of 3′UTR. Construct E contains the complete LPL coding sequence from nt 175 up to nt 1599. All the constructs were cloned in transcription vector pGEM2 [8].
In vitro translation
RNA transcripts were made from an LPL cDNA construct described earlier [8,17]. In vitro translation of RNA transcripts were performed as described previously [8]. Equal quantities of RNA transcripts (0.1 μg) were translated in a rabbit reticulocyte lysate system (Promega) in the presence of [35S]methionine. Reactions were incubated at 30 °C for 1 h and terminated by transferring to 4°C. The products of reactions were analysed on SDS/10% PAGE followed by autoradiography. Images were quantified using Image Quant software (Amersham Biosciences). The control cell extract from cells treated with Lipofectin® alone inhibited translation when compared with a lane marked as no extract, probably due to the constitutive presence of inhibitors (results not shown) also observed in earlier studies [8].
Gel shift and RNase protection analysis
Gel shift and RNase protection analysis were performed as described earlier [18,19]. Overlapping DNA fragments were synthesized using PCR with a T7 transcription promoter added to the 5′ end of the primer. LPL transcript corresponding to nt 1512–1663 was made in the presence of [α-32P]UTP. Cytoplasmic extracts (0.5–2.5 μg protein) were incubated with a specific RNA transcript (50000 d.p.m.) for 20 min, followed by RNase T1 digestion (10 units) for 20 min, then yeast tRNA (0.1 μg) and heparin (5 units) were added to decrease non-specific interactions. The reaction products were separated on 5% non-denaturing acrylamide gels. Gels were dried and analysed by autoradiography.
RNA affinity column preparation
Poly(A) RNA transcripts were generated from PCR products containing the C-terminal 50 nt of the coding sequence and the first 500 nt of the LPL 3′UTR (nt 1512 to 2119), as described previously [20]. A tracer amount of [32P]UTP was also added during transcription, to follow the binding and check the quality of the RNA. The poly(A) RNA was incubated with presoaked poly(T)–Sepharose beads for 4 h, the beads were washed with low salt buffer (20 mM Tris/HCl, pH 7.4, and 20 mM KCl) to remove the unbound excess RNA.
Purification of RNA-binding proteins using the affinity column
Cytoplasmic extracts from control or PKCα antisense-treated adipocytes were prepared as described [8] and incubated with (Oligo-dT)–Sepharose beads hybridized with LPL 3′UTR [Poly(A)] mRNA for 16 h at 4°C, and unbound proteins were washed off using several volumes of Buffer B (50 mM Tris/HCl, pH 7.4, 250 mM sucrose, 35 mM KCl, 10 mM MgCl2, 0.5 mM EDTA and 7 mM 2-mercaptoethanol). Proteins specifically bound to the RNA affinity column were eluted with three washes using the same buffer containing 100 mM KCl, followed by three washes with Buffer B containing 300 mM KCl. Fractions were concentrated and fractionated on SDS/10% PAGE, followed by Western blotting with specific antibody as specified.
Western blot analysis
Fractions eluted from the RNA affinity column were fractionated on SDS/10% PAGE and blotted onto nitrocellulose membranes. To identify the PKA catalytic subunit α or the R subunit protein, membranes were probed with rabbit polyclonal antibody (Transduction Labs, CA, U.S.A.), followed by anti-rabbit HRP (horseradish peroxidase) conjugate (Sigma, St. Louis, MO, U.S.A.). AKAP 121 was detected using a mouse monoclonal antibody (BD Transduction Labs) followed by anti-mouse HRP conjugate. Western blot reaction product was visualized with ECL® (enhanced chemiluminiscence) reagents (Amersham).
Measurement of LPL activity
Heparin-releasable LPL activity was determined as described previously [21]. LPL was measured by incubating adipocytes in DMEM containing 10 units/ml heparin for 60 min at 37 °C. LPL catalytic activity was measured as previously described using a substrate containing 3H-triolein and fetal bovine serum as a source of apoC-II [22]. LPL activity was expressed as nmol of FFA (free fatty acid) released/h per mg of protein.
Measurement of intracellular cAMP levels
cAMP levels were measured using a low pH enzyme immunoassay as follows. Adipocytes were rinsed with cold PBS and lysed with 0.1 M hydrochloric acid, which inhibits the endogenous phosphodiesterase activity. Cell lysates were centrifuged at 600 g for 10 min at 4 °C and supernatants were assayed directly using a competitive immunoassay which uses a polyclonal antibody to cAMP that competes for binding to cAMP in the sample or cAMP bound covalently to an alkaline phosphatase conjugate (Sigma–Aldrich). cAMP levels in cell lysates was measured using a standard curve generated from cAMP standards supplied with the kit.
