

Abstract
Small ubiquitin-related modifier (SUMO) is an ubiquitin-like protein that is covalently attached to a variety of target proteins. Unlike ubiquitination, sumoylation does not target proteins for proteolytic breakdown, but is instead involved in regulating multiple protein functional properties including protein-protein interactions and subcellular targeting, to name a few. Protein sumoylation has been particularly well characterized as a regulator of many nuclear processes as well as nuclear structure, making the characterization of this modification vital for understanding nuclear structure and function. Consequently, there has been intense interest in identifying new proteins that are targets of this modification and determining what role it plays in regulating their functions. This chapter presents methodologies for determining whether a particular protein is a substrate of sumoylation, and for identifying the lysine residue(s) where the modification occurs.
Keywords: Sumoylation, SUMO, SUMO-1, testis, ovary, spermatogenesis, ubc9, HSF1, HSF2
1. Introduction
Small ubiquitin related modifier (SUMO) was discovered as a modifier of mammalian proteins in 1997 (1, 2). SUMO has since been demonstrated to be a modifier of many important proteins, giving this modification a vital role in modulating a large number of important cellular processes (3–5). SUMO proteins are very similar to ubiquitin structurally, but sumoylation does not promote degradation of proteins and instead regulates key functional properties of target proteins. These properties include subcellular localization, protein partnering, and transactivation functions of transcription factors, among others (3–5). Protein sumoylation plays a particularly vital role in regulating many important processes occurring in the nucleus, and although sumoylation can be found on proteins that exist in a number of cellular compartments, most of the sumoylation characterized to date occurs on nuclear proteins (3–5). Indeed, proteins of the SUMO conjugation machinery have been found to be localized to nuclear pore complexes, in addition to other locations in the nucleus.
Sumoylation in the testis and ovary has only relatively recently begun to be investigated, but results already suggest that this post-translational modification plays an important role in the regulation and function of both female and male reproductive tissues. For example, sumoylation has been indicated to be important for regulating granulosa cell apoptosis during the differentiation of these cells (6). The existence of a functional connection between SUMO and the progesterone receptor has been suggested as it playes a role in progesterone receptor actions in the ovary (7). Sumoylation has been linked to XY body structure and function, heterochromatin organization, centromere function, and the expression of genes during spermatogenesis (8–10). A number of steroid receptors, including estrogen, progesterone, and androgen receptors have also been shown to be regulated by sumoylation, supporting the likely broad importance of this post-translational modification for proper control of reproductive processes in both females and males (11–13).
SUMO proteins are covalently attached to lysine residues of proteins, which are generally found within the consensus motifΨKXE where Ψ is a hydrophobic amino acid and X is any residue. Like ubiquitination, the covalent attachment of SUMO to other proteins involves a series of enzymatic steps (Fig. 1), but the proteins involved are distinct from those in the ubiquitin conjugation pathway. First, the SUMO proteins have to undergo proteolytic processing near their C-terminal end to form the mature proteins, a step which is performed by SUMO proteases (Ulp’s). These proteases are dual-functional, as they are also responsible for cleaving SUMO groups from substrate proteins by cleaving the isopeptide bonds by which they are joined (3–5, 14). The mature processed SUMO protein is covalently attached via a thioester bond to the SAE2 (Uba2) subunit of the heterodimeric SUMO E1 activating enzyme in an ATP-dependent reaction (15–18). The SUMO moiety is transferred from the E1 to ubc9, the SUMO E2 enzyme, which then binds to the ΨKXE consensus sequence in target proteins and forms an isopeptide bond between the ε-amino group of the lysine within this sequence and the carboxyl group of the C-terminal glycine of the SUMO polypeptide (19–22). SUMO E3 proteins have been identified that enhance the efficiency of SUMO attachment by interacting with both ubc9 (the E2 enzyme) and the substrate, thereby acting as bridging factors (3–5). Vertebrate cells contain three SUMO paralogs. SUMO-2 and SUMO-3 are very similar to each other in sequence, and have approximately 50% sequence identity with SUMO-1, which is the best characterized of the three vertebrate SUMO proteins.
