

Abstract
Perfluorooctanesulfonamides, such as N-ethyl perfluorooctanesulfonamidoethanol (N-EtFOSE), are large scale industrial chemicals but their disposition and toxicity are poorly understood despite significant human exposure. The hypothesis that subacute exposure to N-EtFOSE, a weak peroxisome proliferator, causes a redox imbalance in vivo was tested using the known peroxisome proliferator, ciprofibrate, as a positive control. Female Sprague-Dawley rats were treated orally with N-EtFOSE, ciprofibrate or corn oil (vehicle) for 21 days, and levels of N-EtFOSE and its metabolites as well as markers of peroxisome proliferation and oxidative stress were assessed in serum, liver and/or uterus. The N-EtFOSE metabolite profile in liver and serum was in good agreement with reported in vitro biotransformation pathways in rats and the metabolite levels decreasing in the order perfluorooctanesulfonate ≫ perfluorooctanesulfonamide ∼ N-ethyl perfluorooctanesulfonamidoacetate ≫ perfluorooctanesulfonamidoethanol ∼ N-EtFOSE. Although N-EtFOSE treatment significantly decreased the growth rate, increased relative liver weight and activity of superoxide dismutases (SOD) in liver and uterus (total SOD, CuZnSOD and MnSOD), a metabolic study revealed no differences in the metabolome in serum from N-EtFOSE-treated and control animals. Ciprofibrate treatment increased liver weight and peroxisomal acyl Co-A oxidase activity in the liver and altered antioxidant enzyme activities in the uterus and liver. According to NMR metabolomic studies, ciprofibrate treated animals had altered serum lipid profiles compared to N-EtFOSE-treated and control animals, whereas putative markers of peroxisome proliferation in serum were not affected. Overall, this study demonstrates the biotransformation of N-EtFOSE to PFOS in rats that is accompanied by N-EtFOSE-induced alterations in antioxidant enzyme activity.
Keywords: Metabolomics, perfluorooctanesulfonamides, perfluorooctanesulfonate, peroxisomal acyl Co-A oxidase, superoxide dismutase
Introduction
Perfluorooctanesulfonic acid (PFOS)-based chemicals are a novel and unique group of environmental contaminants that were manufactured for over 50 years by electrochemical fluorination of octanesulfonyl fluoride (Kissa, 2001; Lehmler, 2005). The perfluorooctanesulfonyl fluoride produced by this process was subsequently converted into N-alkylated perfluorooctanesulfonamide intermediates needed for the large range of industrial and consumer applications, for example coatings for carpets, textiles and paper products. These applications utilize unique properties imparted by the perfluorooctyl chain, including its lipophobic and hydrophobic character, and its resistance towards biological, chemical and thermal degradation.
As a result of their widespread commercial use, perfluorooctanesulfonamides have been detected in various environmental matrices (Lau et al., 2007; Lehmler, 2005), food samples (Tittlemier et al., 2007), wildlife (Giesy and Kannan, 2001; Lau et al., 2007; Lehmler, 2005) and in humans (Calafat et al., 2007; Fromme et al., 2009; Kannan et al., 2004; Kärrman et al., 2006a; Lau et al., 2007; Spliethoff et al., 2008). Several studies suggest that perfluorooctanesulfonamides are either degraded in the environment (Paul et al., 2009) or undergo biotransformation in wildlife and humans (Tomy et al., 2004; Xu et al., 2004; Xu et al., 2006). The ultimate product of these processes is PFOS, a highly persistent and ubiquitous environmental contaminant. As an example, a simplified biotransformation pathway of N-EtFOSE (N-ethyl perfluorooctanesulfonamidoethanol) with the chemical structure and names of putative metabolites in humans and in rats is shown in Figure 1. Due to environmental concerns arising from these observations, the main manufacturer of these compounds in the United States, the 3M Corporation, phased out production of PFOS-based chemicals between 2000 and 2002.
Figure 1.

Simplified biotransformation pathways of N-EtFOSE: PFOS is the major metabolite of N-EtFOSE detected in the liver and serum of female rats. Several other metabolites, including FOSA and N-EtFOSAA, were also detected in agreement with the N-EtFOSE biotransformation pathway proposed by Xu and co-workers (Xu et al., 2004). Major metabolites are shown in bold. Metabolites shown in parentheses were either not detected (N-EtFOSA) or not analyzed (FOSAA). O- and N-glucuronide metabolites are omitted for clarity reasons. N-EtFOSE was administered orally over a three week period, animals were euthanized on day 21 and liver and serum samples were analyzed for selected metabolites as described under Materials and Methods. The chemical names of the metabolites are listed under Methods and Materials.
Despite the ubiquitous environmental contamination with PFOS-based compounds, only limited information is available about the mammalian toxicity of perfluorooctanesulfonamides, including N-EtFOSE. Teratological findings with N-EtFOSE in the rat and rabbit are unremarkable (Case et al., 2001) and comparable to the effect of PFOS (Lau et al., 2007). Acute exposure to N-EtFOSE has no significant effect on markers of peroxisome proliferation in male rats (e.g., changes in liver and body weight, lauroyl CoA oxidase enzyme activity or serum cholesterol) (Berthiaume and Wallace, 2002), whereas subacute exposure for four days causes peroxisome proliferation in male and female rats (Wallace et al., 2001). The structurally related PFOS is also a weak peroxisome proliferator in rats (Berthiaume and Wallace, 2002; Haughom and Spydevold, 1992) and mice (Sohlenius et al., 1993).
Several mechanisms have been proposed for the peroxisome proliferative effect of N-EtFOSE and other PFOS-based chemicals. Human and rodent peroxisome proliferator-activated receptors α (PPARα), a transcription factor that is involved in lipid storage, transport and metabolism, can be activated by perfluorooctanesulfonamides and related compounds (Maloney and Waxman, 1999; Shipley et al., 2004; Takacs and Abbott, 2007; Vanden Heuvel et al., 2006). Another mechanism is the displacement of endogenous ligands (i.e., cellular fatty acids) from rat liver fatty acid-binding protein and subsequent activation of PPARα in tissues by these endogenous ligands (Luebker et al., 2002).
Although N-EtFOSE and PFOS have no effect on endpoints associated with mitochondrial biogenesis in rats (Berthiaume and Wallace, 2002), mechanistic studies suggest that perfluorooctanesulfonamides cause mitochondrial dysfunction, but by different mechanisms and with different potencies (Panaretakis et al., 2001; Starkov and Wallace, 2002). For example, N-EtFOSE, PFOS and perfluorooctanoic acid (PFOA) cause an intrinsic proton leak of the mitochondrial inner membrane, which may be due to a change in the fluidity of the mitochondrial membrane (Hu et al., 2003; Lehmler et al., 2006; Starkov and Wallace, 2002; Xie et al., 2007). Perfluorooctanesulfonamide (FOSA) is a potent uncoupler of oxidative phosphorylation (Schnellmann and Manning, 1990; Starkov and Wallace, 2002), whereas N-ethyl perfluorooctanesulfonamidoacetate (N-EtFOSAA) potently induces the mitochondrial permeability transition (O'Brien and Wallace, 2004; Starkov and Wallace, 2002). Some N-EtFOSE metabolites, such as N-EtFOSAA and perfluorooctanesulfonamidoacetate (FOSAA), stimulate production of reactive oxygen species in isolated mitochondria (O'Brien and Wallace, 2004) and in isolated hepatocytes (Liu et al., 2007). Similarly, PFOA and perfluorodecanoic acid alter redox homeostasis in vitro and in vivo (O'Brien and Wallace, 2004).
In the present study we test the hypothesis that exposure to perfluorooctanesulfonamides causes a redox imbalance in female Sprague-Dawley rats that is associated with peroxisome proliferation and/or mitochondrial dysfunction. N-EtFOSE was selected as a test compound because of its large scale use in commercial products and its occurrence in the environment (Fromme et al., 2009; Lau et al., 2007; Lehmler, 2005). Initially, changes in systemic biomarkers of oxidative stress, mitochondrial dysfunction and peroxisome proliferation were assessed in comparison to ciprofibrate using a NMR metabolomic approach. Ciprofibrate was selected because its hepatotoxicity has been studied in parallel with other perfluorochemicals (Borges et al., 1993; Chen et al., 1994; Glauert et al., 1992; Huang et al., 1994; Wilson et al., 1995). In addition, the activities of peroxisomal acyl Co-A oxidase (AOX) and selected antioxidant enzymes were measured in the liver, a known target organ of perfluorinated compounds. Because of the developmental concerns associated with the exposure to N-EtFOSE and related compounds (Lau et al., 2007), selected enzyme antioxidant activities were also measured in the uterus to assess if an altered redox homeostasis in utero could potentially contribute to the developmental toxicity of perfluorochemicals.
Materials and methods
Chemicals
Perfluorooctanoic acid (PFOA, 98%) and potassium salt of perfluorooctane sulfonic acid (PFOS, >98%) were purchased from TCI (Portland, OR). N-Ethyl perfluorooctanesulfonamidoethanol (N-EtFOSE; >99%), N-ethyl perfluorooctanesulfonamide (N-EtFOSA; >99%, 0.15 % PFOA), N-ethyl perfluorooctanesulfonamidoacetate (N-EtFOSAA; >92%, 7.2 % PFOS and 0.5 % PFOA), perfluorooctanesulfonamidoethanol (FOSE; > 90%, 6.0 % PFOA and 4.2 % PFOA) and perfluorooctanesulfonamide (FOSA; > 75%, 20.1 % PFOS and 4.1% PFOA) were synthesized as described previously (Lehmler et al., 2007). Their PFOS and PFOA contents were determined using a 100 ppb standard solution in methanol as described below. The chemical structure of these compounds with the respective abbreviations is shown in Figure 1. 13C4-PFOS, 13C4-PFOA (99% purity, Wellington Laboratories, Guelph, ON, Canada) were used as surrogate standards. Ciprofibrate was generously provided by Sanofi Pharmaceuticals (Malvern, PA) and corn oil was purchased from Acros Organics (Morris Plains, NJ).
