

Abstract
A sensitive method based on gold nanoparticle-enhanced capillary electrophoresis-chemiluminescence detection was developed for quantifying uric acid in serum. In this work, gold nanoparticles were added into the running buffer of capillary electrophoresis to catalyze the post-column chemiluminescence reaction between luminol and hydrogen peroxide, achieving highly efficient chemiluminescence emission. Negative peaks were produced due to the inhibitory effects on chemiluminescence emission from uric acid eluted from the electrophoretic capillary. The decrease in chemiluminescence intensity was proportional to the concentration of uric acid in the range of 2.5 × 10−7 ~ 1.0 × 10−5 M. Detection limit was 4.6 × 10−8 M uric acid. Ten human serum samples were analyzed by the presented method. Serum level of uric acid was found to be in the range from 204 to 324 μM for healthy subjects (n=5), and from 464 to 497 μM for diabetic patients (n=5). The two groups were significantly different (p < 0.05). The results suggested a potential application of the proposed assay in rapid primary diagnosis of diseases such as diabetes.
Keywords: Uric acid, serum, diabetes, capillary electrophoresis, chemiluminescence, gold nanoparticles
1. Introduction
Uric acid (UA) is a primary end product of purine metabolism. Abnormal levels of UA in human serum and urine are related to several medical conditions such as gout, hyperuricaemia, pneumonia, kidney damage, cardiovascular diseases, and Lesch–Nyhan syndrome [1–5]. A recently study also suggested that serum UA was a strong and independent risk factor for type 2 diabetes [6]. Therefore, quantification of UA in human serum is highly significant for early diagnoses of these diseases.
Methods based on high-performance liquid chromatography (HPLC) [7–9], spectrofluorometry [10], enzymatic assay [11–13], electrochemical methods [14,15] and capillary electrophoresis electrochemical detection (CE-ED) [16,17] have been developed for UA determination in biological specimens. Each technique has its own advantages and disadvantages. Enzymatic methods are attractive due to their high level of selectivity, but this methodology is inherently expensive and poor in reproducibility. Electrochemical methods for the determination of UA are less expensive and less time-consuming than the other methods. However, a major problem of the electrochemical technique is the interference from other electroactive compounds. HPLC methods usually offer a good selectivity, but they have shortcomings including long analysis time. CE is increasingly recognized as an important analytical separation technique. The advantages of CE include short analysis times, high resolution, and the compatibility of very small sample volumes. However, a CE-based method usually requires a highly sensitive detection scheme because only a very minute amount of the sample is injected for analysis.
Nanoparticles (NPs) have found widespread applications in the fields of electronics, catalysis, and biosensors in recent years [18]. Use of NPs to enhance chemical analysis is also gaining research interest. NPs were used as adsorbents to extract/enrich analytes from sample matrices for subsequent MALDI-MS [19] and HPLC-MS [20] analyses. By using gold nanoparticles (AuNPs)-filled capillaries, CE separations were significantly improved [21]. In the work, AuNPs were added to the CE running buffer. They functioned as pseudo-stationary phase, thus improving the separation efficiency and reproducibility for acidic and basic proteins. It has been know for long that Au3+ catalyzes luminol chemiluminescence (CL) reaction. However, AuNPs were found displaying far stronger catalytic activity on luminol CL than Au(III) did [22]. Certain organic compounds containing OH, NH2, and SH groups were observed to inhibit the CL signal of the luminol-H2O2-gold colloids system, promising a sensitive CL determination of these compounds. In situ CL detection of DNA and immunoassay of IgG based on AuNP-catalyzed luminol CL reaction was also reported [23]. We recently reported the first study on AuNP-enhanced CE-CL technique and its analytical potentials [24].
The present study aimed at the biomedical/clinical applications of the highly selective and sensitive AuNP-enhanced CE-CL analytical technique. It is believed that UA levels in physiological fluids are related to certain medical conditions such as diabetes. It was found that trace UA inhibited AuNP-catalyzed luminol-H2O2 CL emission, which led to the development of a novel CE-CL assay of UA in human serum. AuNPs added into running buffer not only enhanced the sensitivity of the detection, but also improved the detection selectivity as only a few components present in the serum matrix produced CE-CL signals. The present AuNP-enhanced CE-CL assay was used to determine UA level in human serum samples taken from healthy subjects and diabetic patients. Significantly different UA levels were found for the two groups. The results suggested a potential application of the proposed assay in rapid primary diagnosis of diabetes.