Fig. 1.
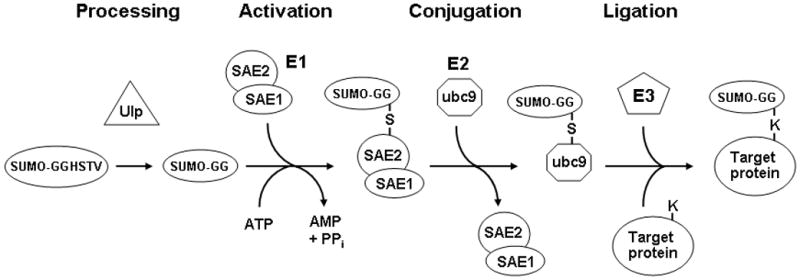
The SUMO conjugation pathway. After they are translated, SUMO proteins must first be processed by a SUMO protease such as Ulp1, which removes four C-terminal residues so that the mature form ends with a glycine. These SUMO proteases are also responsible for removing SUMO groups from proteins. This mature form is then activated in an ATP-dependent manner by forming a thioester bond with a cysteine residue in the SAE2 subunit of the heterodimeric E1 activating enzyme. Following this activation step, the SUMO moiety is transferred to the E2 conjugating enzyme ubc9. In the final step SUMO is transferred in a ligation reaction from ubc9 to substrate proteins, forming an isopeptide bond between the terminal glycine on SUMO and the ε-amino group of a lysine in the target protein. The efficiency of sumoylation of some proteins is enhanced by SUMO ligase E3 proteins, via their ability to bind both ubc9 and the target protein, thereby increasing the kinetics of the SUMO transfer.
In this chapter we describe two different experimental approaches for determining whether a specific protein is sumoylated. The first method employs immunoprecipitation of the protein of interest, either endogenous or transfected epitope-tagged protein, followed by Western blot with SUMO antibodies. The second method involves incubating the protein, either as a 35S-labeled in vitro translation product or purified recombinant protein, with a reconstituted in vitro sumoylation enzymatic reaction, followed by SDS-PAGE and autoradiography or Western blot, respectively, to look for the appearance of higher molecular weight bands indicative of sumoylation. By comparing wild type protein constructs with those containing non-sumoylatable arginine substitutions of candidate target lysine residues, these protocols can also allow identification of those lysine residues where SUMO attachment actually occurs in a given protein. This information then provides the critical reagents for testing the functional consequences of blocking sumoylation of that particular protein. To illustrate the types of data that can be obtained using these methodologies we present figures showing the results of immunoprecipitation and in vitro sumoylation analyses of a transcription factor, heat shock transcription factor-2 (HSF2).
2. Materials
2.1. Detection of Sumoylated Proteins by Immunoprecipitation Analysis
Cells expressing the protein of interest.
Phosphate-Buffered Saline (PBS): 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.5 mM KH2PO4.
Lysis solution: 0.15 M Tris-HCl, pH 6.7, 5% SDS, and 30% glycerol.
N-ethylmaleimide.
Complete protease inhibitor (Roche; Indianapolis, IN).
Protein G-sepharose or protein A-sepharose.
Primary antibody capable of immunoprecipitating protein of interest (or against epitope tag if analyzing a transfected tagged protein), species-matched non-specific IgG, and antibodies against SUMO-1, SUMO-2, or SUMO-3 (Invitrogen).
2x SDS sample buffer: 125 mM Tris-HCl, pH 6.7, 4% SDS, 20% glycerol, 0.2M dithiothreitol, and 0.01% bromophenol blue.
4x SDS sample buffer: 250 mM Tris-HCl, pH 6.7, 8% SDS, 40% glycerol, 0.4M dithiothreitol, and 0.02% bromophenol blue.
Polyacrylamide gel electrophoresis equipment and SDS-PAGE solutions.
Reagents for immunoblotting and detection (non-fat dried milk for blocking, and ECL or other detection system).