Animals and treatment
All experimental protocols were approved by the Institutional Animal Care and Use Committee of The University of Iowa. The study was designed to resemble previous studies investigating the effect of perfluorochemicals, such as perfluorodecanoic acid, on peroxisome proliferation and oxidative stress (Borges et al., 1993; Chen et al., 1994; Glauert et al., 1992; Huang et al., 1994; Wilson et al., 1995). Twenty six sexually mature female Sprague-Dawley rats (10 weeks, 220±20 g; Harlan, Indianapolis, IN) were placed in cages in a controlled environment, maintained at 22°C with a 12 hour light–dark cycle, and with food and water ad libitum. After a one week conditioning period, rats were randomly divided into three treatment groups and treated with the respective test compound by gastric intubation. One group (n=9) received 5.0 mg/kg body weight of N-EtFOSE in corn oil (2 mL/kg body weight) for a period of 21 days (once daily for 5 days/week, no administration on days 6 and 7). The average total N-EtFOSE dose was 16.3±0.7 mg/animal (28.6±1.3 μmol/per animal). The negative control group was treated analogously with 2 ml/kg body weight corn oil (n=9). A third group (n=8) was given a daily dose of ciprofibrate (7.5 mg/kg/day) suspended in 0.5 ml water and methylcellulose for 21 days (Hammer et al., 1998). Both the N-EtFOSE and the ciprofibrate doses were expected to cause general toxicity.
General toxicity and pathological examination
Rats were monitored regularly for clinical signs of toxicity and body weight was recorded daily. The animals were weighed and euthanatized by CO2 asphyxiation three days after the last N-EtFOSE administration (negative control and N-EtFOSE treatment group) or one day after the last ciprofibrate administration (ciprofibrate treatment group). Blood was collected by cardiac puncture, serum was isolated by centrifugation and aliquots were stored at -80 °C. Kidney, liver, spleen and uterus were excised, weighed, and the organ body weight ratios were calculated (Table 1). Small aliquots of the liver and uterus were either homogenized immediately in a 5% 5-sulfosalicylic acid buffer for the determination of total and oxidized glutathione or fixed in a 10% formalin solution. The remaining parts of the liver and uterus were frozen in liquid nitrogen and stored at -80 °C for future measurements. Formalin fixed tissues were embedded in paraffin in the Comparative Pathology Research Laboratory of the Carver College of Medicine at The University of Iowa and hematoxylin and eosin stained sections (thickness: 4 μm) were evaluated in a blinded fashion by a pathologist (J.W.).
Table 1.
Effect of N-EtFOSE on body and organ weights.
| Tissue | Control | N-EtFOSE | Ciprofibratea |
|---|---|---|---|
| Final body weight (g) | 235 ± 11 | 228 ± 9 | 231 ± 9 |
| Growth rateb | 12.7 ± 2.7 | 8.3 ± 0.9** | 9.9 ± 0.6 |
| Organ weight (g) | |||
| Liver | 8.7 ± 1.1 | 9.2 ± 0.08 | 13 ± 0.97*** |
| Kidney | 1.48 ± 0.14 | 1.54 ± 0.09 | 1.69 ± 0.05*** |
| Spleen | 0.60 ± 0.03 | 0.63 ± 0.07 | 0.58 ± 0.04 |
| Uterus | 0.39 ± 0.08 | 0.37 ± 0.07 | 0.31 ± 0.06 |
| Ratio of Organ Weight to Body Weight (%) | |||
| Liver | 3.7 ± 0.3 | 4.1 ± 0.2* | 5.9 ± 0.3*** |
| Kidney | 6.3 ± 0.5 | 6.8 ± 0.4 | 7.3 ± 0.3*** |
| Spleen (× 10-3) | 2.5 ± 0.1 | 2.7 ± 0.2* | 2.5 ± 0.2 |
| Uterus | 1.7 ± 0.4 | 1.6 ± 0.3 | 1.5 ±0.4 |
7.5 mg ciprofibrate/kg/day suspended in 0.5 ml water and methylcellulose was administered by oral gavage continuously for 3 weeks.
Growth rate = body weight (end exposure) – body weight (begin exposure)/body weight (end exposure) × 100.
p < 0.05
p < 0.001
1H NMR analysis of serum
A NMR metabolomic approach was used to assess changes in systemic biomarkers of oxidative stress, mitochondrial dysfunction and/or peroxisome proliferation. In short, serum samples (500 μL) were gently thawed and diluted with 500 μL phosphate buffer in D2O (0.2 M, pH 7.4; 5 mM sodium 3-trimethylsilyl-2,2,3,3,-d4-propionate, TSP) as described previously (Viant, 2007). The pH change was minimal after the dilution. The 1H chemical shifts were referenced internally to TSP at 0.0 ppm. TSP also functioned as a concentration standard with a sample concentration of 2.5 mM. All the NMR experiments were performed on an AVANCE 600 instrument with a conventional 5 mm triple resonance inverse probe (Bruker Biospin, Billerica, MA, USA) at 295±0.2 K. Samples (600 μL) were analyzed in random order by 1-D 1H NMR with water presaturation, 1-D Carr Purcell Meiboom Gill (CPMG) spin-echo and homonuclear two dimensional J-resolved 1H NMR spectroscopy (JRES) (Viant, 2007). The programs from the Bruker standard pulse sequence library were used for these experiments. The residual HOD signal was presaturated in all the experiments during the relaxation period. The experimental parameters are as follows. Single pulse experiment: spectral width (SW), 9 kHz; time domain data points (TD), 64k; 60° pulse width (PW), 5.9us; relaxation delay (RD), 2.5 s; number of scans (NS), 128. CPMG experiment: SW, 9 kHz; TD, 64k; 90° PW, 9.8us; spin echo delay, 200ns; total spin-spin relaxation delay, 80 ms; RD, 3.5 s; NS, 128, dummy scans (DS), 8. 2D HOMO-J: SW (F2), 9 kHz; SW (F1), 63 Hz; TD2, 8k; TD1, 64; 90o PW, 9.8us; RD, 2 s; NS, 8; DS, 8. All the 1D spectra were processed with zero-filling to 128k data points followed by manual phase and baseline (quadratic) corrections. For the 2D spectra, the F1-data were zero-filled to 1024 data points and multiplied by sine-bell functions followed by Fourier transformation. The 2D spectra were then tilted by 45° and symmetrized about the F1 axis. The 1H NMR data were initially analyzed by manually integration of the CPMG and 1H-JRES spectra, followed by Principal Component Analysis (PCA) as a data reduction tool. Subsequently, NMR signals that were significantly different between treatment groups in the CPMG spectra were analyzed in the 1-D 1H NMR spectra with water presaturation and the 1H-JRES spectra to confirm our findings. Representative spectra and a plot of the loadings of each spectral region are shown in Figures S1 to S4 in the Supplementary Material.
Determination of levels of N-EtFOSE and metabolites in liver and serum
Extraction of N-EtFOSE and Metabolites
N-EtFOSE, N-EtFOSA, N-EtFOSAA, FOSE, PFOS, PFOA, and PFOSA were analyzed in rat serum and liver samples using an ion-pairing extraction procedure and were determined by using of a high-performance liquid chromatograph (HPLC) with an electrospray tandem mass spectrometer (ESI-MS/MS) (Kannan et al., 2004). Briefly, 0.2 mL of serum or 1 mL of liver homogenate were added with internal standards (13C4-PFOS, 13C4-PFOA at 5 ng for serum samples and 10 ng for liver samples), 2 mL of 0.25 M sodium carbonate/sodium bicarbonate buffer, and 1 mL of 0.5 M tetrabutylammonium (TBA) hydrogensulfate solution (adjusted to pH 10). After the resulting mixture was vortexed, 5 mL of methyl-tert-butyl ether (MTBE) was added for extraction followed by shaking vigorously for 45 min. The MTBE layer was separated by centrifugation at 3500 rpm for 3 min and then transferred to a polypropylene tube. Another 3 mL of MTBE was added to the aqueous phase and the procedure was repeated. The two MTBE fractions were combined and gently evaporated to dryness using a stream of dry nitrogen and then reconstituted with methanol (0.5 mL methanol for serum samples and 1 mL methanol for liver samples). The samples were vortexed for 60 sec and than transferred to glass GC vials.
Instrumental analysis
N-EtFOSE and its metabolites were analyzed and quantified using an Agilent 1100 series high performance liquid chromatography (HPLC) coupled with an Applied Biosystems API 2000 electrospray triple-quadruple mass spectrometer (ESI-MS/MS). Ten μL of the extract was injected onto a 100 mm × 2.1 mm Betasil C18 column (5 μm; Thermo Electron Corporation, Bellefonte, PA). The mobile phase was 2 mM ammonium acetate/methanol starting at 10% methanol at a flow rate of 300 μL/min. The gradient increased to 99% methanol at 9 min and then was held for 5 min before reversed back to 10% methanol. The tandem mass spectrometer was operated in electrospray negative ionization mode. Target compounds were determined by multiple reactions monitoring (MRM). The MRM transitions were 499>99 for PFOS, 503>99 for 13C4-PFOS, 497.7>77.7 for PFOSA, 413>369 for PFOA, 417>372 for 13C4-PFOA, 583.7>82.9 for N-EtFOSAA, 526.1>169.1 for N-EtFOSA, 542>65 for FOSE and 629.9>59.2 for N-EtFOSE. When possible, multiple daughter ions were monitored for confirmation, but quantitation was based on a single product ion. In all cases, the capillary was held at -4 kV and the desolvation temperature was kept at 400°C.
Limits of quantitation
The limits of quantitation (LOQ) were determined based on the linear range of calibration curves prepared at a concentration range of 0.1 to 100 ng/mL. Concentrations in samples which were at least three-fold greater than the lowest acceptable standard concentration were considered to be valid. A curve point was deemed acceptable if (1) it was back-calculated to be within 30% of the theoretical value when evaluated versus the 1/× weighted curve, and (2) the peak area of the standard was at least 3 times greater than that in the blank. Concentration/dilution factors were not included in the calculation of the LOQ. The LOQ for N-EtFOSE, N-EtFOSA, N-EtFOSAA, FOSA, PFOA was 0.5 ng/mL, and for FOSE and PFOS it was 0.1 and 1.0 ng/mL, respectively.
Quality assurance and quality control
13C4-PFOS and 13C4-PFOA (99% purity, Wellington Laboratories, Guelph, ON, Canada) were spiked as internal standards into all blanks and samples prior to the addition of reagents for extraction. Recoveries of 13C4-PFOS in serum samples and liver samples were 76±27% and 67±22%, respectively; recoveries of 13C4-PFOA in serum samples and liver samples were 116±18% and 125±6%, respectively. PFOA levels (adjusted for recovery of 13C4-PFOA) were 5.4 ± 0.8 and 0.7 ± 0.1 ppb in the liver and serum of N-EtFOSE treated animals. PFOA background levels were 0.7 ± 0.1 ppb in the liver of control animals, whereas not PFOA was detected in control serum.