2. Experimental
2.1 Reagents and apparatus
UA and Luminol were obtained from Sigma (St. Louis, MO, USA). Hydrogen peroxide (H2O2) was obtained from Taopu Chemicals (Shanghai, China), Chlorauric acid (HAuCl4·4H2O) was obtained from National Pharmaceutical Group Chemical Reagents Company (Beijing, China). All other chemicals used in this work were of analytical grade. Water was purified by employing a Milli-Q plus 185 equip from Millipore (Bedford, MA, USA), and used throughout the work. AuNPs approximately 38 nm in diameter were prepared by the trisodium citrate procedure [25]. The electrophoretic buffer was 12 mM Na2HPO4 buffer (pH 9.3, adjusted with 1M NaOH solution) containing 0.12 mM AuNPs (expressed as HAuCl4 mM) and 0.2 mM luminol. The post-column oxidizer solution was 3 mM NaOH solution containing 35 mM H2O2. The stock solution of UA was prepared in 10 mM NaOH solution. All solutions were filtered through 0.45 μm membrane filters before use.
Determination of UA was carried out by using a laboratory-built CE-CL system described previously [24, 26]. Briefly, a high-voltage supply (0–30 kV, Beijing Cailu Science Instrument, Beijing, China) was used to drive the electrophoresis. Uncoated fused silica capillaries (55 cm × 75 μm id, Hebei Optical Fiber, China) were used for the separation. The polyimide on the 5-cm end section of the capillary was burned and removed. After etching with 10% hydrogen fluoride (HF) for 1 h, this end of capillary was inserted into the reaction capillary (320 μm id, Hebei Optical Fiber). A four-way Plexiglass joint held a separation capillary and a reaction capillary in place. The CL solution was siphoned into a tee. The grounding electrode was put in one joint of the tee. The CL solution flowed down to the detection window, which was made by burning 2 cm of the polyimide of the reaction capillary and was placed in front of the photomultiplier tube (PMT, R374 equipped with a C1556-50 DA-type socket assembly, Hamamatsu, Shizuoka, Japan). CL emission was collected by a PMT and was recorded and processed with a computer using a CT-21 Chromatography Data System (Beijing Cailu Science Instrument Company, Beijing, China).
2.2 Preparation of human serum samples
Human blood samples were centrifuged at 2000 rpm for 15 min to obtain serumsamples. These samples were stored at −20 °C until analysis. A 200-μL portion of a serum sample was diluted with 400 μL of acetonitrile and shaken vigorously for 5 min to precipitate out proteins. After centrifuging at 12,000 rpm for 20 min, the supernatant was transferred into an 1.0-ml vial and dried with a N2 stream. The residue was re-dissolved in 200 μL of water. The solution was thoroughly vortexed and then diluted 100 times with the CE running buffer before being injected into the CE-CL system.
2.3 CE-CL assay
New capillary was preconditioned by flushing with 1 M NaOH solution for 30 min before its first use. Between two consecutive injections, the capillary was rinsed sequentially with 100 mM NaOH solution, water and running buffer for 2 min each. The reaction capillary was rinsed with oxidation reagent solution for 2 min. Samples were injected into the capillary by hydrodynamic flow at a height differential of 20 cm for 10 s. Running voltage was 20 kV. Electrophoresis running buffer was 12 mM Na2HPO4 buffer (pH 9.3) containing 0.12 mM AuNP and 0.2 mM luminol. The oxidizer solution was 3 mM NaOH solution containing 35 mM H2O2.
3. Results and discussion
3.1 CL reaction between luminol and hydrogen peroxide in the presence of AuNPs and uric acid
It was found that AuNPs catalyzed the CL reaction between luminol and H2O2, and that UA displayed an inhibitory effect on the catalyzed CL emission. CL inhibition resulted in an inverted CE-CL peak as shown in Fig. 1. The degree of CL suppression was proportional to UA concentration, which formed the basis for UA quantification. To maximize the sensitivity of UA assay, the effects of luminol, H2O2, AuNPs, and NaOH concentrations on the CL inhibitory signal were investigated. In these experiments, a 4.0 × 10−6 M of UA solution was injected into the CE-CL system, and then the CL intensity was recorded. It was noticed that AuNPs had to be added to the CE running buffer instead of to the post-column introduced oxidizing reagent solution in order to observe enhanced CL emission. This suggested that binding of luminol molecules to AuNPs prior to their reaction with the oxidizing agent was essential for CL enhancement. Concentration of AuNPs in the running buffer also affected CL emission. AuNPs at a concentration in the range from 110 to 130 mM increased CL emission by a factor of > 20 compared with AuNPs at a concentration below 50 mM. The influence of luminol concentration (from 2.0 × 10−5 to 4.0 × 10−4 M) on the CE-CL signal was studied while keeping H2O2 concentration at 35 mM, phosphate concentration at 12 mM, and NaOH concentration at 3 mM. It was found that the background signal increased tremendously with the increase in luminol concentration. UA inhibited the CL emission resulting in a negative peak. The peak height also increased slightly with the increase of luminol concentration. However, a higher baseline noise was observed when the concentration was higher than 2.0 × 10−4 M. It was, therefore, chosen for further experiments. Changing the concentration of H2O2 from 10 to 50 mM, it was observed that the relative height of UA peak increased first and then decreased. The maximum signal was obtained when the concentration was 35 mM. In the presence of AuNPs, luminol reacted with H2O2 emitting CL in alkaline solutions. Therefore, a NaOH solution was selected as the CL reaction medium. The effects of NaOH concentration on the analysis of UA were investigated by varying NaOH concentration from 0.5 mM to10 mM. Maximum peak height was recorded when the concentration of NaOH was at 2 mM or above.