Measurement of PKA activity
PKA phosphotransferase activity was measured in adipocytes transfected with and without PKCα antisense oligomers. Cells were lysed in Tris/HCl (50 mM, pH 7.5) containing 1% Triton X-100, 2 mM EDTA, 1 mM PMSF, 1 mM aprotinin, 2 mM leupeptin and 5 mM sodium vanadate. RII β is the predominant PKA subtype in adipocytes, therefore RII β antibody bound to Protein G beads was used to immunoprecipitate PKA activity from cell lysates. The assay is based on the phosphorylation of kempeptide using the transfer of the γ-phosphate of [γ-32P] ATP by PKA. The incubation mixture contained 12.5 mM magnesium, 80 μM ATP, 0.16 μM cAMP and 50 mM kempeptide. The assay was linear for 30 min and up to 20% of ATP is incorporated (Upstate Biotec labs, CA, U.S.A.). Control reactions were run simultaneously with PKA and PKC inhibitors.
Glycerol release assay
Lipolysis was determined by measuring the amount of glycerol released by the cells into the medium. Medium was collected at 72 h and glycerol content was measured using the Free Glycerol Reagent from Sigma.
Results
To demonstrate the post-transcriptional inhibition of LPL by PKCα depletion, 3T3-F442A adipocytes were transfected with 0.5 μM antisense oligomers to PKCα as described in the Experimental section. PKCα mRNA and protein as well as LPL activity and mRNA levels were then measured 72 h after transfection. As shown in Figure 1, PKCα mRNA expression was decreased by 50 ± 10% in PKCα antisense oligomer-treated cells, and there was no change in 18S mRNA (not shown) or LPL expression. The change in PKCα mRNA was accompanied by a 50 ± 10% decrease in PKCα protein as measured by Western blot analysis of cell lysates. Although there was no decrease in LPL mRNA expression in PKCα antisense-treated cells, LPL synthesis and protein expression were inhibited as demonstrated previously [11], and LPL activity was inhibited by 52 ± 12% (Figure 1).
Figure 1. Effect of antisense PKCα on 3T3-F442A adipocytes.
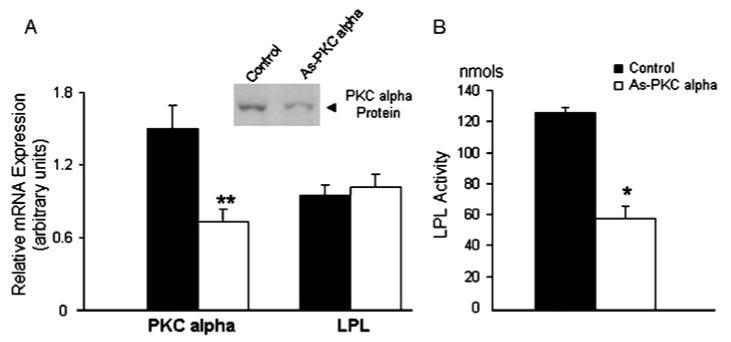
Quantitative measurement of PKCα and LPL mRNA in 3T3-F442A cells. (A) Adipocytes were transfected with or without 0.5 μM PKCα antisense (As-PKC alpha) oligomer, then PKC α and LPL mRNA were measured 72 h following transfection using real-time PCR. PKCα mRNA expression was depleted by 50± 10% (**P < 0.02), and there was no change in LPL mRNA. PKCα protein expression was decreased by 50 ± 10% (P < 0.05) as determined by Western blot analysis of cell lysates. (B) LPL activity expressed as nmol of FFA released/h per mg of protein was inhibited by 50 ± 10% (*P < 0.05). The results represent three separate experiments performed in triplicate expressed as means ± S.D.
As demonstrated previously, the regulation of LPL by PKCα depletion was mediated by transacting factors interacting with the 3′UTR of LPL mRNA [11]. To identify the specific region of the LPL 3′UTR involved in the interaction, LPL constructs were designed with progressive deletions of the 3′UTR, and two constructs lacked the 5′UTR, as described in the Experimental section and shown in Figure 2(A). Cytoplasmic extracts (0.1– 0.25 μg protein) from PKCα antisense or scrambled oligomer-treated cells were incubated with 0.1 μg of LPL transcript in an in vitro translation system in the presence of [35S]methionine. Figure 2 compares LPL mRNA translation in the presence of cytoplasmic extracts from control or antisense PKCα-treated cells. In vitro translation of LPL was inhibited by cytoplasmic extracts from PKCα-depleted cells in all constructs except E, which lacked both the 5′and 3′UTR. Comparing the translation of transcripts C and D, deletion of the 5′UTR in construct D did not alter the inhibition of translation as long as the 39 nt of the 3′UTR was retained. These data indicate that a cytoplasmic fraction from PKCα antisense oligomer-treated cells inhibited LPL translation in vitro and this inhibition was dependent on the first 39 nt of LPL 3′UTR. To demonstrate further the importance of the LPL 3′UTR in the translational regulation by PKCα depletion, a construct was created using an irrelevant coding sequence (luciferase) with LPL nt 1512–1812 cloned 3′ to the luciferase sequence. This sequence of LPL contains 87 nt of LPL coding sequence along with 213 nt of 3′UTR. As shown in Figure 2(B), a cytoplasmic extract from PKCα-depleted cells inhibited the translation in vitro of this construct, indicating that the LPL 3′UTR is able to confer translational regulation in response to PKCα depletion to an irrelevant coding sequence.