Matrix spikes (two liver samples and one serum sample) were performed. Known amounts of mixed PFC standards (10 ng for serum sample and 20 ng each for liver samples) were spiked into sample matrices before extraction and were passed through the entire analytical procedure. Blanks were analyzed by passing Milli-Q water and reagents through the whole analytical procedure. A midpoint calibration standard was injected after every 5 samples to check the stabilization of instrumental response and drift. Calibration standards were injected daily before and after the analysis. The matrix recovery rates from liver were from 55 to 158 %, except for N-EtFOSE and N-EtFOSA in liver in which these two compounds were recovered at 2.5±1% and 18.5±1.5%, respectively (n=2). The recoveries of all target analytes in serum were between 67 and 159%. The reported concentrations in liver and serum were corrected for recoveries.
Enzyme activity assays
Activities of antioxidant enzymes of liver and uterus, expressed per milligram of protein, were assayed by the Antioxidant Enzyme Services of the Radiation and Free Radical Research Core of The University of Iowa's Holden Comprehensive Cancer Center.
Peroxisomal acyl Co-A oxidase activity
AOX activity in liver tissue homogenates was determined fluorometrically using the lauroyl CoA-dependent production of hydrogen peroxide as described previously (Poosch and Yamazaki, 1986). AOX activity is expressed as nmol hydrogen peroxide formed per minute per milligram protein. The protein concentration was determined using Pierce bicinchoninic acid protein determination kit (Pierce, Rockford, IL).
Superoxide dismutase
Total SOD activity was determined using the assay established by Spitz and Oberley (Spitz and Oberley, 1989). In this competitive inhibition assay the superoxide anion radical generated by xanthine-xanthine oxidase is detected by monitoring the reduction of nitroblue tetrazolium colorimetrically at 560 nm. One unit of SOD activity is defined as the amount of protein that yields 50% of maximal inhibition of nitroblue tetrazolium reduction by superoxide anion radicals. Manganese superoxide dismutase (MnSOD) activity was measured by inhibition of copper zinc superoxide dismutase (CuZnSOD) activity in the presence of 5 mM cyanide. CuZnSOD activity was subsequently determined by subtracting MnSOD activity from total SOD activity. Specific activity data for total SOD, MnSOD and CuZnSOD are presented as units of SOD activity per milligram of protein as determined by the method of Lowry et. al. (Lowry et al., 1951).
Catalase
Catalase activity was determined using the colorimetric assay established by Beers and Sizer (Beers and Sizer, 1952). In short, H2O2 was added to tissues homogenate in potassium phosphate buffer (pH 7.8), and the disappearance of H2O2 was measured at 240 nm for 2 min at 15 s intervals. Catalase activity was expressed as milli-k units per milligram of protein as described by Aebi (Aebi, 1984) using protein values obtained from the Lowry protein assay.
Glutathione peroxidases
Total GPx activity was detected using hydroperoxide to determine Se-dependent activity (GPx-1) or cumene hydroperoxide to determine total activity (Lawrence and Burk, 1976). In this assay, GPx catalyzes the oxidation of reduced glutathione to glutathione disulfide by hydroperoxide or cumene hydroperoxide. In the presence of glutathione reductase and NADPH, glutathione disulfide is immediately converted to glutathione with a concomitant oxidation of NADPH to NADP+. The decrease in NADPH was measured at 340 nm and total GPx activity is expressed as milliunits of GPx activity per milligram of protein determined by the method of Lowry.
Levels of glutathione and thiobarbituric acid reactive substances (TBARS)
Total and oxidized glutathione levels were determined in whole liver homogenate using the recycling method of Anderson (Anderson, 1985). Thiobarbituric acid reactive substances (TBARS) were measured fluorometrically in whole liver homogenate according to the method described by Uchiyama and Mihara (Uchiyama and Mihara, 1978).
Statistical analyses
R statistical software (version 2.8.0, http://www.r-project.org) was used for the multivariate and the statistical analyses. Data are presented as mean ± standard deviation if not mentioned otherwise. Outliers were removed prior to statistical analyses based on box plots and the 3 × inter-quartile range criterion. The data were analyzed with ANOVA accompanied by post-hoc Dunnett's test to detect significant differences between treatment groups and control. The results were considered significant if p<0.05. Principal Component Analysis (PCA) as implemented by R statistical software was used to observe groupings of animals from different treatment groups based on the metabolomic profile in serum.
Results
General toxicity and pathology
All rats survived to the end of the study and no clinical findings were observed in female rats from all three treatment groups. Exposure to N-EtFOSE resulted in a significant decrease in the growth rate of female Sprague-Dawley rats compared to controls, whereas ciprofibrate did not significantly alter the growth rate (Table 1). While N-EtFOSE caused a slight, but significant increase in the body weight-adjusted liver weight, the actual liver weight was not significantly increased compared to controls (p=0.41). In contrast, ciprofibrate significantly increased liver and kidney weights compared to controls. Histopathological evaluation of hematoxylin and eosin (H&E) stained liver and uterus slices did not reveal pathological changes in any treatment group (data not shown).
Comparison of the metabolic profile of N-EtFOSE and ciprofibrate in serum
A NMR metabolomic study was performed in an attempt to identify changes in the metabolome of serum resulting from treatment with N-EtFOSE. Initially, CPMG and 1H-JRES NMR spectra were integrated and analyzed by PCA. As illustrated in Figure 2, we retained two principal components (PC) accounting for 64 % (PC1) and 27% (PC2) of the variability in the TSP-adjusted CPMG data. PCA of the CPMG data adjusted for total spectral area gave similar results (data not shown). PC1 separated ciprofibrate-treated animals from control and N-EtFOSE-treated animals. In contrast, no groupings by treatment group were observed using homonuclear two dimensional J-resolved 1H NMR data (data not shown).
Figure 2.
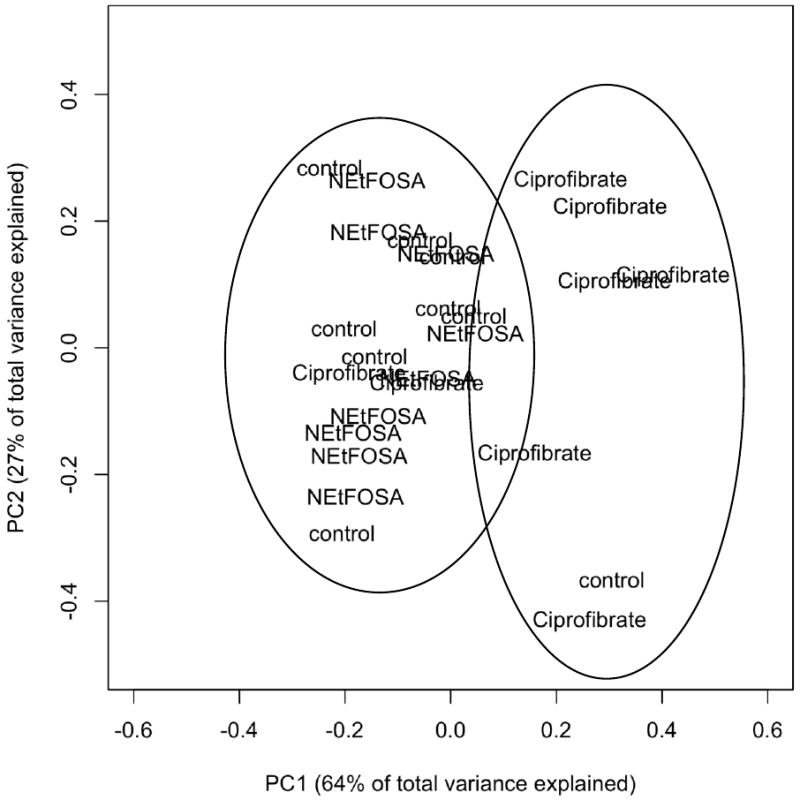
Projections of Principal Component 1 versus Principal Component 2 from the Principal Component Analysis (PCA) of the data from the metabolomic study reveal two groupings: One grouping with ciprofibrate-treated animals and a second grouping of N-EtFOSE-treated and control animal. Serum was mixed in a 1:1 ratio (v/v) with phosphate buffer (pH = 7.4) containing TSP as internal standard and CPMG spectra were recorded on a 600-NMR instrument as described under Materials and Methods. The spectra were integrated manually and adjusted for TSP prior to PCA. Each word-label in the Figure denotes one animal.
Based on the loadings, the signals at δ = 0.96-0.84 (lipid -CH3), 1.34-1.27 (lipid) and 3.33-3.21 ppm (glucose plus -N(CH3)3 of the surface phospholipids (Ala-Korpela, 2008)) were identified as contributing to the groupings in the PCA of the CPMG data. Further statistical analysis showed that these signals were significantly different between ciprofibrate and control animals (p < 0.05), thus suggesting lower levels of lipids with a -N(CH3)3 moiety (e.g., phosphatidylcholine and/or sphingomyelin) in the ciprofibrate treatment group compared to the N-EtFOSE and the negative control groups. Because the CPMG spin-echo sequence is designed to remove broad resonances associated with macromolecules (such as triglycerides and lipid assemblies) we also analyzed the same spectral areas in the water presaturated 1-D 1H NMR and 1H-JRES spectra spectral datasets. Analysis of the 1-D 1H NMR spectra confirmed that animals from the ciprofibrate treatment group have lower lipids levels compared to negative control animals, whereas the 1H-JRES spectra showed no broad lipid signals. The later observation is also consistent with the results of the PCA of this dataset and indirectly confirms a significant difference in lipid levels between ciprofibrate and corn oil-treated animals.
Liver and serum levels of N-EtFOSE and its metabolites
Levels of N-EtFOSE and several putative metabolites were analyzed in liver and serum (Table 2). Levels of N-EtFOSE in serum were in the low ppb range, whereas no N-EtFOSE was detected in serum from control animals. Hepatic N-EtFOSE levels appeared to be in the ppb range. FOSE in both liver and serum samples were also in the low ppb range, and N-EtFOSA was not detected in either matrix. In contrast, levels of N-EtFOSAA were in the low ppm range in the liver and serum. FOSA levels in the liver were also in the low ppm range, but were approximately one order of magnitude lower in serum. The major metabolites detected in both liver and serum was PFOS, with levels that were at least 10-times higher compared to the five perfluorooctanesulfonamides. The liver-to-serum ratios ranged from 1.3 for N-EtFOSAA to 10. 1 for FOSA and decreased in the order FOSA > FOSE > PFOS > N-EtFOSAA (Table 2).