Figure 1.

Analysis of a standard UA solution at 4.0×10−6 M. Electrophoresis running buffer was 12 mM Na2HPO4 buffer at pH 9.3 containing 0.12 mM AuNPs and 0.2 mM luminol; oxidizer solution was 3 mM NaOH solution containing 35 mM H2O2; separation capillary was 50 cm × 75 μm id; voltage applied was 20 kV.
3.2 Analytical figures of merit
Under the selected CE-CL conditions, 5-point calibration curves were prepared by using UA standard solutions ranging from 2.0 × 10−7 to 1.0 × 10−5 M. Linear regression resulted in the following regression equation: H=2.54746 × 105 C + 0.0842 (r2 = 0.9971), where H was peak height (mV) and C was the UA concentration in M. The detection limit was calculated to be 4.0 × 10−8 M (signal/noise = 3). It is worth noting that compared with the previously reported methods for UA quantification, the present method was one of the most sensitive methods (see Table 1). Reproducibility was investigated by analyzing a standard solution of UA at 4.0 × 10−6 M six times. The relative standard deviation (RSD) in peak height was found to be 3.8%.
Table 1.
Comparison of UA assays
| Method* | Linear range (M) | Detection limit (M) | Sample type | Reference |
|---|---|---|---|---|
| CE-ED | 5.0×10−7 ~ 5.0×10−4 | 3.3×10−7 | Human saliva | Guan et al (17) |
| HPLC-UV | NA | 3.7×10−7 | Human urine | Zuo et al (27) |
| HPLC-MS | NA | 9.5×10−8 | Human serum | Dai et al (28) |
| Electrochemistry | 1.0×10−6 ~ 5.0×10−4 | 1.0×10−7 | Human urine | Fang et al (29) |
| Electrochemistry | 1.0×10−6 ~ 3.2×10−5 | 1.0×10−7 | Human urine | Huang et al (30) |
| CE-CL | 2.0×10−7 ~ 1.0×10−5 | 4.0×10−8 | Human serum | This work |
NA: not available.
3.3 Quantification of UA in human serum samples
Ten samples, five from healthy subjects and five from diabetic patients, were analyzed. A typical electropherogram from these analyses is shown as trace 1 in Figure 2. As can be seen, the electropherogram was relatively simple with only four peaks. Considering many endogenous components in this sample matrix including amino acids and biogenic amines could affect luminol-H2O2 CL emission, these results indicated that AuNPs enhanced CL detection offered an improved selectivity. The proposed CE-CL assay of UA was highly selective and also quick with a run time of < 6 min. The analytical results are summarized in Table 2 and 3. Recovery of UA from these samples was studied. UA was spiked to each sample analyzed. Fig. 2 (trace 2) shows the electropherogram obtained from a serum sample spiked with UA. The recovery results (shown in Table 2 and 3) were in the range of 94–105 %. The UA level in the serum samples from healthy subjects was found be in the range of 204 ~ 324 μM. However, it was found in the range from 464 ~ 497 μM for diabetic patients. Varian analysis with the Student-Newman-Kleuls test showed a significance difference in serum UA level between the two groups (P < 0.05).
Figure 2.
