

Abstract
We have developed a model in which inflammation contiguous to and within a dorsal root ganglion (DRG) was generated by local application of complete Freund’s adjuvant (CFA) to the L4 lumbar spinal nerve as it exits from the intervertebral foramen. The periganglionic inflammation (PGI) elicited a marked reduction in withdrawal threshold to mechanical stimuli and an increase in heat pain sensitivity in the ipsilateral hindpaw in the absence of any hindpaw inflammation. The pain sensitivity appeared within hours and lasted for a week. The PGI also induced a prominent increase in IL-1 β and TNF-α levels in the DRG and of cyclooxygenase-2 (COX-2) expression in neurons and satellite cells. A selective COX-2 inhibitor reduced the PGI-induced hyperalgesia. We also show that IL-1 β induces COX-2 expression and prostaglandin release in DRG neurons in vitro in a MAP kinase-dependent fashion. The COX-2 induction was prevented by ERK and p38 inhibitors. We conclude that periganglionic inflammation increases cytokine levels, including IL-1 β, leading to the transcription of COX-2 and prostaglandin production in the affected DRG, and thereby to the development of a dermatomally distributed pain hypersensitivity.
Keywords: Inflammation, Low back pain, Sciatica, Dorsal root ganglion, Cyclooxygenase 2
1. Introduction
Although acute low back pain (LBP) is amongst the most common of medical complaints [3] the pathophysiology of the pain is poorly understood. Acute low back pain may result from mechanical compression of spinal roots and the dorsal root ganglion [8,59], arthritis of intervertebral articular joints and damage or inflammation of paravertebral muscles and tendons [34]. Although intervertebral disc herniation is considered to produce pain primarily by mechanical compression of spinal roots [8], recent data also suggest a role for local inflammation [42]. High levels of IL-1α, IL-1β, IL-6 and TNF-α are present in herniated lumbar discs [53]. Pain radiating from the back down the leg to the foot is a characteristic symptom of lumbar radiculopathy, is present in 25–57% of LBP cases [48] and correlates with the severity of the disease [50]. Radiculopathy may not always be accompanied by clearly defined nerve root compression or sensory or motor deficits [42], implying that other factors, such as inflammation, may contribute in some patients, particularly in the acute phase. However, it is uncertain whether inflammation of the dorsal root or DRG without compression can by itself induce pain hypersensitivity. This is what we have now investigated.
Prostaglandin E2 (PGE2), a major pain enhancing inflammatory mediator [47], is produced from arachidonic acid by a sequence of enzymes that include cyclooxygenase (COX). Tissue damage and inflammation result in a strong local induction of COX-2, the inducible isoform of COX [57]. In addition, peripheral inflammation induces via a central action of IL-1β, COX-2 in spinal cord dorsal horn neurons [23,46,54,61]. Although DRG neurons are capable of expressing COX-2 in response to direct cytokine application in vitro [16], neither peripheral nerve injury nor distal peripheral inflammation induce COX-2 in the DRG in vivo [6]. Nevertheless, since NSAIDs [56] and COX-2 selective inhibitors [28,43] reduce pain in some patients with acute LBP, a COX dependent prostaglandin synthesis is likely in some way and at some location, to contribute to the pain hypersensitivity.
Peripheral inflammation induces a local inflammatory hyperalgesia that is due both to the sensitization of nociceptor peripheral terminals at the site of the inflammation (peripheral sensitization) [27] and of dorsal horn neurons (central sensitization) [60], both of which involve COX-2 induction, the former locally in immune cells [4,49] and the latter in dorsal horn neurons [46]. When the sciatic nerve is treated locally at the mid thigh level with CFA [14] or zymosan [7] this also produces a sustained pain hypersensitivity in the hindpaw in the absence of inflammation in the paw.
We have now investigated whether inflammation of the DRG induces distally radiating pain hypersensitivity. To test this CFA was applied directly to the spinal segmental nerve at its exit from the intervertebral foramen. We find that direct inflammation of the spinal nerve and DRG produces pain hypersensitivity in the distal dermatome and that this is associated with COX-2 induction in the DRG.
2. Methods
Experiments were approved by the Animal Use Committee of the Massachusetts General Hospital and followed the ethical guidelines of International Association for the Study of Pain [63]. Experiments were performed on adult (250–300 g) male Sprague–Dawley rats (Chares River lab, MA).
2.1. Periganglionic inflammation (PGI)
Under 2% of isoflurane general anesthesia a sagittal skin incision (2–3 cm) was made in the lumbar region 1 cm lateral to the midline and the L5 transverse process removed. A small piece (3–5 mm square) of cotton gauze immersed with 10 μl of CFA (Sigma, MO) was placed on the exposed L4 spinal nerve just distal to the DRG. Muscle and skin were closed in two layers with 3–0 silk. All procedure was performed in sterilized technique and animals did not receive postoperative antibiotics or anti-inflammatory drugs. Sham controls received the same procedure with exposure of the nerve to small piece (3–5 mm square) of cotton gauze immersed with 10 μl of Saline. An investigator who was not involved in the experimental procedure checked the animal’s status and general behavior including food intake, activity, fighting and sleep, throughout the experiment.