Table 2.
Concentration of N-EtFOSE and its metabolites in liver and serum from female Sprague Dawley rats.
| Tissue and treatment group | Liver (ppb) | Serum (ppb) | |||
|---|---|---|---|---|---|
| N-EtFOSE metabolites |
N-EtFOSE treatment group (n=9) |
Control animalsa (n=5) |
N-EtFOSE treatment group (n=9) |
Control animalsa (n=5) |
Liver: serum ratio |
| N-EtFOSEb | -c | -c | 177 ± 86 | n.d. | - |
| N-EtFOSAAb | 7,773 ± 2,413 | 8.0 ± 4.3 | 5,808 ± 2,132 | 4.1 ± 1.0 (n=3) | 1.3 |
| N-EtFOSAb | n.d. | n.d. | n.d. | n.d. | - |
| FOSEb | 10.0 ± 6.2 | n.d. | 1.6 ± 0.7 | n.d. | 6.4 |
| FOSAb | 4,470 ± 830 | 8.3 ± 6.0 | 441 ± 107 | 0.1 ± 0.1 (n=4) | 10.1 |
| PFOSd | 96,316 ± 11,452 | 64.7 ± 23.8 | 42,511 ± 11,365 | 8.1 ± 1.0 (n=4) | 2.3 |
Average of samples from the negative control (n=3) and the ciprofibrate (n=2) treatment group. One N-EtFOSE-contaminated animal in the ciprofibrate group was excluded from the data analysis. The number of samples above the LOQ is provided in parentheses where appropriate.
Adjusted for the respective matrix spike recovery in serum (N-EtFOSE = 67%; N-EtFOSAA = 159%; N-EtFOSA = 82%; FOSE = 96%; FOSA = 132%) and in the liver (N-EtFOSAA = 156±3%; N-EtFOSA = 19±2%; FOSE = 57±2%; FOSA = 98±3%). See the experimental part for additional details.
Corrected N-EtFOSE levels in the liver are not reported because of the poor recovery rate (2.5±1%); uncorrected N-EtFOSE levels group were low, with 4.6 ± 4.7 ppb in N-EtFOSE-treated animals and 7.8 ± 1.3 ppb in control animals.
Adjusted for the recovery of 13C4-PFOS.
n.d. = not detected.
Activity of peroxisomal acyl Co-A oxidase
Acyl CoA oxidase (AOX) is the rate limiting enzyme of peroxisomal β oxidation and is induced by peroxisome proliferators, such as ciprofibrate (O'Brien et al., 2005). N-EtFOSE increased peroxisomal AOX activity 16-fold compared to control (Figure 3). However, this increase did not reach statistical significance (p = 0.35). Ciprofibrate, a well known peroxisome proliferator (O'Brien et al., 2005), increased AOX activity in the liver 300-fold.
Figure 3.

AOX activity in liver of female Sprague-Dawley treated with N-EtFOSE was not increased, whereas a significant increase was observed in ciprofibrate-treated animals. Data are presented as mean ± S.E.M.. *** p < 0.001.
Effect of N-EtFOSE and ciprofibrate on enzyme activities and markers of oxidative stress
The effect of the N-EtFOSE on the activities of selected antioxidant enzymes, such as superoxide dismutases (SOD), catalase and glutathione peroxidases (GPx's), was assessed in the uterus and liver of female Sprague-Dawley rats (Figures 4 to 6). In addition, the ratio of reduced to oxidized glutathione (GSH:GSSG ratio) and the levels of TBARS were analyzed. Neither N-EtFOSE nor ciprofibrate had an effect on the GSH:GSSG ratio and TBARS in the liver or the GSH:GSSG ratio in the uterus (data not shown).
Figure 4.

N-EtFOSE increased the activity of total SOD, MnSOD and CuZnSOD in (A) the uterus and (B) the liver. The only exception was MnSOD activity in the uterus. In contrast, ciprofibrate significantly decreased MnSOD activity and increased CuZnSOD activity in the uterus. Otherwise, ciprofibrate did not significantly alter SOD activities. Data are presented as mean ± S.E.M.; * p < 0.05; ** p < 0.01; *** p < 0.001.
Figure 6.

N-EtFOSE significantly decreased the activity of total GPx in the uterus (A), but not the liver (B), and had no effect on GPx-1 activity. Ciprofibrate significantly decreased total GPx activity in the liver and uterus and GPx-1 activity in the liver. Data are presented as mean ± S.E.M.. * p < 0.05; ** p < 0.01; *** p < 0.001.
SODs catalyze the dismutation of the superoxide anion radical to hydrogen peroxide. The two important forms of SOD investigated in this study are CuZnSOD and MnSOD, which are found in the cytosol and mitochondria, respectively. CuZnSOD and MnSOD activities were 11- and 1.5-times higher in the liver of control animals compared to the uterus, respectively. N-EtFOSE-treatment significantly increased the activity of total SOD and CuZnSOD in the uterus and of total SOD, MnSOD and CuZnSOD in the liver of female Sprague-Dawley rats (Figure 4). Ciprofibrate significantly decreased MnSOD activity and increased CuZnSOD activity in the uterus, but had not significant effect on total and CuZnSOD activities in the liver. However, hepatic MnSOD activity tended to be increased in ciprofibrate-treated animals (p = 0.07).
Catalase is a heme-containing antioxidant enzyme that decomposes hydrogen peroxide to water and oxygen. In the present study, catalase activity was approximately 17-times higher in the liver compared to the uterus (424±112 versus 24.3±4.6 milli-k units/mg, respectively). Treatment with N-EtFOSE, but not ciprofibrate, significantly increased catalase activity in the uterus. Catalase activity was not significantly altered in the liver of N-EtFOSE or ciprofibrate-treated animals (Figure 5).
Figure 5.

N-EtFOSE significantly increased the activity of catalase in the uterus but had no significant effect on hepatic catalase activity. Ciprofibrate did not alter catalase activity. Data are presented as mean ± S.E.M.. *** p < 0.001.
GPx's are enzymes that catalyze the decomposition of H2O2 and organic hydroperoxides using reducing equivalents from glutathione. Total GPx (using cumene hydroperoxide as substrate) and GPx-1 (using H2O2 as substrate) activities were 2.8- and 3.4-times higher in the liver of control animals compared to the uterus, respectively. No significant change in total GPx or GPx-1 activity was observed in the liver of N-EtFOSE-treated animals (Figure 6). A significant decrease of total GPx, but not GPx-1 activity, was observed in the uterus. Total GPx activity was significantly decreased in the liver and uterus of ciprofibrate treated animals, whereas GPx-1 activity was significantly decreased only in the liver.
Discussion
The present study investigates the effect of N-EtFOSE on the metabolome of female Sprague-Dawley rats as well as antioxidant capacity indicative of redox homeostasis in selected target organs. Oral exposure to N-EtFOSE resulted in a significant decrease in the growth rate of female Sprague-Dawley rats compared to controls, whereas the ciprofibrate did not significantly alter the growth rate (Table 1). Similarly, N-EtFOSE caused a reduced body weight gain in pregnant rats at doses > 5 mg/kg body weight/day in an earlier study (Case et al., 2001). Two structurally related compounds, N-EtFOSA and PFOS, also significantly affect growth rates in rats (Austin et al., 2003; Cui et al., 2009; Curran et al., 2008; Lau et al., 2003; Seacat et al., 2003; Thibodeaux et al., 2003; Zheng et al., 2009) and mice (Era et al., 2009; Lau et al., 2003; Peden-Adams et al., 2007; Thibodeaux et al., 2003; Yahia et al., 2008) in various acute, subchronic, reproductive and developmental exposure regimes.
N-EtFOSE also caused a slight increase in the weight of the liver. Similar effects of N-EtFOSA (Peden-Adams et al., 2007) and PFOS (Curran et al., 2008; Era et al., 2009; Yahia et al., 2008; Zheng et al., 2009) on liver weight have been reported in rodents. Taken together, the similarity of the effect of PFOS-based chemicals on the growth rate and liver weight suggests a common underlying cause for both effects. As expected based on earlier studies (Borges et al., 1993; Chen et al., 1994), the liver weight in the ciprofibrate group was significantly increased compared to controls due to peroxisome proliferation. Despite the changes in organ weights, histopathological evaluation did not reveal any pathological changes in the liver or uterus of the N-EtFOSE treatment group. In contrast, subacute exposure to PFOS resulted in significant histopathological changes in rats, probably due to the higher total dose employed (Cui et al., 2009).
Because N-EtFOSE cause changes in the growth rate and the liver weight, a 1H NMR metabolomic study was performed to identify markers for N-EtFOSE toxicity. Metabolomics is defined as “the quantitative measurement of the multiparametric response of living systems to pathophysiological stimuli or genetic modification” (Nicholson et al., 2002; Nicholson et al., 1999) and provides information about the state and severity of organ dysfunction. Serum was used in our study because it can be used to assess systemic effects of a toxic insult on lipid metabolism (Coen et al., 2003), mitochondrial dysfunction (Lei et al., 2008), peroxisome proliferation (Sheikh et al., 2007) and, potentially, redox homeostasis (Viant et al., 2005). As shown in Figure 2, PCA of the 1H NMR data revealed no differences between corn oil and N-EtFOSE treated animals, despite the fact that N-EtFOSE treatment caused general toxicity (i.e., decrease in the growth rate and increased liver weight; Table 1) at the dose investigated. Ciprofibrate, a well investigated peroxisome proliferator (Borges et al., 1993; Chen et al., 1994; Glauert et al., 1992; Huang et al., 1994; Wilson et al., 1995), did not affect metabolomic markers typically associated with exposure to other peroxisome proliferators, such as amino acids (Sheikh et al., 2007) or, possibly, tryptophan-NAD+ pathway metabolites (Connor et al., 2004; Delaney et al., 2005; Sheikh et al., 2007). However, ciprofibrate treatment resulted in slightly, but significantly higher phosphatidylcholine and/or sphingomyelin levels compared to corn oil- and N-EtFOSE-treated animals. Since ciprofibrate was only employed for comparison purposes, this change in lipid levels and/or composition was not further investigated.