Figure 2. Effect of S-100 extracts on in vitro translation of specific transcripts.
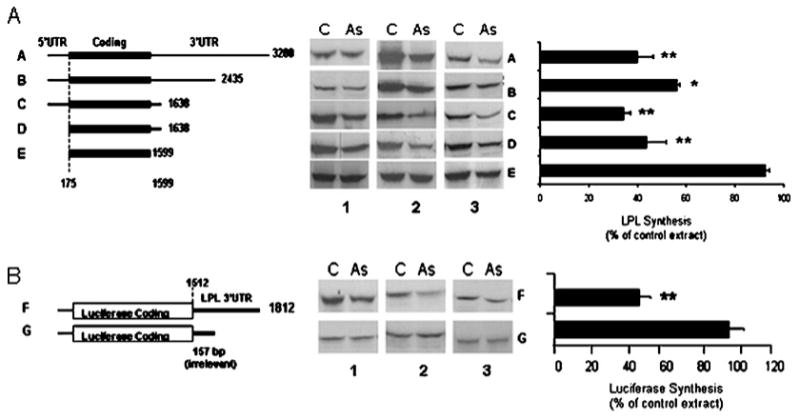
(A) Cytoplasmic extracts from control (C) or PKC antisense (As)-treated cells were added to in vitro translation reactions in the presence of specific LPL transcripts (left). The bar graph represents the means of three similar experiments, expressed as % of control. *P < 0.01, **P < 0.05 compared with the control. (B) Nucleotide sequence 1512–1812 of the LPL 3′UTR or an irrelevant 3′UTR of 157 bases were cloned 3′ to the luciferase coding sequence. These constructs were used by us in earlier studies [9]. The constructs were linearized and transcribed as described in the Experimental section. The transcripts generated were translated by using a rabbit reticulocyte in vitro translation system in the presence of cytoplasmic extracts from control or PKC antisense-treated cells. The bar graph represents the means of three similar experiments expressed as % of control. **P < 0.05 compared with the control.
To assess the binding of cytoplasmic factors in PKCα-depleted cells to sequences on the 3′UTR of LPL mRNA, a [32P]UTP-labelled transcript corresponding to nt 1512–1663 was incubated with 1–5 μg of S-100 extract from control or antisense PKCα-treated adipocytes, and examined on a non-denaturing gel to detect RNA–protein interactions. A specific gel-shifted band, which was protected from RNase T1 digestion, was obtained with the transcript involving LPL nt 1512–1663, demonstrating that PKCα depletion resulted in the interaction of cytoplasmic factors with the proximal 3′UTR (Figure 3). A gel-shifted band was also present when the extract from control (Lipofectin®-treated) cells was used. However, the control cell extract resulted in a much weaker gel-shifted band when compared to the extracts from PKCα antisense-treated cells (Figure 3). The presence of a gel-shifted band from the control cells is consistent with the presence of a constitutively present LPL translational inhibitor which is then increased by the depletion of PKCα.
Figure 3. Assessment of the binding of cytoplasmic factors in PKCα-depleted cells to sequences on the 3′ UTR of LPL.
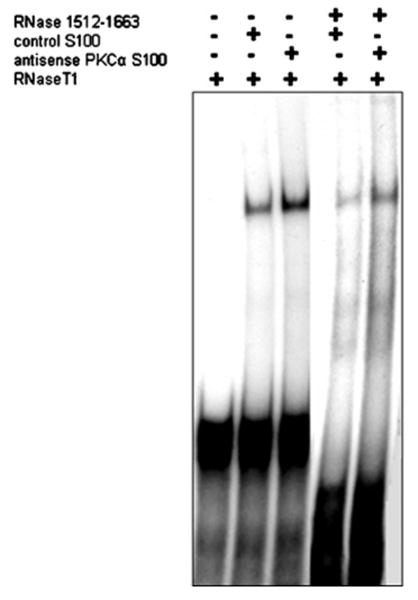
Gel shift and RNase protection analyses have been performed to assess the binding of cytoplasmic factors to sequences on the LPL 3′ UTR. [32P]UTP-labelled transcripts corresponding to nt 1512–1663 of LPL mRNA were incubated with 1–5 μg of S-100 extract from control and antisense PKCα-treated adipocytes, followed by the digestion of unprotected RNA with RNase T1. A specific RNaseT1 protected gel shift was obtained consistently only with the transcript 1512–1663 as is illustrated in the Figure. The control cell extract from cells treated with Lipofectin® alone provided a weaker gel shift.