To asses inflammation related vascular permeability, Evans blue leakage test was performed 3 days after the PGI treatment. Evans blue (1%, 5 ml, Sigma) was intraperitoneally injected under isoflurane anesthesia and bilateral L4 DRGs were taken 2 h after the injection. DRGs were incubated with formamide at 50 °C overnight and Evans blue concentration was determined by absorption spectrometer. To investigate inflammatory reactions of the tissue, L4 DRG and the plantar tissue ipsilateral to the PGI treatment were removed under terminal anesthesia with isoflurane. The tissues were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (PB) at 4 °C, 4 h. The sections were cut at 12 μm and mounted onto saline-coated slides and stained with Mayer’s haematoxylin for 10 min and, subsequently, with eosin for 1 min. Dermal thickness was determined under the microscope.
2.2. Sciatic nerve and peripheral inflammation
Cotton gauze immersed with 10 μl of CFA was applied onto the sciatic nerve at the popliteal fossa [14] and peripheral inflammation induced by an intraplantar injection of 100 μl CFA in separate groups of animals, under isoflurane anesthesia [1].
2.3. Behavioral assessments
To measure mechanical sensitivity, unrestricted rats were placed in a clear plastic chamber on an elevated wire grid and allowed to acclimate. Withdrawal responses to mechanical stimulation were determined using a calibrated von Frey monofilament set (1, 1.4, 2, 4, 6, 8, 10, 15, 26, 60 gm) (Stoelting company, IL) [11]. The threshold was taken as the lowest force that evoked clear withdrawal response at least twice in 10 applications. The medial and lateral sides as well as the middle part of the plantar surface (see Fig. 2A) were stimulated. For thermal sensitivity, animals were placed in Perspex boxes and the hindpaw exposed to a beam of radiant heat (Ugo Basile Inc, Italy) [22]. Withdrawal latency time was recorded with a minimal value of 0.5 s. The mean of three measurements taken between 10 and 15 min apart was taken as the latency to paw withdrawal. To investigate the contribution of COX-2 to the behavioral hyperalgesia in PGI animals, rofecoxib, a selective COX-2 inhibitor (1 mg/kg in 1 ml) [6,18] was administrated subcutaneously 3 days after the PGI treatment. Mechanical and thermal sensitivity was measured before and 30 min after the rofecoxib treatment.
Fig. 2.

Pain hypersensitivity in the hindpaw following PGI. (A) Sites tested on the paw surface are indicated by a, b, and c. (B) Mechanical thresholds (von Frey) of these three sites on the ipsilateral plantar surface after PGI. (C) Time course of the change in mechanical threshold in area a. (D) Changes in withdrawal time to a noxious radiant heat stimulus of the ipsilateral hindpaw after PGI. N = 7 for each group. *p < 0.05 vs. sham operation group. **p < 0.01 vs. sham operation group.
2.4. Gene expression
RNA was prepared after tissue homogenization in Trizol (Invitrogen, CA), quantified using a QUBIT fluorimeter (Invitrogen) and integrity assessed using a standard RNAse free agarose gel. Expression of IL-1β and TNF-α was quantified by real-time PCR. Reverse transcription was carried out with SuperScript First-Strand Synthesis (Invitrogen) using 1 μg of total RNA. The PCRs were assembled in 96 well plates (Applied Biosystems, MA) with 0.5 μl of cDNA and a final primer concentration of 0.5 μM. Quantitative PCR was performed using an ABI Prism 7000 real-time PCR system (Applied Biosystems) and was analyzed using SDS 7000 software. The house-keeping gene GAPDH was used as a loading control. Primers for IL-1β (5′-GGCTGAC AGACCCCAAAAGAT-3′ and 5′-CTTGTCCAGATGCTGCTGTGA-3′) were designed using Primer Express 2.0 (Applied Biosystems), and primers forTNF-α(5′-AAGCCTGTAGCCCACGTCGTA-3′ and5′-GGCACCACTAG TTGGTTGTCTTTG-3′) were purchased from Takara Bio Inc (Japan). COX-2 mRNA was measured by RNase protection assay using a radiolabeled riboprobe generated by PCR from rat spinal cord complementary DNA using primers 5′-GCAAATCCTTGCTGTTCCAACCCA-3′ and 5′-TTGGGGATCCGGGATGAACTCTCT-3′, and subsequently cloned into pCRII vector (Invitrogen). The plasmid was linearized and an antisense probe synthesized using Sp6 RNA polymerase, and labeled with [32P]UTP (800 μCi/mmol; NEN, MA). RNase protection assays (RPAs) were performed using the RPA III (Ambion, TX) protocol [46]. 10 μg RNA samples were hybridized with labeled probe overnight at 42 °C and were then digested with RNase A/RNase T1 mix in RNase digestion buffer for 30 min at 37 °C. Finally, the samples were separated on denaturing acrylamide gel and exposed to X-rayfilms. A β-actinprobe wasused asa loading control. Signaldensity was calculated by NIH Image-J software with Windows XP computer.