In addition to the analysis of endogenous metabolites in serum, several putative N-EtFOSE metabolites were selected based on established biotransformation pathway of perfluorooctanesulfonamides (Tomy et al., 2004; Xu et al., 2004) and their levels in liver and serum determined (Table 2). The objective of the chemical analysis was to confirm the formation of these metabolites in vivo and to further understand the biological effects of N-EtFOSE treatment. Phase II metabolites, such as O- or N-glucuronides were not analyzed because suitable methods for the extraction and detection of this class of metabolites were not available. However, based on several in vitro studies (Xu et al., 2004; Xu et al., 2006), these metabolites are most likely formed in vivo. We also did not determine FOSAA levels, a major FOSE metabolite in vitro (Xu et al., 2004), because of the unavailability of an analytical standard.
The metabolites observed in both liver and serum support the biotransformation pathway proposed by Xu and co-workers for N-EtFOSE (Xu et al., 2004). As shown in Figure 1, N-EtFOSE was either oxidized to N-EtFOSAA or dealkylated to FOSE. N-EtFOSA, another possible dealkylation product of N-EtFOSE, was not detected in liver or serum. Similarly, the formation of N-EtFOSA from N-EtFOSE was not observed in in vitro metabolism studies (Xu et al., 2004). Subsequently, FOSE was further dealkylated to FOSA. Analogously, N-EtFOSA, a perfluorooctanesulfonamide that was commercially sold as a delayed-action insecticide, has been shown to be rapidly dealkylated to FOSA by liver microsomes from several species (Tomy et al., 2004; Xu et al., 2004) as well as in rats (Arrendale et al., 1989; Manning et al., 1991) and sheep (Vitayavirasuk and Bowen, 1999). FOSA, which has a half-life of approximately 100 hours in rats (Manning et al., 1991) and an elimination phase half-life of 63.4 hours in sheep (Vitayavirasuk and Bowen, 1999), was finally hydrolyzed to PFOS, the ultimate metabolite of N-EtFOSE. Although the hydrolysis of perfluorooctanesulfonamides (such as N-EtFOSA or FOSA) to PFOS has been documented in vitro (Tomy et al., 2004; Xu et al., 2004), this study for the first time documents the in vivo metabolism of N-EtFOSE to PFOS. In fact, PFOS was the major N-EtFOSE metabolite observed and accounted for approximately 9.5±1.2% of the total N-EtFOSE dose in the serum and liver (Table 3). The other N-EtFOSE metabolites accounted for only 1.1±0.3% of the total dose.
Table 3.
Liver and serum levels of N-EtFOSE and its metabolites in female Sprague Dawley rats expressed as percentage of the total N-EtFOSE dose (%TD).a
| N-EtFOSE metabolitesb | %TD in Liver | %TD in Serumc |
|---|---|---|
| N-EtFOSE | -d | 0.42 ± 0.20 (× 10-3) (n=5) |
| N-EtFOSAA | 0.43 ± 0.13 | 0.38 ± 0.17 |
| N-EtFOSA | n.d. | n.d. |
| FOSE | 0.58 ± 0.35 (× 10-3) | 0.07 ± 0.03 (× 10-3) |
| FOSA | 0.29 ± 0.06 | 25.3 ± 6.1 (× 10-3) |
| PFOSe | 6.23 ± 0.95 | 3.86 ± 0.86 |
| Total | 6.96 ± 0.92 | 3.69 ± 0.91 |
The total dose of N-EtFOSE for each animal was calculated as
where 5 is the daily N-EtFOSE dose of 5.0 mg/kg body weight, b.w.day is the body weight on each day of N-EtFOSE administration and n is the number of days of N-EtFOSE administration (n = 3 × 5 days).
Adjusted for the respective matrix spike recovery in serum (N-EtFOSE = 67%; N-EtFOSAA = 159%; N-EtFOSA = 82%; FOSE = 96%; FOSA = 132%) and in the liver (N-EtFOSE = 2.5±1%; N-EtFOSAA = 156±3%; N-EtFOSA = 19±2%; FOSE = 57±2%; FOSA = 98±3%) if not stated otherwise. See the experimental part and Table 2 for additional details.
Calculated assuming a serum volume of 4.86 mL/100 mg for female Sprague-Dawley rats (Probst et al., 2006).
d %TD of N-EtFOSE in the liver is not reported because of the poor recovery rate (2.5±1%); also see Table 2.
e Adjusted for the recovery of 13C4-PFOS.
The mean liver-to-serum ratios for several metabolites N-EtFOSE (Table 2) were calculated to assess their affinity for the liver compared to blood. The liver-to-serum ratios for these compounds suggest a preferential accumulation in the liver relative to serum, with a rank order of FOSA > FOSE > PFOS > N-EtFOSAA. However, the mean liver-to-serum ratio for FOSA (and possibly other perfluorooctanesulfonamides) needs to be interpreted with caution. Kärrman and co-workers report a mean plasma-to-whole blood ratio of 0.2 for FOSA in human blood (Kärrman et al., 2006b). Similarly, the plasma-to-whole blood ratio was 0.04 in sheep (Vitayavirasuk and Bowen, 1999). These values suggest that FOSA binds to red blood cells, which are removed during the preparation of plasma and, as in our study, serum. Therefore, our serum analyses probably underestimate the FOSA levels in whole blood and, as a consequence, overestimate the liver-to-blood ratio. In contrast, PFOS has mean plasma-to-whole blood ratios of 1.2 and 1.4, respectively, and plasma (or serum) levels are a good approximation of whole blood levels (Kärrman et al., 2006b).
Several earlier studies have determined liver-to-serum ratios for PFOS. Seacat and co-workers reported liver-to-serum ratios ranging from approximately 3:1 to 12:1 in male and female rats (Seacat et al., 2003), which is higher compared to the ratio of 2.3:1 reported in the present study. Seacat and co-workers report that the liver-to-serum ratio was independent of the PFOS dose and consistently lower in female rats. Recently, Curran and colleagues reported much higher liver-to-serum ratios ranging from 20:1 to 46:1 in female and 35:1 to 51:1 in male rats (Curran et al., 2008). In that study, rats were treated for 28 days with a diet containing 2-100 mg PFOS/kg diet. Interestingly, the liver-to-serum ratio of PFOS in untreated animals was 1.3:1 for female and 2.0:1 for male rats, which is more consistent with the study by Seacat et al. (2003) and our study. In contrast to rats, the mean liver-to-serum ratio of PFOS in female and male human liver samples was approximately 1.3:1 (Olsen et al., 2003b), which is comparable to ratios of 1:1 to 2:1 in cynomolgus monkeys (Seacat et al., 2002). A study using a single pooled liver and blood samples suggests a somewhat higher liver-to-tissue ratio of 2.7:1 for PFOS (Maestri et al., 2006).
In addition to providing an insight into the metabolism and disposition of perfluorooctanesulfonamides, the present study allows a comparison with human tissue and serum levels. Biomonitoring studies from around the world indicate that occupationally and non-occupationally exposed populations have measurable serum concentrations of PFOS and related sulfonamides (for an extensive review see (Fromme et al., 2009)). The concentrations of perfluorooctanesulfonamides are typically an order of magnitude lower compared to concentrations of PFOS in humans (Kannan et al., 2004; Olsen et al., 2003a). Similarly, PFOS levels in our study are one order of magnitude higher compared to other perfluorooctanesulfonamides, such as FOSA and N-EtFOSAA (Table 2). Several recent biomonitoring studies demonstrated that FOSA is the major perfluorooctanesulfonamide found in humans, with levels typically in the low part per billion range (Calafat et al., 2007; Kärrman et al., 2006a; Spliethoff et al., 2008). There are also reports that N-EtFOSAA and structurally related perfluorooctanesulfonamides are present in human blood in low part per billion range. However, human blood levels of perfluorooctanesulfonamides are declining since the phase-out of the production of PFOS-based chemicals in the United States in 2002 (Calafat et al., 2007; Spliethoff et al., 2008). These levels are three orders of magnitude lower compared to the FOSA and N-EtFOSAA levels (4,470 and 7,773 ppb, respectively) determined in the present study.
In parallel to the metabolomic study and the investigation of the metabolite profile, the effect of N-EtFOSE on antioxidant capacity indicative of redox homeostasis was investigated in the liver and uterus. As a weak peroxisome proliferator, N-EtFOSE may induce the generation of reactive oxygen species and cause oxidative stress in specific tissues. This hypothesis is supported by a few in vivo and in vitro studies which suggested that perfluorooctanesulfonamides and related compounds activate PPARα (Maloney and Waxman, 1999; Shipley et al., 2004; Takacs and Abbott, 2007; Vanden Heuvel et al., 2006), cause hepatic peroxisome proliferation (Berthiaume and Wallace, 2002; O'Brien et al., 2005; Wallace et al., 2001), interfere with mitochondrial function and induce the production of reactive oxygen species (O'Brien et al., 2005; Schnellmann, 1990; Schnellmann and Manning, 1990; Starkov and Wallace, 2002). Therefore, we hypothesized that N-EtFOSE and/or its metabolites might also alter the activity of antioxidant enzymes, such as SODs, GPx's and catalase.
Hepatic levels of AOX activity were determined to assess if a peroxisome proliferating effect of N-EtFOSE could be responsible for changes in the redox homeostasis of the liver and uterus of female Sprague-Dawley rats. While subacute exposure to N-EtFOSE caused peroxisome proliferation in rats (Wallace et al., 2001), acute administration did not have an effect on biological markers of peroxisome proliferation (Berthiaume and Wallace, 2002). In the present study, N-EtFOSE slightly increased peroxisomal AOX activity, a marker of peroxisome proliferation (Figure 3). However, this increase was not statistically significant. In contrast, ciprofibrate, a well known peroxisome proliferator (O'Brien et al., 2005), significantly increased AOX activity in the liver. These observations, together with the high hepatic PFOS levels (Table 2), suggests that the slight increase in peroxisomal AOX activity may be due to PFOS, a potent peroxisome proliferator, and not N-EtFOSE.