2.2. Detection of Sumoylated Proteins by In Vitro Sumoylation Analysis
Plasmid containing open reading frame of protein of interest oriented to be expressed from a T7 promoter in the vector.
pGEX-SUMO-1, pQE30-SUMO-1, pGEX-Ubc9, and pGEX-SAE2/SAE1 bicistronic expression construct, or purified SUMO, ubc9, SAE1/SAE2 available commercially (e.g. AE Biotech International).
Ampicillin and LB media.
IPTG (isopropyl-β-D-thio-galactopyranoside).
PMSF (phenylmethylsulfonyl fluoride).
Glutathione-agarose and Ni-NTA-agarose.
In vitro translation kit (e.g Promega TNT T7 Quick for PCR DNA kit).
10x sumoylation buffer: 500 mM Tris-HCl (pH 7.6), 500 mM KCl, 50 mM MgCl2, 10 mM DTT, and 10 mM ATP.
Creatine phosphokinase.
Creatine phosphate.
Inorganic pyrophosphatase.
Purified SAE1/SAE2 heterodimer (E1) (AEI international).
Purified ubc9 (E2).
Purified 6xHis-SUMO-1
French press (Thrermo Scientific).
Polyacrylamide gel electrophoresis equipment and SDS-PAGE solutions.
Whatman paper, X-ray film, and cassettes for detection of 35S-labeled proteins in sumoylation reaction.
Quickchange Site Directed Mutagenesis Kit (Stratagene).
3. Methods
3.1. Detection of Sumoylated Proteins by Immunoprecipitation Analysis
In this protocol, proteins to be tested for sumoylation are immunoprecipitated using lysis buffers designed to block the action of desumoylating enzymes (see Note 1) and then subjected to Western blot analysis using anti-SUMO antibodies to look for the appearance of a band with a size consistent with a sumoylated form of the protein. Although the theoretical molecular weight of the SUMO proteins is approximately 11 kDa, the size increase for each SUMO added on SDS-PAGE is typically in the range of 15–17 kDa. In the case of a protein with multiple sumoylation sites, or where SUMO chains form on a lysine target site (see Note 2), multiples of this size increase are expected, sometimes yielding very large shifts in mobility. Multiple bands representing different sumoylation states of the protein are also possible. This approach can be used to analyze endogenous proteins, or transfected proteins containing an epitope tag (FLAG, myc, etc.) (see Note 3). The transfection approach can also be used to determine the lysine residue(s) to which the SUMO group is attached, by comparing the sumoylation of wildtype constructs to ones in which candidate lysines have been changed to non-sumoylatable arginines. Two different lysis conditions are described, one using SDS to inhibit desumoylase enzymes and the other containing N-ethylmaleimide, a chemical inhibitor of these enzymes.
Tissue culture cells are grown in media appropriate for that cell line or primary cell type, and typically at least 1×106 cells are needed for each immunoprecipitation.
For harvesting place the plate of cells on ice, remove media by aspiration and add 1ml of ice-cold PBS to the plate. Scrape cells off plates with a cell scraper, transfer to a 1.5 ml microcentrifuge tube.
Collect cells by centrifugation at 13,000× g for 30 seconds at room temperature, and remove supernatant by aspiration.
Lyse cells in 150μl of lysis solution, which is then diluted 1:10 in PBS/0.5% NP40 plus complete protease inhibitor (Roche). If the lysate is highly viscous, perform sonication until viscosity is reduced. Centrifuge the lysates at 16,000× g, 10 min, 4°C to remove cellular debris. Alternatively, the cells can be lysed in any of the standard immunoprecipitation lysis buffers (e.g. RIPA, etc.) known to extract the protein of interest, providing the lysis solution is supplemented with 20 mM N-ethylmaleimide (desumoylase inhbitor, freshly dissolved).
While the cell lysate is being centrifuged, prepare 30μl of 50% protein G-Sepharose (or protein A-sepharose if that is preferential for the particular antibody) as per manufacturer’s instructions. After protein G-Sepharose has been prepared add the above cell lysate to the protein G-Sepharose and rotate at 4°C for 30 min to preclear the lysates to reduce non-specific binding.