2.5. In situ hybridization histochemistry
The L4 DRG was removed under terminal anesthesia and kept in −80 °C. Sections were cut at 12 μm and mounted onto silane-coated slides. Sections were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (PB) at 4 °C 10 min and were washed three times with 0.1 M phosphate buffered saline (PBS) for 5 min. Then the sections were acetylated in 0.25% acetic anhydride in 0.1 M triethanolamine for 10 min at room temperature and were hybridized with riboprobe diluted in hybridization buffer (Sigma) at 60 °C for 12 h in a humidified chamber. Sections were washed in 5 × SSC for 10 min, 0.2 × SSC for 15 min, and 0.1 × SSC for 30 min at 60 °C and incubated with anti-DIG-AP antibody (1:1000, Roche, Switzerland) in DIG buffer 1 (100 mM Tris–HCl, pH 7.5, 150 mM NaCl) for 12 h at 4 °C. Signals were visualized by incubating the sections with 4.5 μl/ml of 5-bromo-4-chloro-3-indolyl-phosphate and 3.5 μl/ml of 4-Nitroblue tetrazolium chloride in DIG buffer 3 (100 mM Tris–HCl, pH 7.5, 100 mM NaCl, 50 mM MgCl2).
2.6. Immunohistochemistry
Under terminal anesthesia, rats were perfused with normal saline followed by 4% paraformaldehyde in 0.1 M PB. The L4 DRG was removed and postfixed for 2 h in the same fixative solution, then cryoprotected in 20% sucrose in 0.1 M PBS for 24 h at 4 °C. Sections were cut and mounted as above. After washing with 0.1 M PBS, sections were incubated with rabbit anti COX-2 antibody (Tocris, MI; 1:1000) for 2 days at 4 °C. The sections were incubated with biotinylated anti rabbit secondary antibody (Vector Laboratories, CA; 1:1000) for 2 h at room temperature. Signals were visualized with Vectastain ABC systems (Vector) following manufacturers’ instruction.
Fluorescent immunohistochemistry was performed for double staining. Sections were incubated with rabbit anti COX-2 antibody and mouse anti NeuN (Chemicon, CA; 1:400) or GFAP (Chemicon; 1:100) or CD11b (Chemicon; 1:100) in 0.1MPBS for 2 days at 4 °C. The sections were then washed and incubated with Rhodamine conjugated anti rabbit and FITC conjugated anti mouse antibodies (Chemicon; 1:100).
2.7. DRG Culture
Lumbar and thoracic DRGs were harvested from freshly dissected naïve adult rats (180–250 g) and placed into HBSS (Life Technologies), digested with 5 mg/ml collagenase 1 mg/ml Dispase II (Roche) for 120 min, 0.25% trypsin (GibcoBRL, MD) for 8 min at 37 °C. Ganglia were then washed in DMEM (GibcoBRL, MD) and triturated through a flame-polished pipette ~ 10 times, and the suspension was centrifuged through 15% BSA (fatty acid free; Sigma) in DMEM. The pellet was resuspended in Neurobasal (GibcoBRL) containing B27 supplement (GibcoBRL), penicillin and streptomycin (Sigma), 10 μM Ara-C and 100 ng/ml 2.5 S NGF (Promega, WI). Cells were preplated onto uncoated tissue culture dishes for 1.5 h at 37 °C. Nonadherent cells were then recovered from the dishes by gentle pipetting and plated on poly-lysine (Sigma)-coated glass slides or plastic dishes. Cells were grown at 37 °C in 5% CO2 for 48 h [2]. DRG cultures were pretreated with the MAP kinase p38 inhibitor SB203580 (Calbiochem, CA, 20 μM) and/or the MAP kinase kinase (ERK) inhibitor-2 PD98059 (Sigma, 50 μM) 20 min before the IL-1β treatment. Cultures were incubated for an additional 6 h with IL-1β (Sigma, 10 ng/ml), DRG neurons were then harvested for further investigations.
2.8. Statistics
Statistical analysis was performed with one-way ANOVA followed by Turkey’s multiple comparison test or repeated-two way ANOVA followed by Bonferroni post test (comparison of sham vs. PGI in the three foot paw regions). Significance was determined as p < 0.05.
3. Results
We set out to produce inflammation in proximity to the DRG with minimal compression or damage to its vascular supply, and without a laminectomy. To do this, we applied 10 μl of CFA on a small piece of cotton gauze to the L4 lumbar spinal segmental nerve, exposed by the removal of the transverse process, as it emerged from the intervertebral foramen (Fig. 1A). Immunohistochemistry for CD11b, a marker for macrophages [44], revealed very few macrophages in naïve DRGs but a significant infiltration at the site of CFA application and proximal to this throughout the DRG, when measured 3 days following the treatment (Fig. 1B). Hematoxylin–Eosin (HE) staining of the DRG and nerve root taken from PGI animals showed no indication of gross mechanical compression (Fig. 1C). Wet weight of the ipsilateral L4 DRG from PGI treated animals did not differ significantly compared to contralateral L4 DRG (4.2 ± 0.7 vs. 3.7 ± 0.6 mg, n = 5 each). The Evans blue leakage test showed a similar amount of extravascular Evans blue level in the psi- and contralateral L4 DRGs (49.7 ± 4.7 vs. 44.9 ± 7.7 μg/mg of tissue, n = 5 each). Macroscopically, however, there was visible edema in the connective tissue near the spinal nerve (Fig. 1).
Fig. 1.