The activity of antioxidant enzymes (SODs, GPx's and catalase), levels of cellular antioxidants (reduced to oxidized glutathione ratio) and a marker of oxidative stress (TBARS) were investigated to assess changes in the redox homeostasis. While the N-EtFOSE treatment did not affect the ratio of reduced to oxidized glutathione in the liver and uterus or levels of TBARS in the liver, it had effects on the activity of antioxidant enzymes. Most consistent were changes in the activity of SOD caused by N-EtFOSE and/or its metabolites (Figure 4). N-EtFOSE treatment increased the activity of total SOD and CuZnSOD in the uterus and of total SOD, MnSOD and CuZnSOD in the liver of female Sprague-Dawley rats. Similarly, perfluorodecanoic acid increased total hepatic SOD activities in female Sprague-Dawley rats (Glauert et al., 1992), and PFOS increased SOD activity in primary cultured hepatocytes from freshwater tilapia (Liu et al., 2007). In contrast, PFOS and PFOA had no apparent effect on mRNA levels of SODs in rats (Guruge et al., 2006; Hu et al., 2005; Martin et al., 2007) or mice (Rosen et al., 2008). Ciprofibrate did not significantly alter SOD activities in the liver, which is in agreement with an earlier study that did not show a change in total SOD activity over a 546 day period (Glauert et al., 1992).
N-EtFOSE treatment significantly decreased total GPx activity in the uterus, but otherwise had no effect on total GPx or GPx-1 activity in the liver or uterus (Figure 6). In earlier studies with perfluorodecanoic acid, hepatic GPx activities were also not altered in the liver of female (Glauert et al., 1992) or male (Chen et al., 1990) Sprague-Dawley rats compared to controls. Several recent toxicogenomic studies reported no effect of PFOS and PFOA on hepatic GPx mRNA levels in rats (Guruge et al., 2006; Hu et al., 2005) or mice (Rosen et al., 2008) but an increase in GPx-1 mRNA levels in common cormorants (Nakayama et al., 2008). However, in other studies PFOA or perfluorodecanoic acid decreased (Glauert et al., 1992) or increased (Chen et al., 1990; Olsson et al., 1993) hepatic GPx activities, possible due to the different experimental conditions and time points under investigation. In contrast to the N-EtFOSE treatment group, total GPx activity was significantly decreased in the ciprofibrate group in the uterus. Total GPx and GPx-1 activity in ciprofibrate-treated animals were also significantly decreased in the liver, which is in agreement with earlier studies (Glauert et al., 1992; Goel et al., 1986; O'Brien et al., 2005).
Like other peroxisome proliferators (O'Brien et al., 2005), perfluorinated compounds, such as PFOS and PFOA, have been shown in some studies to mildly increase hepatic catalase mRNA expression and activity in rodents due to the increase in the levels and activity H2O2-producing enzyme AOX (O'Brien et al., 2005). In our study, treatment with N-EtFOSE significantly increased catalase activity in the uterus, but had no effect of catalase activity in the liver (Figure 5). Catalase activity was also not altered in the liver and uterus of ciprofibrate-treated animals, which is in contrast to some earlier studies showing an increase in hepatic catalase activity (Glauert et al., 1992).
In conclusion, the administration of N-EtFOSE resulted in a complex metabolite profile, with levels decreasing in the order PFOS > FOSA ∼ N-EtFOSAA > FOSE ∼ N-EtFOSE, and caused only few changes in the biological endpoints under investigation, such as a decreased growth rate, an increased liver weight, an increased hepatic and uterine SOD activity, and an increased uterine catalase activity. Overall, the general toxicity of N-EtFOSE in our study resembles the toxicity of PFOS. Because PFOS is the major N-EtFOSE metabolite in liver and serum (Table 2), this similarity between both compounds suggests that, as mentioned above, PFOS (and not another N-EtFOSE metabolite) is likely responsible for the toxicity observed in our study. The effect of PFOS (and perfluorooctanesulfonamides) on antioxidant enzyme levels and activities, in particular SOD, is poorly investigated and appear to be conflicting: While PFOS caused an increase in SOD activity in primary cultured hepatocytes of freshwater tilapia (Liu et al., 2007), a gene profiling study did not report changes in SOD gene expression in rats (Guruge et al., 2006). In contrast, some metabolites, such as N-EtFOSAA and FOSAA, have been shown to increase the production of reactive oxygen species by isolated mitochondria (O'Brien and Wallace, 2004), which could result in an induction of antioxidant enzymes, such as SOD. Therefore, dose-response studies with N-EtFOSE, PFOS and other relevant metabolites are needed to identify which N-EtFOSE metabolite(s) caused the increase in SOD activity in the present study. Furthermore, it will be important to determine if the increase is SOD activity is a result of increased production of superoxide anion radicals. Most likely, such studies will lead to the identification of new and more sensitive markers of N-EtFOSE toxicity and, thus, are crucial for the assessment of the human health risk associated with exposure to N-EtFOSE in particular and PFOS-based chemicals in general.
Supplementary Material
Acknowledgments
We thank Brett A. Wagner, Garry R. Buettner and The University of Iowa ESR Facility for invaluable support and Rama Rao V.V.V.N.S. for the synthesis of the perfluorooctanesulfonamides. The work was supported by a grant from the University of Iowa Center for Health Effects of Environmental Contamination (CHEEC). Additional support was provided by grants ES05605, ES013661 and ES012475 from the National Institute of Environmental Health Sciences, NIH, and by the Kentucky Agricultural Experiment Station. DRS and MCC were supported by NIH P30-CA086862.
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
References
- Aebi H. Catalase in vitro. In: Packer L, editor. Methods in Enzymology. Academic Press; 1984. pp. 121–126. [DOI] [PubMed] [Google Scholar]
- Ala-Korpela M. Critical evaluation of 1H NMR metabonomics of serum as a methodology for disease risk assessment and diagnostics. Clin Chem Lab Med. 2008;46:27–42. doi: 10.1515/CCLM.2008.006. [DOI] [PubMed] [Google Scholar]
- Anderson ME. Tissue glutathione. In: Greenwald R, editor. Methods for oxygen radical research. CRC Press; Boca Raton: 1985. pp. 317–323. [Google Scholar]
- Arrendale RF, Stewart JT, Manning R, Vitayavirasuk B. Determination of GX 071 and its major metabolite in rat blood by cold on-column injection capillary GC/ECD. J Agric Food Chem. 1989;37:1130–1135. [Google Scholar]
- Austin ME, Kasturi BS, Barber M, Kannan K, MohanKumar PS, MohanKumar SM. Neuroendocrine effects of perfluorooctane sulfonate in rats. Environ Health Perspect. 2003;111:1485–1489. doi: 10.1289/ehp.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers RF, Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- Berthiaume J, Wallace KB. Perfluorooctanoate, perflourooctanesulfonate, and N-ethyl perfluorooctanesulfonamido ethanol; peroxisome proliferation and mitochondrial biogenesis. Toxicol Lett. 2002;129:23–32. doi: 10.1016/s0378-4274(01)00466-0. [DOI] [PubMed] [Google Scholar]
- Borges T, Peterson RE, Pitot HC, Robertson LW, Glauert HP. Effect of the peroxisome proliferator perfluorodecanoic acid on the promotion of two-stage hepatocarcinogenesis in rats. Cancer Lett. 1993;72:111–120. doi: 10.1016/0304-3835(93)90019-6. [DOI] [PubMed] [Google Scholar]
- Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL. Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003-2004 and comparisons with NHANES 1999-2000. Environ Health Perspect. 2007;115:1596–1602. doi: 10.1289/ehp.10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case MT, York RG, Christian MS. Rat and rabbit oral developmental toxicology studies with two perfluorinated compounds. Int J Toxicol. 2001;20:101–109. doi: 10.1080/10915810151115236. [DOI] [PubMed] [Google Scholar]
- Chen H, Huang Cy, Wilson MW, Lay LT, Robertson LW, Chow CK, Glauert HP. Effect of the peroxisome proliferators ciprofibrate and perfluorodecanoic acid on hepatic cell proliferation and toxicity in Sprague-Dawley rats. Carcinogenesis. 1994;15:2847–2850. doi: 10.1093/carcin/15.12.2847. [DOI] [PubMed] [Google Scholar]
- Chen LC, Borges T, Glauert HP, Knight SAB, Sunde RA, Schramm H, Oesch F, Chow CK, Robertson LW. Modulation of selenium-dependent gluthathione peroxidase by perfluorodecanoic acid in rats: Effect of dietary selenium. Nutrition, Pharmacology and Toxicology. 1990:298–304. doi: 10.1093/jn/120.3.298. [DOI] [PubMed] [Google Scholar]
- Coen M, Lenz Eva M, Nicholson Jeremy K, Wilson Ian D, Pognan F, Lindon John C. An integrated metabonomic investigation of acetaminophen toxicity in the mouse using NMR spectroscopy. Chem Res Toxicol. 2003;16:295–303. doi: 10.1021/tx0256127. [DOI] [PubMed] [Google Scholar]
- Connor SC, Hodson MP, Ringeissen S, Sweatman BC, McGill PJ, Waterfield CJ, Haselden JN. Development of a multivariate statistical model to predict peroxisome proliferation in the rat, based on urinary 1H-NMR spectral patterns. Biomarkers. 2004;9:364–385. doi: 10.1080/13547500400006005. [DOI] [PubMed] [Google Scholar]