Pellet the protein G-Sepharose by centrifugation (16,000× g, 20 seconds, 4°C) and transfer the supernatant to a new tube. At this point take 30μl of the supernatant, place in a separate tube with 10 μl 4x SDS sample buffer and label “input”.
Divide the remainder of the supernatant into 2 equal amounts in separate 1.5 ml centrifuge tubes. To one of the tubes add a sufficient amount of primary antibody and to the other tube add a species-matched non-specific IgG. Place samples on rotator at 4°C for 1hr after which add 20 μl of protein G-Sepharose that has been washed with PBS, and rotate at 4°C for 3 hours.
Collect beads by centrifugation (16,000× g, 10 seconds, 4°C) and discard supernatant.
Wash the beads 4 times with PBS/0.5% NP40 plus complete protease inhibitor, or other lysis buffer if another was chosen, collecting the beads by centrifugation after each wash (16,000× g, 10 sec, 4°C). Add 30μl 2x SDS sample buffer to beads after removing supernatant from final wash.
Boil the samples for 5 minutes and separate on SDS-PAGE gel. Separate immunoprecipitated proteins by SDS-polyacrylamide gel electrophoresis.
Transfer proteins to nitrocellulose or nylon membrane and subject to Western blotting using antibodies against SUMO-1, SUMO-2, or SUMO-3 (commercially available). This methodology can also be used to examine the sumoylation state of an epitope-tagged version of the protein of interest being expressed in transfected cells (see Note 3). The results of immunoprecipitation analysis to examine sumoylation of the HSF2 protein in HeLa cell extracts is shown in Fig 2.
Fig. 2.

Detection of sumoylation by immunoprecipitation/SUMO-1 Western blot. HSF2 protein was immunoprecipitated from extracts of HeLa cells followed by Western blot using anti-SUMO-1 antibodies. The positions of molecular weight standards are indicated on the left side of the panel. This figure is from our published work (24), used with permission.
3.2 In vitro Sumoylation Assay
In this protocol the protein of interest is in vitro translated (typically with 35S-methionine incorporation) and then incubated in a reaction containing the SUMO E1 and E2 enzymes and SUMO-1, followed by SDS-PAGE and autoradiography to determine whether a lower mobility band appears that would be consistent with a sumoylated form of the target protein. Because of the high concentrations of SUMO E1 and E2 enzymes used in this in vitro sumoylation reaction, the need for SUMO E3 proteins is diminished and thus sumoylation can be detected without their addition. Performing the in vitro sumoylation reaction using mutants of the protein in which candidate sumoylation site lysine residues are changed to non-sumoylatable arginines can be used to determine the site(s) at which SUMO attachment is occurring. The expression and purification of the recombinant proteins required for the in vitro sumoylation assay is described in subheadings 3.2.1, and the protocol for performing assay itself is described in subheading 3.2.3.
3.2.1. Expression of Recombinant SUMO, ubc9 (SUMO E2), and SAE1/SAE2 (SUMO E1) Heterodimer
The SUMO proteins are expressed and purified as fusion proteins with GST or 6xHis at the N-terminal end of the SUMO proteins, and these affinity tags do not need to be removed prior to using these SUMO proteins for the sumoylation reaction. The size difference of GST-SUMO vs. 6xHis-SUMO can also provide a useful control for the sumoylation reaction as it yields a predictable size difference between the sizes of the sumoylation products (e.g. see Figure 3). The SUMO E1 is a heterodimer of SAE1/SAE2, and is active when expressed in E. coli from a bicistronic contruct of GST-SAE2 and untagged SAE1; the two proteins complex and can be purified using glutathione-agarose affinity chromatography (23). The SUMO E2 can be expressed which appears to be more active, at least in our hands when the GST tag is removed by thrombin cleavage. The following is the general protocol for expressing and purifying these recombinant proteins needed for the in vitro sumoylation assay. Once purified these recombinant proteins can be stored at −80°C for extended periods of time.
Fig. 3.