To identify the protein(s) involved with LPL mRNA binding following PKCα depletion, an RNA affinity column was designed to take advantage of the affinity of the induced RNA-binding proteins for specific sequences on the LPL 3′UTR. As described in the Experimental section, the cytoplasmic extracts from control and PKCα antisense-treated cells were applied to an Oligo-dT–Sepharose column that contained the relevant RNA sequence of the LPL 3′UTR (nt 1512–1663) hybridized via a poly(A) tail. The elution from the RNA affinity column was examined by Western blotting for the presence of potential RNA-binding proteins involved in LPL regulation. In previous studies, the treatment of adipocytes with adrenaline resulted in an inhibition of translation, also involving the proximal 3′UTR. This inhibition of translation was due to the formation of a complex involving the PKA C subunit α, RIIβ and AKAP121 [10]. In the Western blots from PKCα-depleted cells, PKA Cα was detected prominently in the low salt elution fractions, and small amounts of PKA Cα were also detected in control extracts, consistent with the low level of translational inhibition that is present in control cell extracts (Figure 4). AKAP121 was also detected essentially exclusively in the column fractions from the PKCα antisense-treated cells and required a higher salt concentration for elution (Figure 4). The PKA-R subunit was detected in both the control and the PKCα-treated extracts (Figure 4). The above results demonstrate an increased binding of PKA Cα to the LPL RNA following PKCα antisense treatment. To establish whether PKCα depletion resulted in PKA activation, we measured cAMP levels and PKA activity in adipocytes. As shown in Figure 5(A), adrenaline treatment increased PKA activation by 60 ± 5%. PKCα was depleted from adipocytes using two methods: long-term (16 h) treatment with PMA or treatment with antisense oligomers to PKCα. PKCα depletion using PMA or antisense oligomers resulted in an 65 ± 2% and 70 ± 2% increase in PKA activity respectively. Thus PKCα depletion activated PKA to a similar extent as direct stimulation of PKA with adrenaline. Adrenaline treatment of adipocytes causes an increase in cAMP levels, which mediates activation of PKA; therefore we measured cAMP levels in adipocytes following PKCα depletion with antisense oligomers and adrenaline treatment. cAMP levels were increased 2-fold, from 3.8 ± 2 to 6.2 ± 2 pmol, following PKCα depletion, and 1.5-fold, from 3.8 ± 2 to 5.5 ± 0.5, with adrenaline as shown in Figure 5(B), accompanied by a 2-fold increase in lypolysis or glycerol release, which is a functional consequence of the inhibition of PKCα or adrenaline treatment, also shown in Figure 5(B). When cells were treated with antisense oligomers to PKCα or adrenaline, intracellular cAMP levels increased, which caused the increase in PKA activity and resulted in an increase in lipolysis, as assessed by adipocyte glycerol release, and an inhibition of LPL activity (Figure 5B). Although there was a 2-fold increase in PKA activation in PKCα-depleted cells, there was no corresponding increase in PKA Cα or RIIβ subunits mRNA expression (results not shown).
Figure 4. Purification of the RNA-binding complex following PKCα depletion and Western blot analysis.
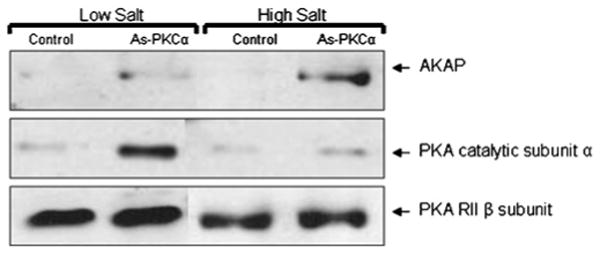
To purify the RNA-binding complex from adipocytes following PKCα depletion, an RNA affinity column consisting of nt 1512–2119 of human LPL mRNA with a poly(A) tail was prepared. Cytoplasmic extracts from control or PKCα antisense (As-PKCα)-treated adipocytes were incubated with (Oligo-dT)–Sepharose beads hybridized with the LPL 3′UTR [poly(A)] mRNA for 4–6 h at 4°C, and unbound proteins were washed off using several volumes of Buffer B. Proteins specifically bound to the RNA affinity column were eluted with three washes using the same buffer containing 100 mM KCl, followed by three washes with buffer containing 300 mM KCl. Fractions were concentrated and fractionated on SDS/10% PAGE, followed by Western blotting with specific antibody as indicated. The results represent one of three similar experiments.
Figure 5. Relationship between PKCα depletion and PKA activation in adipocytes.
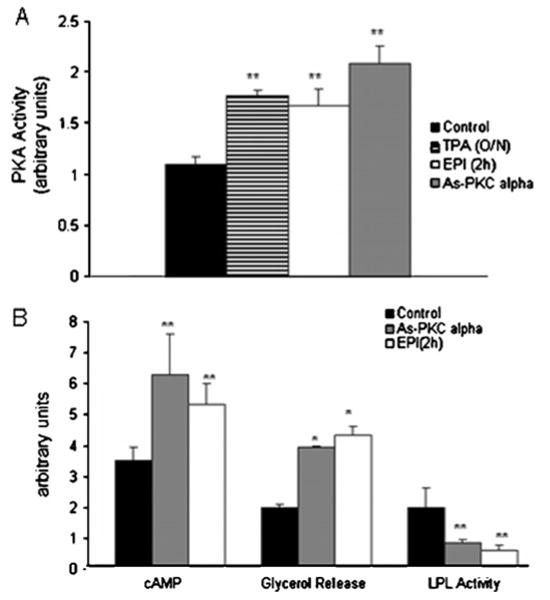
Adrenaline (EPI; 10−5 M) increased PKA activation by 60 ± 5%, 16 h of PMA (TPA; 100 nM) treatment also stimulated PKA activation by 65 ± 2%. PKA activation was increased by 75 ± 15% following treatment with PKCα antisense (As-PKC alpha) oligomers. The results represent the means of three independent experiments done in triplicate. (B) The functional consequence of the inhibition by PKCα and adrenaline treatment. The depletion of PKCα or adrenaline treatment resulted in an increase in intracellular cAMP levels and in increased lipolysis as well as inhibition of heparin-releasable LPL activity. Intracellular cAMP concentration was measured in cell lysates using an enzyme immunoassay as described in the Experimental section. Change in lipolysis was measured by quantifing glycerol released into the medium, also as described in the Experimental section. LPL activity is expressed as nmol of FFA released/h per mg of protein. The results represent the means of four independent experiments. *P < 0.01, **P < 0.05 compared with the untreated control.