- Cui L, Zhou Qf, Liao Cy, Fu Jj, Jiang Gb. Studies on the toxicological effects of PFOA and PFOS on rats using histological observation and chemical analysis. Arch Environ Contam Toxicol. 2009;56:338–349. doi: 10.1007/s00244-008-9194-6. [DOI] [PubMed] [Google Scholar]
- Curran I, Hierlihy SL, Liston V, Pantazopoulos P, Nunnikhoven A, Tittlemier S, Barker M, Trick K, Bondy G. Altered fatty acid homeostasis and related toxicologic sequelae in rats exposed to dietary potassium perfluorooctanesulfonate (PFOS) J Toxicol Environ Health A. 2008;71:1526–1541. doi: 10.1080/15287390802361763. [DOI] [PubMed] [Google Scholar]
- Delaney J, Hodson MP, Thakkar H, Connor SC, Sweatman BC, Kenny SP, McGill PJ, Holder JC, Hutton KA, Haselden JN, Waterfield CJ. Tryptophan-NAD+ pathway metabolites as putative biomarkers and predictors of peroxisome proliferation. Arch Toxicol. 2005;79:208–223. doi: 10.1007/s00204-004-0625-5. [DOI] [PubMed] [Google Scholar]
- Era S, Harada KH, Toyoshima M, Inoue K, Minata M, Saito N, Takigawa T, Shiota K, Koizumi A. Cleft palate caused by perfluorooctane sulfonate is caused mainly by extrinsic factors. Toxicology. 2009;256:42–47. doi: 10.1016/j.tox.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Fromme H, Tittlemier SA, Völkel W, Wilhelm M, Twardella D. Perfluorinated compounds - Exposure assessment for the general population in western countries. Int J Hyg Environ Health. 2009 doi: 10.1016/j.ijheh.2008.04.007. In Press. [DOI] [PubMed] [Google Scholar]
- Giesy JP, Kannan K. Global distribution of perfluorooctane sulfonate in wildlife. Environ Sci Technol. 2001;35:1339–1342. doi: 10.1021/es001834k. [DOI] [PubMed] [Google Scholar]
- Glauert HP, Srinivasan S, Tatum VL, Chen LC, Saxon DM, Lay LT, Borges T, Baker M, Chen LH, Robertson LW. Effects of the peroxisome proliferators ciprofibrate and perfluorodecanoic acid on hepatic cellular antioxidants and lipid peroxidation in rats. Biochem Pharmacol. 1992;43:1353–1359. doi: 10.1016/0006-2952(92)90513-i. [DOI] [PubMed] [Google Scholar]
- Goel SK, Lalwani ND, Reddy JK. Peroxisome proliferation and lipid peroxidation in rat liver. Cancer Res. 1986;46:1324–1330. [PubMed] [Google Scholar]
- Guruge KS, Yeung LWY, Yamanaka N, Miyazaki S, Lam PKS, Giesy JP, Jones PD, Yamashita N. Gene expression profiles in rat liver treated with perfluorooctanoic acid (PFOA) Toxicol Sci. 2006;89:93–107. doi: 10.1093/toxsci/kfj011. [DOI] [PubMed] [Google Scholar]
- Hammer TA, Sandvik AK, Waldum HL. Potentiating hypergastrinemic effect by the peroxisome proliferator ciprofibrate and omeprazole in the rat. Scand J Gastroenterol. 1998;33:595–599. doi: 10.1080/00365529850171855. [DOI] [PubMed] [Google Scholar]
- Haughom B, Spydevold O. The mechanism underlying the hypolipemic effect of perfluorooctanoic acid (PFOA), perfluorooctane sulphonic acid (PFOSA) and clofibric acid. Biochim Biophys Acta. 1992;1128:65–72. doi: 10.1016/0005-2760(92)90258-w. [DOI] [PubMed] [Google Scholar]
- Hu W, Jones PD, Celius T, Giesy JP. Identification of genes responsive to PFOS using gene expression profiling. Environ Toxicol Pharmacol. 2005;19:57–70. doi: 10.1016/j.etap.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Hu Wy, Jones PD, DeCoen W, King L, Fraker P, Newsted J, Giesy JP. Alterations in cell membrane properties caused by perfluorinated compounds. Comp Biochem Physiol C Toxicol Pharmacol. 2003;135C:77–88. doi: 10.1016/s1532-0456(03)00043-7. [DOI] [PubMed] [Google Scholar]
- Huang Cy, Wilson MW, Travis Lay L, Chow CK, Robertson LW, Glauert HP. Increased 8-hydroxydeoxyguanosine in hepatic DNA of rats treated with the peroxisome proliferators ciprofibrate and perfluorodecanoic acid. Cancer Lett. 1994;87:223–8. doi: 10.1016/0304-3835(94)90226-7. [DOI] [PubMed] [Google Scholar]
- Kannan KC, S, Falandysz J, Fillmann G, Kumar KS, Loganathan BG, Mohd MA, Olivero J, Van Wouwe N, Yang JH, Aldous KM. Perfluorooctanesulfonate and related fluorochemicals in human blood from several countries. Environ Sci Technol. 2004;38:4489–4495. doi: 10.1021/es0493446. [DOI] [PubMed] [Google Scholar]
- Kärrman A, Mueller JF, van Bavel B, Harden F, Toms LM, Lindström G. Levels of 12 perfluorinated chemicals in pooled australian serum, collected 2002-2003, in relation to age, gender, and region. Environ Sci Technol. 2006a;40:3742–3748. doi: 10.1021/es060301u. [DOI] [PubMed] [Google Scholar]
- Kärrman A, van Bavel B, Järnberg U, Hardell L, Lindström G. Perfluorinated chemicals in relation to other persistent organic pollutants in human blood. Chemosphere. 2006b;64:1582–1591. doi: 10.1016/j.chemosphere.2005.11.040. [DOI] [PubMed] [Google Scholar]
- Kissa E. Fluorinated surfactants and repellents. Vol. 97. Marcel Dekker; New York: 2001. (Surfactant Science). [Google Scholar]
- Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A, Seed J. Perfluoroalkyl acids: A review of monitoring and toxicological findings. Toxicol Sci. 2007;99:366–394. doi: 10.1093/toxsci/kfm128. [DOI] [PubMed] [Google Scholar]
- Lau C, Thibodeaux JR, Hanson RG, Rogers JM, Grey BE, Stanton ME, Butenhoff JL, Stevenson LA. Exposure to perfluorooctane sulfonate during pregnancy in rat and mouse. II: postnatal evaluation. Toxicol Sci. 2003;74:382–392. doi: 10.1093/toxsci/kfg122. [DOI] [PubMed] [Google Scholar]
- Lawrence RA, Burk RF. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem Biophys Res Commun. 1976;71:952–958. doi: 10.1016/0006-291x(76)90747-6. [DOI] [PubMed] [Google Scholar]
- Lehmler HJ. Synthesis of environmentally relevant fluorinated surfactants-a review. Chemosphere. 2005;58:1471–1496. doi: 10.1016/j.chemosphere.2004.11.078. [DOI] [PubMed] [Google Scholar]
- Lehmler HJ, Rao VVVNSR, Nauduri D, Vargo JD, Parkin S. Synthesis and structure of environmentally relevant perfluorinated sulfonamides. J Fluorine Chem. 2007;128:595–607. doi: 10.1016/j.jfluchem.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmler HJ, Xie W, Bothun GD, Bummer PM, Knutson BL. Mixing of perfluorooctanesulfonic acid (PFOS) potassium salt with dipalmitoyl phosphatidylcholine (DPPC) Colloids Surf B. 2006;51:25–29. doi: 10.1016/j.colsurfb.2006.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei R, Wu C, Yang B, Ma H, Shi C, Wang Q, Wang Q, Yuan Y, Liao M. Integrated metabolomic analysis of the nano-sized copper particle-induced hepatotoxicity and nephrotoxicity in rats: A rapid in vivo screening method for nanotoxicity. Toxicol Appl Pharmacol. 2008;232:292–301. doi: 10.1016/j.taap.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Liu C, Yu K, Shi X, Wang J, Lam PKS, Wu RSS, Zhou B. Induction of oxidative stress and apoptosis by PFOS and PFOA in primary cultured hepatocytes of freshwater tilapia (Oreochromis niloticus) Aquat Toxicol. 2007;82:135–143. doi: 10.1016/j.aquatox.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr L, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Luebker DJ, Hansen KJ, Bass NM, Butenhoff JL, Seacat AM. Interactions of fluorochemicals with rat liver fatty acid-binding protein. Toxicology. 2002;176:175–185. doi: 10.1016/s0300-483x(02)00081-1. [DOI] [PubMed] [Google Scholar]
- Maestri L, Negri S, Ferrari M, Ghittori S, Fabris F, Danesino P, Imbriani M. Determination of perfluorooctanoic acid and perfluorooctanesulfonate in human tissues by liquid chromatography/single quadrupole mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:2728–2734. doi: 10.1002/rcm.2661. [DOI] [PubMed] [Google Scholar]
- Maloney EK, Waxman DJ. trans-Activation of PPARa and PPARy by structurally diverse environmental chemicals. Toxicol Appl Pharmacol. 1999;161:209–218. doi: 10.1006/taap.1999.8809. [DOI] [PubMed] [Google Scholar]
- Manning RO, Bruckner JV, Mispagel ME, Bowen JM. Metabolism and disposition of sulfluramid, a unique polyfluorinated insecticide, in the rat. Drug Metab Dispos. 1991;19:205–211. [PubMed] [Google Scholar]
- Martin MT, Brennan RJ, Hu W, Ayanoglu E, Lau C, Ren H, Wood CR, Corton JC, Kavlock RJ, Dix DJ. Toxicogenomic study of triazole fungicides and perfluoroalkyl acids in rat livers predicts toxicity and categorizes chemicals based on mechanisms of toxicity. Toxicol Sci. 2007;97:595–613. doi: 10.1093/toxsci/kfm065. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Iwata H, Tao L, Kannan K, Imoto M, Kim EY, Tashiro K, Tanabe S. Potential effects of perfluorinated compounds in common cormorants from Lake Biwa, Japan: an implication from the hepatic gene expression profiles by microarray. Environ Toxicol Chem. 2008;27:2378–2386. doi: 10.1897/07-614.1. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Connelly J, Lindon JC, Holmes E. Innovation: Metabonomics: a platform for studying drug toxicity and gene function. Nat Rev Drug Discovery. 2002;1:153–161. doi: 10.1038/nrd728. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Lindon JC, Holmes E. “Metabonomics”: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 1999;29:1181–1189. doi: 10.1080/004982599238047. [DOI] [PubMed] [Google Scholar]
- O'Brien ML, Spear BT, Glauert HP. Role of oxidative stress in peroxisome proliferator-mediated carcinogenesis. Crit Rev Toxicol. 2005;35:61–88. doi: 10.1080/10408440590905957. [DOI] [PubMed] [Google Scholar]