Analysis of SUMO-1 modification by reconstituted in vitro sumoylation reaction. (A) In vitro translated 35S-labeled HSF2 protein was incubated with HeLa cytosol (as a source of E1), Ubc9, SUMO-1, SUMO-2 or with various combinations of each of these, and then subjected to SDS-PAGE followed by autoradiography. The positions of unmodified and SUMO-modified HSF2 are indicated to the right of the panel. (B) In vitro translated 35S-labeled HSF2 protein was subjected to the in vitro SUMO-1 modification assay using either 6xHis-SUMO-1 or GST-SUMO-1 as the SUMO-1 substrate for the reaction. This figure is from our published work (24), used with permission.
Transform expression construct into DH10B E. coli cells using standard molecular biology methods.
Plate cells on LB plates containing ampicillin and incubate overnight at 37°C.
Select single colonies and grow overnight at 37°C in LB media containing ampicillin.
Inoculate individual liters of LB containing ampicillin with aliquots (5 mL) of overnight culture and grow cells to an O.D.600nm = 0.6–0.8.
Induce the cells with IPTG (1mM) for 3 hours.
After 3 hr induction harvest cells by centrifugation (4,000× g; 5 min; 4°C) and resuspend pellet in PBS (4°C) then repellet cells. At this point the cell pellet can be extracted or stored at −80°C.
To extract protein from the cells they are resuspended in PBS (4°C) with PMSF (phenylmethylsulfonyl fluoride) to a final concentration of 1mM.
Pass cells through a French press at 10,000 to 14,000 psi, and repeat to ensure complete lysis.
Centrifuge the cell lysate (30,000× g; 1 h; 4°C) and retain the supernatant.
Purify proteins using standard nondenaturing affinity chromatography techniques suitable for their fusion tags (glutathione agarose affinity chromatography for GST-fusion proteins and Ni-NTA-agarose affinity chromatography for 6xHis-fusion proteins).
Check purified proteins by Coomassie blue staining of a SDS-PAGE gel (GST-SUMO-1= 38kDa, GST-Ubc9=44kDa, and 6xHis-SUMO-1=14kDa).
For the in vitro sumoylation assay the GST-Ubc9 needs to be thrombin cleaved to remove the GST-tag. This can be done following manufacturer’s protocols. Check thrombin cleavage of GST-Ubc9 by Coomassie staining of a SDS-PAGE gel (Ubc9=18 kDa).
3.2.2 In Vitro Sumoylation of Rabbit Reticulocyte System Translated Proteins
The in vitro sumoylation assay uses 35S-methionine radiolabeled protein as the substrate. We generate radiolabeled protein using the TNT T7 Quick for PCR DNA kit following the manufacturers instructions. Described below is the SUMO modification procedure to be utilized with radiolabeled translated proteins.
Translate target protein fresh before each sumoylation assay following the manufacturer’s instructions. Place freshly translated protein on ice until step 3.
For each sumoylation reaction prepare a mixture containing the following (on ice): 1 μl 10x sumoylation buffer, 0.4 units creatine phosphokinase, 10 mM creatine phosphate, 0.6 units inorganic pyrophosphatase, 100 ng purified SAE1/SAE2 heterodimer (E1), 400 ng purified ubc9 (E2), 1μg purified 6xHis-SUMO-1, and H2O to 10 μl. As a negative control, make another mix identical to the one above except that it lacks SUMO protein. If desired a third mix can be made in which GST-SUMO-1 is added instead of 6xHis-SUMO.
Set up the reactions (no SUMO control, +6xHis-SUMO, and +GST-SUMO) by adding 2μl in vitro translated protein to the 10μl reaction mixes, and then incubate at 37°C for 1 hr.
Terminate the reaction by adding 12 μl 2x SDS sample buffer. Store at −20°C until gel electrophoresis.
Boil the samples for 5 minutes and separate on SDS-PAGE gel. After electrophoresis the gel is fixed for 10 minutes in SDS-PAGE fixing solution (50% methanol/10% acetic acid), then dried on Whatman paper, and finally placed on x-ray film. 35S emissions are low energy and so it is advisable not to leave plastic wrap between the dried gel and the X-ray film if possible, as this can increase required exposure times.