Periganglionic inflammation (PGI). (A) Procedure for surgical treatment of PGI. After identifying L5 process (A-1), the process was cut to visualize L4 nerve (A-2). Cotton gauze with CFA or saline was put onto the nerve (A-3). Acute inflammation of the L4 spinal nerve produced by CFA application immediately distal to the L4 DRG. (B) Immunohistochemistry for CD11b, a marker for activated macrophages shows the accumulation of immune cells at the site of the CFA application and in the DRG. Red CD11b, blue DAPI nuclear staining. CD11b positive cells were not observed in DRGs from naive animals. (C) Hematoxylin–Eosin (HE) staining of the DRG from PGI animals. No sign of mechanical compression was observed. Arrows indicate inflammation site. Scale bar = 500 μm. (D) HE staining of the skin of the hindpaw from PGI and naive animals. No inflammatory reaction (inflammatory cell accumulation or tissue edema) was observed in the paw. Scale bar = 200 μm.
3.1. Behavioral experiments
All animals that received the CFA treatment contiguous to the DRG developed a marked hypersensitivity of the ipsilateral hindpaw, which itself displayed no erythema or swelling. HE staining of the plantar surface of the hindpaw failed to reveal any inflammatory reaction (immune cell infiltration and edema) in the skin tissue of the paw following PGI (Fig. 1D). Dermal thickness of the paw treated with PGI was not significantly different from naive control (286 ± 35 μm in naive vs. 288 ± 15 μm in PGI). No autotomy or weight loss was observed, and sleep wake cycles and social interaction with other rats were not affected, indicating minimal systemic distress.
Mechanical stimulation was applied to three areas of the hindpaw (Fig. 2A) to investigate distribution changes in sensitivity in different dermatomes affected by the CFA application. Hypersensitivity to innocuous mechanical stimulation developed across the entire plantar surface including the medial, middle and lateral areas of the hindpaw 3 and 5 days following PGI. Hypersensitivity was most robust in the middle surface area (Fig. 2B). A time course study of the mechanical hypersensitivity measured at the middle plantar surface revealed that the mechanical threshold, 20.8 ± 3.1 g at baseline, decreased 3 days following the establishment of the PGI to 4.29 ± 0.6 g, p < 0.01 (Fig. 2C). The mechanical threshold was still reduced 7 days following the treatment but returned to baseline at 14 days. The withdrawal latency for radiant noxious heat in naive controls was 20.7 ± 0.7 s. and this began to decrease 14 h following CFA treatment of the spinal nerve with a significant reduction at 3 and 7 days (Fig. 2D) and a return to baseline at 14 days. The hindpaw contralateral to the treatment did not show any change in mechanical or thermal sensitivity and animals that received sham operations (exposure of spinal nerve but no CFA) did not show any altered hypersensitivity (Fig. 2).
3.2. COX-2 expression
No COX-2 mRNA was found in naive DRGs by RNase protection but a robust increase in COX-2 mRNA was detected in the ipsilateral L4 DRG 3 days after the PGI (Fig. 3A). Increased COX-2 mRNA was also detected in the L5 DRG and to a much less extent in the L3 DRG. DRGs from sham animals as well as those with intraplantar or perisciatic CFA induced inflammation showed a minimal increase in COX-2 mRNA. In the spinal cord, a significant induction of COX-2 mRNA was observed after peripheral inflammation but not after PGI (Fig. 3B). Moreover, quantitative RT-PCR showed significant increases in mRNA levels for the pro-inflammatory cytokines IL-1β (Fig. 3C) and TNF-α (Fig. 3D) in the DRG in response to PGI.
Fig. 3.

COX-2 and inflammatory cytokine induction in the DRG following PGI. (A) COX-2 mRNA levels measured in L3–L5 DRGs, 3 days after PGI and compared to a sham operation or hindpaw inflammation. (B) Hindpaw inflammation but not PGI-induced COX-2 mRNA in the dorsal horn (DH). (C) Increase in IL-1β mRNA in L4 DRG of PGI treated animals. (D) Increase in TNF-α mRNA in L4 DRG of PGI treated animals. *p < 0.05 vs. sham control. **p < 0.01 vs. sham control. N = 4 for each group of animals.
Fig. 4 shows COX-2 mRNA and protein expression in the DRG following PGI. In response to PGI COX-2 mRNA was detected in both neuronal and non-neuronal cells. Non-neuronal COX-2 expression was first detected at 6 h and continued for 3 days. Neuronal COX-2 mRNA could first be detected at 1 day and was still present at 7 days. Consistent with the increased mRNA expression, COX-2 protein was also detected in the DRG 3 days following PGI in neurons as well as perineuronal connective tissue (Fig. 4B). Double labeling immunohistochemistry of COX-2 with neuronal and glial markers confirmed expression of COX-2 within NeuN positive neurons, GFAP positive satellite cells and CD11b positive macrophages (Fig. 4C). After inflammation of the sciatic nerve in the popliteal fossa produced by perineural CFA administration, a small amount of non-neuronal COX-2 expression was observed in the DRG, but none in neurons 3 days following the treatment. We conclude that the inflammation needs to be adjacent to the ganglion in order to induce COX-2 expression in DRG neurons.
Fig. 4.