To demonstrate further the relationship between the depletion of PKCα mRNA and PKA activation, 3T3-F442A adipocytes were transfected with 0.5 μM PKCα antisense oligomers, and PKCα mRNA expression as well as PKA activity was measured at 24, 48 and 72 h after transfection. At 24 h after transfection, there was no significant change in PKCα mRNA or PKA activity. Over the course of 72 h, PKCα mRNA expression increased in control cells, however this increase was not significant. PKCα mRNA was inhibited 45 ± 10% at 48 h and 55 ± 10% at 72 h after transfection of PKCα antisense oligomers, and PKA activity increased 2-fold by 100 ± 15% at both time points (Figure 6). Thus inhibition of PKCα resulted in an increase in PKA activity. Together, this would be predicted to result in a decrease in adipose lipid accumulation.
Figure 6. PKCα mRNA depletion is accompanied by activation of PKA.
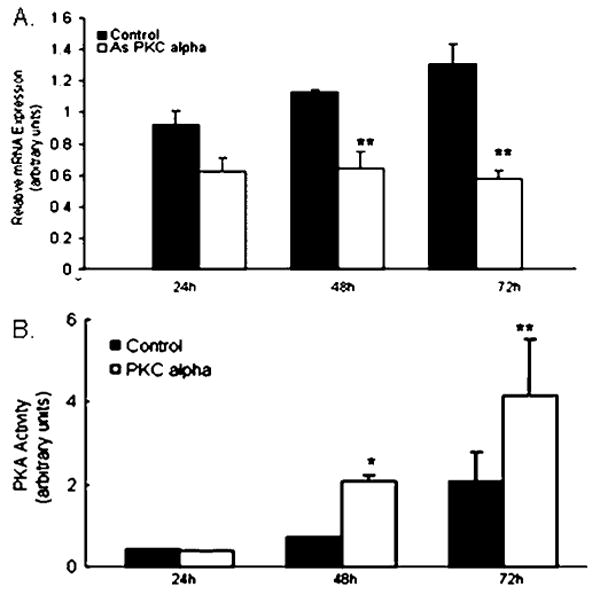
3T3-F442A adipocytes were transfected with 0.5 μMPKCα antisense (As PKC alpha) oligomers, and PKCα mRNA expression as well as PKA activity was measured at 24, 48 and 72 h after transfection. (A) PKCα mRNA expression was inhibited by 45 ± 10% at 48 h and by 55 ± 10% at 72 h after transfection. (B) PKA activity increased 2-fold by 100 ± 15% at both time points. The results represent the means of at least three experiments done in triplicate. *P < 0.01, **P < 0.05 compared with the untreated control.
These results suggest that PKCα depletion inhibits LPL through activation of PKA. Hence, the addition of adrenaline to PKCα-depleted cells may be expected to have no additional effects, since PKA may be maximally stimulated. To examine this, adipocytes transfected with PKCα antisense were treated with adrenaline, and LPL activity, cAMP levels and PKA activity were measured. LPL activity was inhibited by 60 ± 5% (P < 0.05) following treatment with PKCα antisense oligomers or adrenaline treatment, and the combined treatment with both PKCα antisense and adrenaline did not significantly increase the inhibitory effect. In addition, the combined treatment of adipocytes with both PKCα antisense oligomer and adrenaline did not significantly increase cAMP levels or PKA activation (Table 1).
Table 1. Combined effects of PKCα depletion followed by epinephrine treatment.
Results are expressed as means ± S.D. of three separate experiments performed in duplicate.
| PKCα depletion | Epinephrine treatment | PKCα depletion and epinephrine treatment | |
|---|---|---|---|
| LPL activity (% of control) | 60 ± 5 | 62 ± 5 | 65 ± 10 |
| cAMP (% of control) | 70 ± 10 | 65 ± 5 | 70 ± 10 |
| PKA activation (% of control) | 80 ± 10 | 65 ± 10 | 70 ± 5 |
Discussion
LPL regulates the hydrolysis of circulating triacylglycerols and provides FFAs for uptake by adipose tissue and muscle [23]. Adipose tissue LPL is particularly subject to regulation by numerous dietary and hormonal factors and is sensitive to pathophysiological changes that occur during diabetes. Studies on subjects with varying degrees of insulin sensitivity have demonstrated reduced LPL activity in adipose tissue and postheparin plasma in insulin-resistant and diabetic subjects [24,25], and improved diabetes control increased adipose LPL activity [26].