One important experiment to do to give confidence in the in vitro sumoylation assay is to do a reconstitution test where each of the components required for sumoylation (E1, ubc9, SUMO-1) are individually left out of the reaction and compared to a reaction where all components are present (Fig. 3A). As shown in this figure, such an experiment can also reveal the relative efficiency of sumoylation of your target protein by different SUMO proteins (e.g. SUMO-1 vs. SUMO-2). Another experiment which increases confidence that your protein is indeed being sumoylated vs. being targeted by some other modification is to compare the effect of using of 6xHis-SUMO vs. GST-SUMO as the donor SUMO, because this gives a predictable size shift between the sumoylated reactions (Fig. 3B).
To identify the sumoylation site in the protein of interest site directed mutagenesis is done to change candidate lysine (changed to arginine) in the sumoylation consensus sequence (ΨKXE) to non-sumoylatable arginine residues utilizing protocols such as the Quickchange Site Directed Mutagenesis Kit. The extent of sumoylation of the wild type vs. lysine-to-arginine mutant can then be compared using either the transfection/immunoprecipitation approach or the in vitro sumoylation assay. Results of such an experiment using the in vitro sumoylation assay are shown in Fig 4.
Fig. 4.

In vitro translated 35S-labeled wildtype HSF2 protein and the HSF2 SUMO-1 consensus site mutants K82R, K139R, and K151R were used as substrates for in vitro SUMO-1 modification reactions. The positions of unmodified and SUMO-modified HSF2 proteins are indicated to the right of the panel. This figure is from our published work (24), used with permission.
Acknowledgments
We are very grateful to Mike Matunis (Johns Hopkins), Ron Hay (University of Dundee), and Chris Lima (Sloan Kettering Institute) for providing constructs and reagents.
Footnotes
SUMO-modified proteins are highly susceptible to SUMO proteases. The SDS in the lysis buffer described in this protocol inactivates the SUMO proteases allowing for easier detection of sumoylated proteins. However, a common complication with the SDS-lysis method is that the cell lysates tend to be very viscous and sticky due to genomic DNA in the lysate. This problem is remedied by brief sonication which shears the DNA and makes the samples easier to manipulate. SUMO proteases can also be inhibited by the addition isopeptidase inhibitor N-ethylmaleimide (20mM) to standard lysis buffers such as NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40) if another lysis buffer besides the SDS-lysis is more desirable.
The size of putative sumoylated forms of a protein on SDS-PAGE will depend on how many different SUMO attachment sites the protein has. In addition, SUMO-2 and SUMO-3 have been reported to form polymeric chains reminiscent of ubiquitin, which could result in large increases in size for a sumoylated protein on SDS-PAGE compared to the non-sumoylated form (23). Thus, it is possible to observe bands that are multiples of the approximate 15–17 kDa size of each SUMO unit, as well as multiple bands representing different sumoylation states.
Investigating sumoylation may also be done using cells transfected with fusion-tagged plasmid constructs of the protein thought to be sumoylated with immunoprecipitation utilizing fusion tag antibodies which are available from commercial sources (e.g. GFP, FLAG, Myc, and 6xHis tags). In these types of experiments it is advisable to co-transfect the cells with a SUMO expression construct (often this is epitope-tagged) to ensure that sufficient SUMO protein is present in the transfected cells to allow for efficient sumoylation of the transfected target protein being tested.