COX-2 expression in the DRG after PGI. (A) COX-2 mRNA detected by in situ hybridization. COX-2 mRNA is first detected in small cells 6 h after the periganglionic CFA treatment. Three days after treatment, relatively strong COX-2 expression is observed in neurons. Scale bar = 50 μm. (B) Immunohistochemical study showing the distribution of COX-2 positive cells in the DRG 3 days after the PGI treatment, compared to sham. COX-2 positive cells are detected within (red square) and adjacent to the DRG (blue square). Scale bar = 1000 μm. (C) Higher magnification of two areas in B. Top: COX-2 expression observed in the connective tissue (area in the blue square in B). Bottom: COX-2 expression in neuronal and satellite cells in the DRG (area in the red square in B). Arrows indicate COX-2 positive satellite cells. Scale bar = 50 μm. (D) Neuronal COX-2 expression. Double immunohistochemistry for COX-2 (red) with NeuN, neuronal cell marker (green) revealed that COX-2 co-localizes with the neuronal cell marker NeuN in the DRG. Scale bar = 1200 μm. (E) Non-neuronal COX-2 expression. A single confocal optical section analysis of COX-2 expression (red) with GFAP (green) or CD11b (green) positive cells. COX-2 expression co-localizes with GFAP and CD11b. The COX-2/GFAP images were taken within the DRG, while the COX-2/CD11b images were taken in an area adjacent to the DRG. Scale bar = 50 μm.
3.3. COX-2 inhibition and PGI
The reduction in mechanical threshold and the increase in heat sensitivity in the hindpaw produced by PGI were significantly reversed by systemic administration of the COX-2 selective inhibitor rofecoxib (1 mg/kg), (Fig. 5A and B). Three days after PGI treatment thermal response latency (14.74 ± 0.76 s) and mechanical threshold (6.4 ± 1.1 g) were significantly reduced from baseline. Rofecoxib (1 mg/kg), subcutaneously injected 30 min prior to the behavioral testing, successfully reversed the thermal response time (18.31 ± 0.72 s, n = 5, p < 0.01) and mechanical threshold (17.0 ± 3.8 g, n = 5, p < 0.01) to the pre-PGI basal levels.
Fig. 5.

A selective COX-2 inhibitor rofecoxib (1 mg/kg administrated subcutaneously) reduced the mechanical (A) and thermal (B) pain hypersensitivity produced by PGI. Behavioral analysis was performed 3 days after the PGI procedure, and the effect of rofecoxib was assessed 30 min after administration. N = 7 in each group. ++: p < 0.01 vs. pre-treatment baseline. **p < 0.01 vs. saline injected control.
3.4. Signal transduction involving COX-2 induction after PGI
In order to assess whether IL-1β can induce COX-2 expression in DRG neurons, and study the signal transduction pathway involved, we analyzed the effect of IL-1β on cultured DRG neurons from unoperated animals. A robust induction of COX-2 mRNA and protein occurred in cultured DRG neurons in response to the application of IL-1β (10 ng/ml, 20 min) (Fig. 6A and B) that was reduced in an additive manner by PD98059, an ERK kinase inhibitor, and SB203580, a p38 MAP kinase inhibitor (Fig. 6A). The induction of COX-2 by IL-1β in DRG neuron cultures led to an increase in PGE2 levels in the culture media that was prevented by treatment with the p38 and ERK inhibitors (Fig. 6C).
Fig. 6.
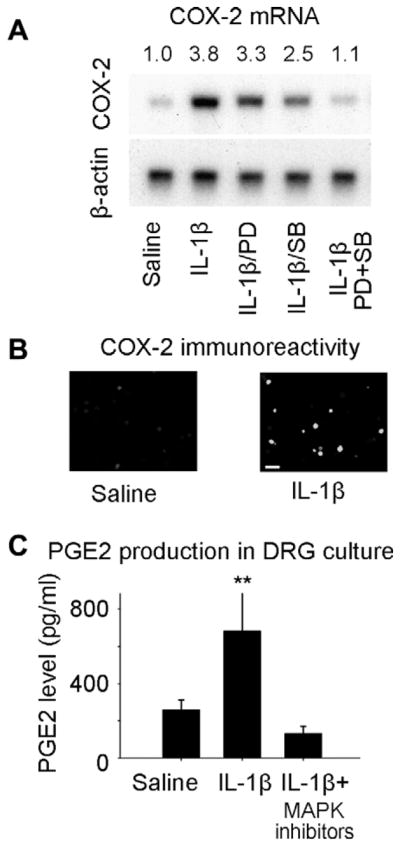
(A) Signal pathways involved in COX-2 induction in DRG neurons cultured from na rats 6 h after exposure to IL-1β. (A) Application of IL-1β to cultured DRG neurons produced an increase in COX-2 mRNA that was partially blocked by administration either of SB203580, a p38 MAP kinase inhibitor or of PD98059, an ERK kinase inhibitor. Co-application of the p38/ERK inhibitors blocked COX-2 induction almost completely. The COX-2/β-actin signal density ratio is presented above the image. (B) COX-2 immunoreactivity in cultured DRG neurons after IL-1β. Scale bar = 50 μm (C) Prostaglandin levels in the media of cultured DRG neurons were increased by IL-1β in a p38/ERK dependent fashion.
4. Discussion
We find that inflammation of the proximal L4 spinal nerve and the contiguous DRG, constituting what we term periganglionic inflammation (PGI), produces an increase in heat pain sensitivity and a reduction in mechanical threshold in the ipsilateral hindpaw that lasts for days. PGI also induces a robust induction of COX-2 in the DRG, in contrast with peripheral inflammation of either the hindpaw or sciatic nerve, indicating that these models differ substantially, and that COX-2 induction in DRG neurons requires direct exposure to locally produced inflammatory mediators such as IL-1β and TNF-α. The induction of COX-2 in DRG neurons occurs in response to exposure to IL-1β, and is mediated by ERK and p38 MAPK. ERK and p38 MAPK are phosphorylated by IL-1β to activate COX-2 transcription in several different non-neuronal cell lines [17,37].