Post-transcriptional regulation of gene expression is important in much of the physiological regulation of LPL [27]. LPL activity is low during periods of fasting because hormones such as adrenaline, which stimulate cAMP-mediated activation of PKA, inhibit LPL activity [6,23]. The mechanism of LPL inhibition by adrenaline in adipose tissue is complex and studies in 3T3-L1 cultured mouse adipocytes demonstrated that the inhibition of LPL translation involved the interaction of an RNA-binding complex with the 3′UTR of LPL mRNA [8]. The components of the RNA-binding complex were identified as the catalytic and regulatory subunits of the holoenzyme PKA as well as the PKA anchor protein AKAP121 [10,20].
LPL translation in adipocytes is also regulated by changes in PKC. Although the acute activation of PKC by the phorbol ester PMA did not affect LPL, the depletion of PKC isoforms by prolonged PMA treatment, or the inhibition of PKCα mRNA and protein expression with an antisense oligomer to PKCα resulted in an inhibition of LPL translation [11,28]. This effect was specific to PKCα and depletion of other PKC isoforms did not inhibit LPL activity [11]. Although previous studies demonstrated the translational regulation of LPL by PKCα depletion, and suggested the presence of RNA binding protein(s) involving the 3′UTR of the LPL mRNA, the nature of the binding proteins, and the precise region of the 3′UTR involved in regulation, was unknown.
In the present study we hypothesized that there could be an interaction between the PKA and PKCα-mediated mechanism of regulation of LPL translation. Other functional interactions between PKA and PKC isoforms in the regulation of mitogenic and anti-proliferative pathways have been reviewed recently [12]. To identify the region on the LPL 3′UTR and the RNA-binding proteins involved in PKCα-depletion mediated translational regulation of LPL, we used in vitro translation with a number of specific LPL mRNA constructs. Cytoplasmic extracts from adipocytes treated with antisense oligomers to PKCα were added to an in vitro translation reaction and demonstrated that the minimum essential region required for translational inhibition was the first 39 nt of LPL 3′UTR. Neither the 5′UTR nor the distal 3′UTR participated in the translational inhibition of LPL, and the addition of LPL 3′UTR sequence to an irrelevant coding region conveyed translational regulation in response to PKCα depletion. In addition, an RNA fragment spanning nt 1512–1663 of LPL cDNA provided a specific RNase T1-protected gel shift in the presence of the cytoplasmic extract from PKCα-depleted adipocytes. These results identify the essential role of the proximal 3′UTR in PKCα-mediated regulation of LPL translation. To identify the proteins involved in PKCα depletion-mediated translational inhibition, an affinity column was prepared using the same region of the LPL mRNA. Because this region of the LPL mRNA is also involved in adrenaline-mediated translational inhibition, we examined the elution from this column for PKA subunits and AKAP 121. PKCα-depleted extracts contained easily identifiable quantities of both the PKA Cα subunit and AKAP121, which were bound to the RNA affinity column. We have demonstrated the specificity of the LPL 3′UTR in earlier studies using an irrelevant mRNA sequence linked to Oligo-dT–Sepharose [20]. In addition, the PKCα-depleted cells demonstrated higher levels of cAMP and PKA activity, along with higher levels of lipolysis, all of which is consistent with an activation of PKA. The time course of the depletion of PKCα mRNA following treatment with specific antisense oligomers was similar to the time course of PKA activation. Together, these results suggest that PKCα depletion results in an inhibition of LPL translation at least in part through an activation of PKA, and the formation of the same RNA binding complex demonstrated previously for PKA [20]. A number of functional interactions between the PKA and PKC pathways have been described previously. In hepatocytes, insulin-induced activation of PKCα decreased secretin-stimulated cAMP and PKA activity [29]. In addition to our studies of LPL, other studies have demonstrated inverse interactions between PKA and PKC in mitogenic and anti-proliferative pathways [12,13]. Other examples of these opposite effects of PKA and PKC occur in the RAF1/MEK [mitogen-activated protein kinase/ERK (extracellular-signal-regulated kinase) kinase]/ERK pathway where PKCα activation directly phosphorylates and activates Raf1 [30], whereas PKA activation blocks Raf1 activity by blocking its interaction with Ras [14].
Although PKC isoforms are important in many aspects of lipid and carbohydrate metabolism, the role of PKC in the physiological regulation of LPL is not clear. PKCα is a typical PKC isoform that is stimulated by DAG (diacylglycerol). In the environment of insulin resistance, obesity and diabetes, blood FFAs and DAG levels are chronically elevated. Although an acute elevation of DAG would probably stimulate PKCα, it is possible that the continuous elevation of DAG may result in a depletion of adipocyte PKCα. Indeed, the decrease in LPL activity that is found in diabetes is in part due to a decrease in LPL translation, which is improved by treatment of the metabolic condition [26]. Since poorly-controlled diabetes is associated with elevated FFA levels, this translational mechanism for LPL inhibition in diabetes suggests a possible involvement of the PKC pathway.