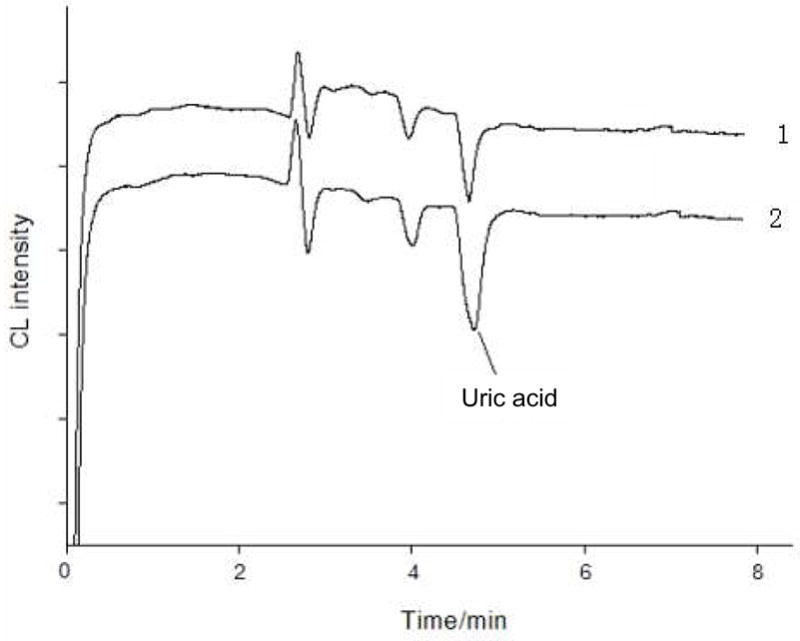
Electropherograms obtained from the separation of a human serum sample (trace 1) and the sample spiked with UA at 1.5 × 10−6 M (trace 2). CE-CL conditions were as in Figure 1.
Table 2.
Analytical results of UA in serum samples from healthy subjects
| Sample (No) | Founda (μM) | RSD (%,n=5) | Added (μM) | Total founda (μM) | Recovery (%) | UA Conc. In serum (mM) |
|---|---|---|---|---|---|---|
| 1 | 2.04 | 2.5 | 1.5 | 3.51 | 98 | 0.204 |
| 2 | 3.24 | 2.9 | 1.5 | 4.82 | 105 | 0.324 |
| 3 | 2.38 | 3.4 | 1.5 | 3.91 | 102 | 0.238 |
| 4 | 2.61 | 2.7 | 1.5 | 4.17 | 104 | 0.261 |
| 5 | 2.49 | 3.1 | 1.5 | 3.94 | 97 | 0.249 |
Average of five determinations.
Table 3.
Analytical results of UA in serum samples from diabetic patients
| Sample (No) | Founda (μM) | RSD (%, n=5) | Added (μM) | Total founda (μM) | Recovery (%) | UA Conc. in serum (mM) |
|---|---|---|---|---|---|---|
| 1 | 4.68 | 2.8 | 1.5 | 6.12 | 96 | 0.468 |
| 2 | 4.87 | 2.5 | 1.5 | 6.45 | 105 | 0.487 |
| 3 | 4.64 | 3.9 | 1.5 | 6.05 | 94 | 0.464 |
| 4 | 4.80 | 4.1 | 1.5 | 6.35 | 103 | 0.480 |
| 5 | 4.97 | 3.4 | 1.5 | 6.40 | 95 | 0.497 |
Average of five determinations.
4. Concluding remarks
In the present work, a new method based on AuNP-enhanced CE-CL technique was developed for the determination of UA in serum samples. AuNPs were added into the running buffer of CE to catalyze the post-column chemiluminescence reaction between luminol and hydrogen peroxide, achieving highly efficient chemiluminescence emission. The presence of AuNPs not only enhanced the sensitivity of the detection, but also improved the selectivity as only a few endogenous components in the serum sample matrix produced CE-CL signals. Sample analyses carried out in this work showed that serum UA level for diabetic patients was significantly different from that for healthy subjects. These results indicated that the CE-CL assay proposed in this work was promising as a diagnostic tool for certain diseases such as diabetes.
Acknowledgments
The financial support from National Natural Science Foundation of China (NSFC, Grant No. 20665002 and 20875019 to SZ) and US National Institutes of Health (S06GM08047 to YML) is gratefully acknowledged.