- O'Brien TM, Wallace KB. Mitochondrial permeability transition as the critical target of N-acetyl perfluorooctane sulfonamide toxicity in vitro. Toxicol Sci. 2004;82:333–340. doi: 10.1093/toxsci/kfh244. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Church TR, Miller JP, Burris JM, Hansen KJ, Lundberg JK, Armitage JB, Herron RM, Medhdizadehkashi Z, Nobiletti JB, O'Neill EM, Mandel JH, Zobel LR. Perfluorooctanesulfonate and other fluorochemicals in the serum of American Red Cross adult blood donors. Environ Health Perspect. 2003a;111:1892–1901. doi: 10.1289/ehp.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GW, Hansen KJ, Stevenson LA, Burris JM, Mandel JH. Human donor liver and serum concentrations of perfluorooctanesulfonate and other perfluorochemicals. Environ Sci Technol. 2003b;37:888–891. doi: 10.1021/es020955c. [DOI] [PubMed] [Google Scholar]
- Olsson U, Sundberg C, Andersson K, De Pierre JW. Further studies on the involvement of selenium in peroxisome proliferation in rat liver. Comparison of effects of clofibric acid and perfluorooctanoic acid and the pharmacokinetics of [14C]clofibrate. Biochem Pharmacol. 1993;46:1805–1810. doi: 10.1016/0006-2952(93)90586-l. [DOI] [PubMed] [Google Scholar]
- Panaretakis T, Shabalina IG, Grander D, Shoshan MC, DePierre JW. Reactive oxygen species and mitochondria mediate the induction of apoptosis in human hepatoma HepG2 cells by the rodent peroxisome proliferator and hepatocarcinogen, perfluorooctanoic acid. Toxicol Appl Pharmacol. 2001;173:56–64. doi: 10.1006/taap.2001.9159. [DOI] [PubMed] [Google Scholar]
- Paul AG, Jones KC, Sweetman AJ. A first global production, emission, and environmental inventory for perfluorooctane sulfonate. Environ Sci Technol. 2009;43:386–392. doi: 10.1021/es802216n. [DOI] [PubMed] [Google Scholar]
- Peden-Adams MM, EuDaly JG, Dabra S, EuDaly A, Heesemann L, Smythe J, Keil DE. Suppression of humoral immunity following exposure to the perfluorinated insecticide sulfluramid. J Toxicol Environ Health A. 2007;70:1130–1141. doi: 10.1080/15287390701252733. [DOI] [PubMed] [Google Scholar]
- Poosch MS, Yamazaki RK. Determination of peroxisomal fatty acyl-CoA oxidase activity using a lauroyl-CoA-based fluorometric assay. Biochim Biophys Acta. 1986;884:585–593. doi: 10.1016/0304-4165(86)90211-4. [DOI] [PubMed] [Google Scholar]
- Probst RJ, Lim JM, Bird DN, Pole GL, Sato AK, Claybaugh JR. Gender differences in the blood volume of conscious Sprague-Dawley rats. J Am Assoc Lab Anim Sci. 2006;45:49–52. [PMC free article] [PubMed] [Google Scholar]
- Rosen MB, Abbott BD, Wolf DC, Corton JC, Wood CR, Schmid JE, Das KP, Zehr RD, Blair ET, Lau C. Gene profiling in the livers of wild-type and PPARa-null mice exposed to perfluorooctanoic acid. Toxicol Pathol. 2008;36:592–607. doi: 10.1177/0192623308318208. [DOI] [PubMed] [Google Scholar]
- Schnellmann RG. The cellular effects of a unique pesticide sulfluramid (N-ethylperfluorooctane sulfonamide) on rabbit renal proximal tubules. Toxicol In Vitro. 1990;4:71–74. doi: 10.1016/0887-2333(90)90012-i. [DOI] [PubMed] [Google Scholar]
- Schnellmann RG, Manning RO. Perfluorooctane sulfonamide: a structurally novel uncoupler of oxidative phosphorylation. Biochim Biophys Acta. 1990;1016:344–348. doi: 10.1016/0005-2728(90)90167-3. [DOI] [PubMed] [Google Scholar]
- Seacat AM, Thomford PJ, Hansen KJ, Clemen LA, Eldridge SR, Elcombe CR, Butenhoff JL. Sub-chronic dietary toxicity of potassium perfluorooctanesulfonate in rats. Toxicology. 2003;183:117–131. doi: 10.1016/s0300-483x(02)00511-5. [DOI] [PubMed] [Google Scholar]
- Seacat AM, Thomford PJ, Hansen KJ, Olsen GW, Case MT, Butenhoff JL. Subchronic toxicity studies on perfluorooctanesulfonate potassium salt in cynomolgus monkeys. Toxicol Sci. 2002;68:249–264. doi: 10.1093/toxsci/68.1.249. [DOI] [PubMed] [Google Scholar]
- Sheikh K, Camejo G, Lanne B, Halvarsson T, Landergren MR, Oakes ND. Beyond lipids, pharmacological PPARα activation has important effects on amino acid metabolism as studied in the rat. Am J Physiol Endocrinol Metab. 2007;292:E1157–E1165. doi: 10.1152/ajpendo.00254.2006. [DOI] [PubMed] [Google Scholar]
- Shipley JM, Hurst CH, Tanaka SS, DeRoos FL, Butenhoff JL, Seacat AM, Waxman DJ. Trans-activation of PPARα and induction of PPARα target genes by perfluorooctane-based chemicals. Toxicol Sci. 2004;80:151–160. doi: 10.1093/toxsci/kfh130. [DOI] [PubMed] [Google Scholar]
- Sohlenius AK, Eriksson AM, Hogstrom C, Kimland M, DePierre JW. Perfluorooctane sulfonic acid is a potent inducer of peroxisomal fatty acid beta-oxidation and other activities known to be affected by peroxisome proliferators in mouse liver. Pharmacol Toxicol. 1993;72:90–93. doi: 10.1111/j.1600-0773.1993.tb00296.x. [DOI] [PubMed] [Google Scholar]
- Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- Spliethoff HM, Tao L, Shaver SM, Aldous KM, Pass KA, Kannan K, Eadon GA. Use of newborn screening program blood spots for exposure assessment: declining levels of perluorinated compounds in New York State infants. Environ Sci Technol. 2008;42:5361–5367. doi: 10.1021/es8006244. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Wallace KB. Structural determinants of fluorochemical-induced mitochondrial dysfunction. Toxicol Sci. 2002;66:244–252. doi: 10.1093/toxsci/66.2.244. [DOI] [PubMed] [Google Scholar]
- Takacs ML, Abbott BD. Activation of mouse and human peroxisome proliferator-activated receptors (α, β/δ, γ) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci. 2007;95:108–117. doi: 10.1093/toxsci/kfl135. [DOI] [PubMed] [Google Scholar]
- Thibodeaux JR, Hanson RG, Rogers JM, Grey BE, Barbee BD, Richards JH, Butenhoff JL, Stevenson LA, Lau C. Exposure to perfluorooctane sulfonate during pregnancy in rat and mouse. I: maternal and prenatal evaluations. Toxicol Sci. 2003;74:369–381. doi: 10.1093/toxsci/kfg121. [DOI] [PubMed] [Google Scholar]
- Tittlemier SA, Pepper K, Seymour C, Moisey J, Bronson R, Cao XL, Dabeka RW. Dietary exposure of Canadians to perfluorinated carboxylates and perfluorooctane sulfonate via consumption of meat, fish, fast foods, and food items prepared in their packaging. J Agric Food Chem. 2007;55:3203–3210. doi: 10.1021/jf0634045. [DOI] [PubMed] [Google Scholar]
- Tomy GT, Tittlemier SA, Palace VP, Budakowski WR, Braekevelt E, Brinkworth L, Friesen K. Biotransformation of N-ethyl perfluorooctanesulfonamide by rainbow trout (Onchorhynchus mykiss) liver microsomes. Environ Sci Technol. 2004;38:758–762. doi: 10.1021/es034550j. [DOI] [PubMed] [Google Scholar]
- Uchiyama M, Mihara M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem. 1978;86:271–278. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- Vanden Heuvel JP, Thompson JT, Frame SR, Gillies PJ. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: A comparison of human, mouse, and rat peroxisome proliferator-activated receptor-α, -β, and -γ, liver × receptor-β, and retinoid × receptor-α. Toxicol Sci. 2006;92:476–489. doi: 10.1093/toxsci/kfl014. [DOI] [PubMed] [Google Scholar]
- Viant MR. Revealing the metabolome of animal tissues using 1H nuclear magnetic resonance spectroscopy. In: Weckwerth W, editor. Metabolomics - Methods and protocols. Humana Press Inc.; 2007. pp. 229–246. [DOI] [PubMed] [Google Scholar]
- Viant MR, Lyeth BG, Miller MG, Berman RF. An NMR metabolomic investigation of early metabolic disturbances following traumatic brain injury in a mammalian model. NMR Biomed. 2005;18:507–516. doi: 10.1002/nbm.980. [DOI] [PubMed] [Google Scholar]
- Vitayavirasuk B, Bowen JM. Pharmacokinetics of sulfluramid and its metabolite desethylsulfluramid after intravenous and intraruminal administration of sulfluramid to sheep. Pestic Sci. 1999;55:719–725. [Google Scholar]
- Wallace KB, Luebker DJ, Butenhoff JL, Seacat AM. Perfluorooctane sulfonate and 2-(N-ethylperfluorooctanesulfonamido)-ethyl alcohol are peroxisome proliferators in rats, but not guinea pigs. Toxicologist. 2001:1657. [Google Scholar]
- Wilson MW, Lay LT, Chow CK, Tai HH, Robertson LW, Glauert HP. Altered hepatic eicosanoid concentrations in rats treated with the peroxisome proliferators ciprofibrate and perfluorodecanoic acid. Arch Toxicol. 1995;69:491–497. doi: 10.1007/s002040050203. [DOI] [PubMed] [Google Scholar]
- Xie W, Kania-Korwel I, Bummer PM, Lehmler HJ. Effect of potassium perfluorooctanesulfonate, perfluorooctanoate and octanesulfonate on the phase transition of dipalmitoylphosphatidylcholine (DPPC) bilayers. Biochim Biophys Acta. 2007;1768:1299–1308. doi: 10.1016/j.bbamem.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Krenitsky DM, Seacat AM, Butenhoff JL, Anders MW. Biotransformation of N-ethyl-N-(2-hydroxyethyl)perfluorooctanesulfonamide by rat liver microsomes, cytosol, and slices and by expressed rat and human cytochromes P450. Chem Res Toxicol. 2004;17:767–775. doi: 10.1021/tx034222x. [DOI] [PubMed] [Google Scholar]
- Xu L, Krenitsky DM, Seacat AM, Butenhoff JL, Tephly TR, Anders MW. N-glucuronidation of perfluorooctanesulfonamide by human, rat, dog, and monkey liver microsomes and by expressed rat and human UDP-glucuronosyltransferases. Drug Metab Dispos. 2006;34:1406–1410. doi: 10.1124/dmd.106.009399. [DOI] [PubMed] [Google Scholar]
- Yahia D, Tsukuba C, Yoshida M, Sato I, Tsuda S. Neonatal death of mice treated with perfluorooctane sulfonate. J Toxicol Sci. 2008;33:219–226. doi: 10.2131/jts.33.219. [DOI] [PubMed] [Google Scholar]
- Zheng L, Dong GH, Jin YH, He QC. Immunotoxic changes associated with a 7-day oral exposure to perfluorooctanesulfonate (PFOS) in adult male C57BL/6 mice. Arch Toxicol. 2009 doi: 10.1007/s00204-008-0361-3. in press. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.