Periganglionic inflammation also induces the transcription of several regeneration-associated genes [33] as a result of a local induction of IL-6 and LIF [10,55], and produces MAP kinase activation [40]. DRG compression to model spinal stenosis, with a rod inserted into the intervertebral foramen, promotes increased spontaneous electrical activity of DRG neurons and thermal and mechanical hyperalgesia that appears to be mediated by the induction and action of cytokines and chemokines in DRG neurons [9,24,51,58]. Dorsal root compression with a silastic tube or steel rod induces a loss of myelinated fibers [26,62], increases spontaneous electrical activity [24], and produces behavioral hyperalgesia [59]. These different studies indicate that local inflammation and compression of the dorsal root and DRG can elicit diverse changes in response to local production of inflammatory signaling molecules.
Proteoglycans in the nucleus pulposus (NP) elicit immune reactions [35] resulting in immune cell invasion [45] and immunoglobulin deposition [21]. Herniated discs contain many pro-inflammatory cytokines such as IL-1α, IL-1β, IL-6 and TNFα [12,25,53]. Homogenated nucleus pulposus injection into the epidural space provokes an inflammatory response [35], increases BDNF expression in the DRG [39] and produces behavioral hyperalgesia [29,30]. In patients with chemical radiculitis [42], radicular pain is present in the absence of signs of nerve compression [41].
We applied CFA to the distal side of DRG. Although this approach will not mimic the direct contact of herniated disc contents to dorsal roots, as occurs most commonly with intervertebral prolapse, this approach avoids the risk of damage to dorsal roots or spinal cord that may be produced by laminectomy and exposure of the subarachnoid space. Nonetheless, we observed a dermatomally distributed pain hypersensitivity in the hindpaw. Based on these findings we suggest that the local inflammatory changes in the DRG that result from a lateral or foraminal disc prolapse [15] and any other causes of periganglionic inflammation, may be sufficient to produce a radicular pattern of pain hypersensitivity with a COX-2 component.
Nonetheless, we do not rule out that inflammation-induced elevations in mechanical tissue pressure might also contribute to the pathogenesis of the pain hypersensitivity we observe in the PGI model. The spinal nerve inflammation may also directly contribute to changes in axonal excitability and thereby to the pain phenotype.
The onset of thermal hyperalgesia in the PGI model occurs earlier (already significant 14 h post injury) than mechanical hyperalgesia, and tends to recover earlier. While our data point to COX-2 induction contributing to both mechanical and thermal hyperlagesia, changes in the DRG and spinal cord may contribute differentially to these two forms of hyperalgesia, resulting in relatively different time course profiles.
COX-2 inhibitors and NSAIDs are frequently prescribed for patients with acute low back pain [5]. A systematic review of 51 randomized controlled trials concluded that NSAID treatment is efficacious for symptomatic relief of acute low back pain [56] and selective COX-2 inhibitors have a similar efficacy [43]. We suggest that the target for the action of the NSAIDs and COX-2 inhibitors is likely to include COX-2 induced in the DRG in those patients where a prolapsed disk has induced inflammation, as well as inflammatory reactions in joints and soft tissue in patients with axial low back pain. Moreover, our data are consistent with a study suggesting that the induction of COX-2 following nerve injury contributes to pain hypersensitivity in humans [13]. We do not know if the inflammation of a dorsal root proximal to the DRG induces COX-2 in the DRG and how close to the DRG such radicular inflammation needs to be.
Inflammation in the vicinity of the DRG with subsequent release of cytokines (e.g. IL-1β, TNF-α), chemokines and inflammatory mediators such as prostanoids and bradykinin, are likely to contribute to pain hypersensitivity in the PGI model by local alterations, such as increased spontaneous activity of DRG neurons, as well as by distant changes due, for example, to changed levels of proteins transported to peripheral and central terminals. The absence of any sign of inflammation at the peripheral terminals following PGI suggests that inflammation close to the DRG is sufficient to produce pain hypersensitivity in skin areas innervated by the DRG.
Radiculopathy patients frequently report radiating pain below the knee and in some cases to the foot [48]. Far lateral disc herniations, which constitute 7–12% of all herniations and directly impact the DRG, include particularly severe radicular pain and unremitting leg pain is frequently present [15]. There is very little formal documentation though, on whether mechanical or thermal allodynia occurs on the foot or leg in patients with acute lumbar radiculopathy. In a recent study using quantitative sensory testing (QST) on 12 patients with chronic radicular pain (with evidence of nerve damage), one patient had tactile allodynia on the foot [19]. It is not possible to assay spontaneous radiating pain in rats, but the animals do display allodynia in the hindpaw in our model, and indeed in all rodent models of radiculopathy using compression, nerve injury or inflammation [20,31,32,36,38,52]. A prospective QST study in patients with acute radicular low back pain, with and without evidence of axonal damage due to compression, is needed to determine if thermal or mechanical allodynia does occur in the foot and if local inflammation may be sufficient. This would help assess if our rodent model is a useful surrogate for the pathophysiology and sensory manifestations of acute radiculopathy in patients.