The reciprocal processes of adipogenesis and enzymatic hydrolysis of stored lipid in adipose tissue is a highly regulated process. Catecholamines trigger lipolysis in adipose tissue through up-regulation of adenyl cyclase and PKA activation, resulting in the suppression of LPL-mediated lipogenesis and the release of FFAs for utilization by peripheral tissues during fasting and exercise. The PKC pathway probably modulates this response, and the results in the present study suggest that the depletion of PKCα also resulted in the increase in cAMP levels and activation of PKA, which subsequently triggers lipolysis and inhibits LPL in adipocytes. This process may further augment the release of FFAs from adipocytes in the hyperglycaemic, hypoinsulinaemic state.
Acknowledgments
We wish to thank Ms Xiaohua Qiu for her expert technical assistance in cell culture and suggestions during the course of the experiments. This work was supported by a Merit Review Grant from the Veterans Administration (to G.R.), and R37 DK 39176 from the National Institute of Health (to P.A.K.).
Abbreviations used
- AKAP
A-kinase-anchoring protein
- DAG
diacylglycerol
- DMEM
Dulbecco's modified Eagle's medium
- FFA
free fatty acid
- LPL
lipoprotein lipase
- PKA
protein kinase A
- PKC
protein kinase C
- UTR
untranslated region
References
- 1.Brunzell JD, Hazzard WR, Porte D, Jr, Bierman EL. Evidence for a common, saturable, triglyceride removal mechanism for chylomicrons and very low density lipoproteins in man. J Clin Invest. 1973;52:1578–1585. doi: 10.1172/JCI107334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merkel M, Eckel RH, Goldberg IJ. Lipoprotein lipase: genetics, lipid uptake, and regulation. J Lipid Res. 2002;43:1997–2006. doi: 10.1194/jlr.r200015-jlr200. [DOI] [PubMed] [Google Scholar]
- 3.Preiss-Landl K, Zimmermann R, Hammerle G, Zechner R. Lipoprotein lipase: the regulation of tissue specific expression and its role in lipid and energy metabolism. Curr Opin Lipidol. 2002;13:471–481. doi: 10.1097/00041433-200210000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Zechner R. The tissue-specific expression of lipoprotein lipase: implications for energy and lipoprotein metabolism. Curr Opin Lipidol. 1997;8:77–88. doi: 10.1097/00041433-199704000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Enerbäck S, Gimble JM. Lipoprotein lipase gene expression: Physiological regulators at the transcriptional and post-transcriptional level. Biochim Biophys Acta. 1993;1169:107–125. doi: 10.1016/0005-2760(93)90196-g. [DOI] [PubMed] [Google Scholar]
- 6.Eckel RH, Jensen DR, Schlaepfer IR, Yost TJ. Tissue-specific regulation of lipoprotein lipase by isoproterenol in normal-weight humans. Am J Physiol. 1996;271:1280–1286. doi: 10.1152/ajpregu.1996.271.5.R1280. [DOI] [PubMed] [Google Scholar]
- 7.Ong JM, Saffari B, Simsolo RB, Kern PA. Epinephrine inhibits lipoprotein lipase gene expression in rat adipocytes through multiple steps in posttranscriptional processing. Mol Endocrinol. 1992;6:61–69. doi: 10.1210/mend.6.1.1738372. [DOI] [PubMed] [Google Scholar]
- 8.Yukht A, Davis RC, Ong JM, Ranganathan G, Kern PA. Regulation of lipoprotein lipase translation by epinephrine in 3T3-L1 cells - importance of the 3′ untranslated region. J Clin Invest. 1995;96:2438–2444. doi: 10.1172/JCI118301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ranganathan G, Vu D, Kern PA. Translational regulation of lipoprotein lipase by epinephrine involves a transacting binding protein interacting with the 3′ untranslated region. J Biol Chem. 1997;272:2515–2519. doi: 10.1074/jbc.272.4.2515. [DOI] [PubMed] [Google Scholar]
- 10.Ranganathan G, Pokrovskaya I, Ranganathan S, Kern PA. Role of A kinase anchor proteins in the tissue specific regulation of lipoprotein lipase. Mol Endocrinol. 2005;19:2527–2534. doi: 10.1210/me.2005-0144. [DOI] [PubMed] [Google Scholar]
- 11.Ranganathan G, Song W, Dean N, Monia B, Barger SW, Kern PA. Regulation of lipoprotein lipase by protein kinase Cα in 3T3-F442A adipocytes. J Biol Chem. 2002;277:38669–38675. doi: 10.1074/jbc.M206917200. [DOI] [PubMed] [Google Scholar]