Abbreviations used
- AuNP
gold nanoparticle
- CE
capillary electrophoresis
- CL
chemiluminescence
- UA
uric acid
References
- 1.Harper HA. Review of Physiological Chemistry. 13. Lange Medical Publications; Los Altos, CA: 1977. p. 406. [Google Scholar]
- 2.Heptinstall RH. Pathology of the Kidney. 2. Little Brown; Boston, MA: 1966. Gout; p. 495. [Google Scholar]
- 3.Ellerbe P, Cohen A, Welch MJ, White VE. Clin Chem. 1988;34:2280–2282. [PubMed] [Google Scholar]
- 4.Mazzali M, Kim YG, Hughes J, Lan HY, Kivlighn S, Johnson RJ. Am J Hypertens. 2000;13:S36–S37. [Google Scholar]
- 5.Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R. J Am Soc Nephrol. 2008;9:2251–2253. doi: 10.1681/ASN.2008010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dehghan A, Hofman A, Hoek MV, Witteman JCM, Sijbrands EJG. Diabetes Care. 2008;31:361–362. doi: 10.2337/dc07-1276. [DOI] [PubMed] [Google Scholar]
- 7.Perello J, Sanchis P, Grases F. J Chromatogr B. 2005;824:175–180. doi: 10.1016/j.jchromb.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 8.Dai X, Fang X, Zhang C, Xu R, Xu B. J Chromatogr B. 2007;857:287–295. doi: 10.1016/j.jchromb.2007.07.035. [DOI] [PubMed] [Google Scholar]
- 9.Sakuma R, Nishina T, Kitamura M. Clin Chem. 1987;33:1427–1433. [PubMed] [Google Scholar]
- 10.Martinez-Pérez D, Ferrer ML, Mateo CR. Anal Biochem. 2003;322:238–242. doi: 10.1016/j.ab.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 11.Akyilmaz E, Sezgintürk MK, Dinçkaya E. Talanta. 2003;61:73–79. doi: 10.1016/S0039-9140(03)00239-X. [DOI] [PubMed] [Google Scholar]
- 12.Fossati P, Prencipe L, Berti G. Clin Chem. 1980;26:227–234. [PubMed] [Google Scholar]
- 13.Cunningham K, Keaveny TV. Clin Chim Acta. 1978;86:217–221. doi: 10.1016/0009-8981(78)90135-3. [DOI] [PubMed] [Google Scholar]
- 14.Raj CR, Ohsaka T. J Electroanal Chem. 2003;540:69–77. [Google Scholar]
- 15.Ndamanisha JC, Guo L. Biosensors and Bioelectronics. 2008;23:1680–1685. doi: 10.1016/j.bios.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 16.Boughton JL, Robinson BW, Strein TG. Electrophoresis. 2002;23:3705–3710. doi: 10.1002/1522-2683(200211)23:21<3705::AID-ELPS3705>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 17.Guan Y, Chu Q, Ye J. Anal Bioanal Chem. 2004;380:913–917. doi: 10.1007/s00216-004-2848-y. [DOI] [PubMed] [Google Scholar]
- 18.Rosi NL, Mirkin CA. Chem Rev. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 19.Vanderpuije BNY, Han G, Rotello VM, Vachet RW. Anal Chem. 2006;78:5491–5496. doi: 10.1021/ac0604181. [DOI] [PubMed] [Google Scholar]
- 20.Song Y, Zhao S, Tchounwou P, Liu YM. J Chromatogr A. 2007;1166:79–84. doi: 10.1016/j.chroma.2007.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu CJ, Su CL, Tseng WL. Anal Chem. 2006;78:8004–10. doi: 10.1021/ac061059c. [DOI] [PubMed] [Google Scholar]
- 22.Zhang ZF, Cui H, Lai CZ, Liu LJ. Anal Chem. 2005;77:3324–3329. doi: 10.1021/ac050036f. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Hu J, Jin Y, Yao X, Li J. Clin Chem. 2006;52:1958–1961. doi: 10.1373/clinchem.2006.071399. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Z, Niu T, Song Y, Liu YM. Electrophoresis. 2009;30:1059–1065. doi: 10.1002/elps.200800283. [DOI] [PubMed] [Google Scholar]
- 25.Frens G. Nature (Physical Sciences) 1973;241:20–22. [Google Scholar]
- 26.Zhao SL, Xie C, Lu X, Song YR, Liu YM. Electrophoresis. 2005;26:1744–1749. doi: 10.1002/elps.200410338. [DOI] [PubMed] [Google Scholar]
- 27.Zuo Y, Wang C, Zhou J, Sachdeva A, Ruelos VC. Anal Sci. 2008;24:1589–1592. doi: 10.2116/analsci.24.1589. [DOI] [PubMed] [Google Scholar]
- 28.Dai X, Fang X, Zhang C, Xu R, Xu B. J Chromatogr B. 2007;857:287–295. doi: 10.1016/j.jchromb.2007.07.035. [DOI] [PubMed] [Google Scholar]
- 29.Fang B, Jiao S, Li M, Tao H. Anal Bioanal Chem. 2006;386:2117–22. doi: 10.1007/s00216-006-0873-8. [DOI] [PubMed] [Google Scholar]
- 30.Huang X, Li Y, Wang P, Wang L. Anal Sci. 2008;24:1563–1568. doi: 10.2116/analsci.24.1563. [DOI] [PubMed] [Google Scholar]