In conclusion, we have demonstrated that inflammation contiguous to the DRG is sufficient to produce a dermatomally distributed pain hypersensitivity that is contributed to by a MAPK mediated induction of COX-2 in the DRG. PGI may model the inflammatory component of acute degenerative disc disease and some of the pathophysiological changes underlying the generation of some forms of NSAID and COX-2 inhibitor sensitive acute low back pain including sciatica, and provide thereby a means for screening for potential therapeutic interventions.
Acknowledgments
C.J.W. was supported by the NIH (NS38253 and NS039518). F.A. was supported by JSPS (18791094). The authors are grateful to Ms. Mika Sasaki for her technical assistance and Dr. Richard Mannion for discussion.
Footnotes
Conflict of interest
The authors do not have a conflict of interest. T.S. is currently employed by Wyeth.
References
- 1.Amaya F, Oh-hashi K, Naruse Y, Iijima N, Ueda M, Shimosato G, et al. Local inflammation increases vanilloid receptor 1 expression within distinct subgroups of DRG neurons. Brain Res. 2003;963:190–6. doi: 10.1016/s0006-8993(02)03972-0. [DOI] [PubMed] [Google Scholar]
- 2.Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J, et al. The voltage-gated sodium channel Na(v)1 9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26:12852–60. doi: 10.1523/JNEUROSCI.4015-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersson GB. Epidemiological features of chronic low-back pain. Lancet. 1999;354:581–5. doi: 10.1016/S0140-6736(99)01312-4. [DOI] [PubMed] [Google Scholar]
- 4.Appleton I, Tomlinson A, Mitchell JA, Willoughby DA. Distribution of cyclooxygenase isoforms in murine chronic granulomatous inflammation. Implications for future anti-inflammatory therapy. J Pathol. 1995;176:413–20. doi: 10.1002/path.1711760413. [DOI] [PubMed] [Google Scholar]
- 5.Arnau JM, Vallano A, Lopez A, Pellise F, Delgado MJ, Prat N. A critical review of guidelines for low back pain treatment. Eur Spine J. 2006;15:543–53. doi: 10.1007/s00586-005-1027-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broom DC, Samad TA, Kohno T, Tegeder I, Geisslinger G, Woolf CJ. Cyclooxygenase 2 expression in the spared nerve injury model of neuropathic pain. Neuroscience. 2004;124:891–900. doi: 10.1016/j.neuroscience.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, et al. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral perisciatic immune activation in rats. Pain. 2001;94:231–44. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- 8.Cornefjord M, Sato K, Olmarker K, Rydevik B, Nordborg C. A model for chronic nerve root compression studies. Presentation of a porcine model for controlled, slow-onset compression with analyses of anatomic aspects, compression onset rate, and morphologic and neurophysiologic effects. Spine. 1997;22:946–57. doi: 10.1097/00007632-199705010-00003. [DOI] [PubMed] [Google Scholar]
- 9.Cuellar JM, Montesano PX, Carstens E. Role of TNF-alpha in sensitization of nociceptive dorsal horn neurons induced by application of nucleus pulposus to L5 dorsal root ganglion in rats. Pain. 2004;110:578–87. doi: 10.1016/j.pain.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 10.Curtis R, Scherer SS, Somogyi R, Adryan KM, Ip NY, Zhu Y, et al. Retrograde axonal transport of LIF is increased by peripheral nerve injury: correlation with increased LIF expression in distal nerve. Neuron. 1994;12:191–204. doi: 10.1016/0896-6273(94)90163-5. [DOI] [PubMed] [Google Scholar]
- 11.Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–58. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 12.Doita M, Kanatani T, Harada T, Mizuno K. Immunohistologic study of the ruptured intervertebral disc of the lumbar spine. Spine. 1996;21:235–41. doi: 10.1097/00007632-199601150-00015. [DOI] [PubMed] [Google Scholar]
- 13.Durrenberger PF, Facer P, Gray RA, Chessell IP, Naylor A, Bountra C, et al. Cyclooxygenase-2 (Cox-2) in injured human nerve and a rat model of nerve injury. J Peripher Nerv Syst. 2004;9:15–25. doi: 10.1111/j.1085-9489.2004.09104.x. [DOI] [PubMed] [Google Scholar]
- 14.Eliav E, Herzberg U, Ruda MA, Bennett GJ. Neuropathic pain from an experimental neuritis of the rat sciatic nerve. Pain. 1999;83:169–82. doi: 10.1016/s0304-3959(99)00102-5. [DOI] [PubMed] [Google Scholar]
- 15.Epstein NE. Foraminal and far lateral lumbar disc herniations: surgical alternatives and outcome measures. Spinal Cord. 2002;40:491–500. doi: 10.1038/sj.sc.3101319. [DOI] [PubMed] [Google Scholar]
- 16.Fehrenbacher JC, Burkey TH, Nicol GD, Vasko MR. Tumor necrosis factor alpha and interleukin-1beta stimulate the expression of cyclooxygenase II but do not alter prostaglandin E2 receptor mRNA levels in cultured dorsal root ganglia cells. Pain. 2005;113:113–22. doi: 10.1016/j.pain.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 17.Fiebich BL, Mueksch B, Boehringer M, Hull M. Interleukin-1beta induces cyclooxygenase-2 and prostaglandin E(2) synthesis in human neuroblastoma cells: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB. J Neurochem. 2000;75:2020–8. doi: 10.1046/j.1471-4159.2000.0752020.x. [DOI] [PubMed] [Google Scholar]