- 12.Maioli E, Torricelli C, Fortino V. Functional interactions of protein kinase A and C in signalling networks: a recapitulation. Cell Mol Life Sci. 2006;63:637–641. doi: 10.1007/s00018-005-5541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortino V, Torricelli C, Gardi C, Valacchi G, Rossi PS, Maioli E. ERKs are the point of divergence of PKA and PKC activation by PTHrP in human skin fibroblasts. Cell Mol Life Sci. 2002;59:2165–2171. doi: 10.1007/s000180200015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dumaz N, Marais R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem. 2003;278:29819–29823. doi: 10.1074/jbc.C300182200. [DOI] [PubMed] [Google Scholar]
- 15.Dean NM, McKay R, Miraglia L, Geiger T, Müller M, Fabbro D, Bennett CF. Antisense oligonucleotides as inhibitors of signal transduction: development from research tools to therapeutic agents. Biochem Soc Trans. 1996;24:623–629. doi: 10.1042/bst0240623. [DOI] [PubMed] [Google Scholar]
- 16.Ranganathan G, Ong JM, Yukht A, Saghizadeh M, Simsolo RB, Pauer A, Kern PA. Tissue-specific expression of human lipoprotein lipase. Effect of the 3′-untranslated region on translation. J Biol Chem. 1995;270:7149–7155. doi: 10.1074/jbc.270.13.7149. [DOI] [PubMed] [Google Scholar]
- 17.Wion KL, Kirchgessner TG, Lusis AJ, Schotz MC, Lawn RM. Human lipoprotein lipase complementary DNA sequence. Science. 1987;235:1638–1641. doi: 10.1126/science.3823907. [DOI] [PubMed] [Google Scholar]
- 18.Kern PA, Ranganathan G, Yukht A, Ong JM, Davis R. The translational regulation of lipoprotein lipase by thyroid hormone is via a cytoplasmic repressor that interacts with the 3′ untranslated region. J Lipid Res. 1996;37:2332–2340. [PubMed] [Google Scholar]
- 19.Ranganathan G, Li C, Kern PA. The translational regulation of lipoprotein lipase in diabetic rats involves the 3′-untranslated region of the lipoprotein lipase mRNA. J Biol Chem. 2000;275:40986–40991. doi: 10.1074/jbc.M008775200. [DOI] [PubMed] [Google Scholar]
- 20.Ranganathan G, Phan D, Pokrovskaya ID, McEwen JE, Li C, Kern PA. The translational regulation of lipoprotein lipase by epinephrine involves an RNA binding complex including the catalytic subunit of protein kinase A. J Biol Chem. 2002;277:43281–43287. doi: 10.1074/jbc.M202560200. [DOI] [PubMed] [Google Scholar]
- 21.Ranganathan S, Kern PA. Thiazolidinediones inhibit lipoprotein lipase activity in adipocytes. J Biol Chem. 1998;273:26117–26122. doi: 10.1074/jbc.273.40.26117. [DOI] [PubMed] [Google Scholar]
- 22.Nilsson-Ehle P, Schotz MC. A stable radioactive substrate emulsion for assay of lipoprotein lipase. J Lipid Res. 1976;17:536–541. [PubMed] [Google Scholar]
- 23.Eckel RH. Adipose tissue lipoprotein lipase. In: Borensztajn J, editor. Lipoprotein Lipase. Evener; Chicago: 1987. pp. 79–132. [Google Scholar]
- 24.Maheux P, Azhar S, Kern PA, Chen YD, Reuven GM. Relationship between insulin-mediated glucose disposal and regulation of plasma and adipose tissue lipoprotein lipase. Diabetologia. 1997;40:850–858. doi: 10.1007/s001250050759. [DOI] [PubMed] [Google Scholar]
- 25.Reynisdottir S, Angelin B, Langin D, Lithell H, Eriksson M, Holm C, Arner P. Adipose tissue lipoprotein lipase and hormone-sensitive lipase: contrasting findings in familial combined hyperlipidemia and insulin resistance syndrome. Arterioscler Thromb Vasc Biol. 1997;17:2287–2292. doi: 10.1161/01.atv.17.10.2287. [DOI] [PubMed] [Google Scholar]
- 26.Simsolo RB, Ong JM, Saffari B, Kern PA. Effect of improved diabetes control on the expression of lipoprotein lipase in human adipose tissue. J Lipid Res. 1992;33:89–95. [PubMed] [Google Scholar]
- 27.Doolittle MH, Ben-Zeev O, Elovson J, Martin D, Kirchgessner TG. The response of lipoprotein lipase to feeding and fasting. Evidence for posttranslational regulation. J Biol Chem. 1990;265:4570–4577. [PubMed] [Google Scholar]
- 28.Ranganathan G, Kaakaji R, Kern PA. Role of protein kinase C in the translational regulation of lipoprotein lipase in adipocytes. J Biol Chem. 1999;274:9122–9127. doi: 10.1074/jbc.274.13.9122. [DOI] [PubMed] [Google Scholar]
- 29.Lesage GD, Marucci L, Alvaro D, Glaser SS, Benedetti A, Marzioni M, Patel T, Francis H, Phinizy JL, Alpini G. Insulin inhibits secretin-induced ductal secretion by activation of PKCα and inhibition of PKA activity. Hepatology. 2002;36:641–651. doi: 10.1053/jhep.2002.35537. [DOI] [PubMed] [Google Scholar]
- 30.Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme DRU. Protein kinase Cα activates RAF-1 by direct phosphorylation. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]