References
- 1.Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- 2.Matunis MJ, Wu J, Blobel G. SUMO-1 modification and its role in targeting the Ran GTPase-activating protein, RanGAP1, to the nuclear pore complex. J Cell Biol. 1998;140:499–509. doi: 10.1083/jcb.140.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 4.Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 5.Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Shao R, Rung E, Weijdegard B, Billig H. Induction of apoptosis increases SUMO-1 protein expression and conjugation in mouse periovulatory granulosa cells in vitro. Mol Reprod Dev. 2006;73:50–60. doi: 10.1002/mrd.20386. [DOI] [PubMed] [Google Scholar]
- 7.Shao R, Zhang FP, Rung E, Palvimo JJ, Huhtaniemi I, Billig H. Inhibition of small ubiquitin-related modifier-1 expression by luteinizing hormone receptor stimulation is linked to induction of progesterone receptor during ovulation in mouse granulosa cells. Endocrinology. 2004;145:384–392. doi: 10.1210/en.2003-0527. [DOI] [PubMed] [Google Scholar]
- 8.Rogers RS, Inselman A, Handel MA, Matunis MJ. SUMO modified proteins localize to the XY body of pachytene spermatocytes. Chromosoma. 2004;113:233–243. doi: 10.1007/s00412-004-0311-7. [DOI] [PubMed] [Google Scholar]
- 9.Vigodner M, Morris PL. Testicular expression of small ubiquitin-related modifier-1 (SUMO-1) supports multiple roles in spermatogenesis: silencing of sex chromosomes in spermatocytes, spermatid microtubule nucleation, and nuclear reshaping. Dev Biol. 2005;282:480–492. doi: 10.1016/j.ydbio.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 10.Vigodner M, Ishikawa T, Schlegel PN, Morris PL. SUMO-1, human male germ cell development, and the androgen receptor in the testis of men with normal and abnormal spermatogenesis. Am J Physiol Endocrinol Metab. 2006;290:E1022–1033. doi: 10.1152/ajpendo.00527.2005. [DOI] [PubMed] [Google Scholar]
- 11.Wu F, Mo YY. Ubiquitin-like protein modifications in prostate and breast cancer. Front Biosci. 2007;12:700–711. doi: 10.2741/2094. [DOI] [PubMed] [Google Scholar]
- 12.Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol. 2006;102:11–21. doi: 10.1016/j.jsbmb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Faus H, Haendler B. Post-translational modifications of steroid receptors. Biomed Pharmacother. 2006;60:520–528. doi: 10.1016/j.biopha.2006.07.082. [DOI] [PubMed] [Google Scholar]
- 14.Mukhopadhyay D, Dasso M. Modification in reverse: the SUMO proteases. Trends Biochem Sci. 2007;32:286–295. doi: 10.1016/j.tibs.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 1997;16:5509–5519. doi: 10.1093/emboj/16.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desterro JM, Rodriguez MS, Kemp GD, Hay RT. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J Biol Chem. 1999;274:10618–10624. doi: 10.1074/jbc.274.15.10618. [DOI] [PubMed] [Google Scholar]
- 17.Gong L, Li B, Millas S, Yeh ET. Molecular cloning and characterization of human AOS1 and UBA2, components of the sentrin-activating enzyme complex. FEBS Lett. 1999;448:185–189. doi: 10.1016/s0014-5793(99)00367-1. [DOI] [PubMed] [Google Scholar]
- 18.Okuma T, Honda R, Ichikawa G, Tsumagari N, Yasuda H. In vitro SUMO-1 modification requires two enzymatic steps, E1 and E2. Biochem Biophys Res Commun. 1999;254:693–698. doi: 10.1006/bbrc.1998.9995. [DOI] [PubMed] [Google Scholar]
- 19.Desterro JM, Thomson J, Hay RT. Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 1997;417:297–300. doi: 10.1016/s0014-5793(97)01305-7. [DOI] [PubMed] [Google Scholar]
- 20.Johnson ES, Blobel G. Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J Biol Chem. 1997;272:26799–26802. doi: 10.1074/jbc.272.43.26799. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem. 2001;276:12654–12659. doi: 10.1074/jbc.M009476200. [DOI] [PubMed] [Google Scholar]
- 22.Sampson DA, Wang M, Matunis MJ. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J Biol Chem. 2001;276:21664–21669. doi: 10.1074/jbc.M100006200. [DOI] [PubMed] [Google Scholar]
- 23.Tatham MH, Jaffray E, Vaughan OA, Desterro JM, Botting CH, Naismith JH, Hay RT. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 2001;276:35368–35374. doi: 10.1074/jbc.M104214200. [DOI] [PubMed] [Google Scholar]
- 24.Goodson ML, Hong Y, Rogers R, Matunis MJ, Park-Sarge OK, Sarge KD. Sumo-1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J Biol Chem. 2001;276:18513–18518. doi: 10.1074/jbc.M008066200. [DOI] [PubMed] [Google Scholar]