- 18.Francischi JN, Chaves CT, Moura AC, Lima AS, Rocha OA, Ferreira-Alves DL, et al. Selective inhibitors of cyclo-oxygenase-2 (COX-2) induce hypoalgesia in a rat paw model of inflammation. Br J Pharmacol. 2002;137:837–44. doi: 10.1038/sj.bjp.0704937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freynhagen R, Rolke R, Rutjes AK, Baron R, Tolle TR, Schu S, et al. Pseudoradicular and radicular low-back pain – a disease continuum rather than different entities? Rebuttal: reply to the letter “Cheese and Chalk? Missing the real anatomy” by Breck McKay. Pain. 2008;137:230–1. [Google Scholar]
- 20.Gu X, Yang L, Wang S, Sung B, Lim G, Mao J, et al. A rat model of radicular pain induced by chronic compression of lumbar dorsal root ganglion with SURGIFLO. Anesthesiology. 2008;108:113–21. doi: 10.1097/01.anes.0000296073.16972.13. [DOI] [PubMed] [Google Scholar]
- 21.Habtemariam A, Gronblad M, Virri J, Seitsalo S, Ruuskanen M, Karaharju E. Immunocytochemical localization of immunoglobulins in disc herniations. Spine. 1996;21:1864–9. doi: 10.1097/00007632-199608150-00005. [DOI] [PubMed] [Google Scholar]
- 22.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 23.Hay CH, Trevethick MA, Wheeldon A, Bowers JS, de Belleroche JS. The potential role of spinal cord cyclooxygenase-2 in the development of Freund’s complete adjuvant-induced changes in hyperalgesia and allodynia. Neuroscience. 1997;78:843–50. doi: 10.1016/s0306-4522(96)00598-2. [DOI] [PubMed] [Google Scholar]
- 24.Hu SJ, Xing JL. An experimental model for chronic compression of dorsal root ganglion produced by intervertebral foramen stenosis in the rat. Pain. 1998;77:15–23. doi: 10.1016/S0304-3959(98)00067-0. [DOI] [PubMed] [Google Scholar]
- 25.Igarashi A, Kikuchi S, Konno S, Olmarker K. Inflammatory cytokines released from the facet joint tissue in degenerative lumbar spinal disorders. Spine. 2004;29:2091–5. doi: 10.1097/01.brs.0000141265.55411.30. [DOI] [PubMed] [Google Scholar]
- 26.Jancalek R, Dubovy P. An experimental animal model of spinal root compression syndrome: an analysis of morphological changes of myelinated axons during compression radiculopathy and after decompression. Exp Brain Res. 2007;179:111–9. doi: 10.1007/s00221-006-0771-5. [DOI] [PubMed] [Google Scholar]
- 27.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–10. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 28.Katz N, Ju WD, Krupa DA, Sperling RS, Bozalis RD, Gertz BJ, et al. Efficacy and safety of rofecoxib in patients with chronic low back pain: results from two 4-week, randomized, placebo-controlled, parallel-group, double-blind trials. Spine. 2003;28:851–8. doi: 10.1097/01.BRS.0000059762.89308.97. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami M, Tamaki T, Hayashi N, Hashizume H, Nishi H. Possible mechanism of painful radiculopathy in lumbar disc herniation. Clin Orthop Relat Res. 1998:241–51. [PubMed] [Google Scholar]
- 30.Kawakami M, Tamaki T, Weinstein JN, Hashizume H, Nishi H, Meller ST. Pathomechanism of pain-related behavior produced by allografts of intervertebral disc in the rat. Spine. 1996;21:2101–7. doi: 10.1097/00007632-199609150-00009. [DOI] [PubMed] [Google Scholar]
- 31.Kirita T, Takebayashi T, Mizuno S, Takeuchi H, Kobayashi T, Fukao M, et al. Electrophysiologic changes in dorsal root ganglion neurons and behavioral changes in a lumbar radiculopathy model. Spine. 2007;32:E65–72. doi: 10.1097/01.brs.0000252202.85377.96. [DOI] [PubMed] [Google Scholar]
- 32.LaCroix-Fralish ML, Rutkowski MD, Weinstein JN, Mogil JS, Deleo JA. The magnitude of mechanical allodynia in a rodent model of lumbar radiculopathy is dependent on strain and sex. Spine. 2005;30:1821–7. doi: 10.1097/01.brs.0000174122.63291.38. [DOI] [PubMed] [Google Scholar]
- 33.Lu X, Richardson PM. Inflammation near the nerve cell body enhances axonal regeneration. J Neurosci. 1991;11:972–8. doi: 10.1523/JNEUROSCI.11-04-00972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manchikanti L, Singh V, Pampati V, Damron KS, Barnhill RC, Beyer C, et al. Evaluation of the relative contributions of various structures in chronic low back pain. Pain Physician. 2001;4:308–16. [PubMed] [Google Scholar]
- 35.McCarron RF, Wimpee MW, Hudkins PG, Laros GS. The inflammatory effect of nucleus pulposus. A possible element in the pathogenesis of low-back pain. Spine. 1987;12:760–4. doi: 10.1097/00007632-198710000-00009. [DOI] [PubMed] [Google Scholar]
- 36.Mizuno S, Takebayashi T, Kirita T, Tanimoto K, Tohse N, Yamashita T. The effects of the sympathetic nerves on lumbar radicular pain: a behavioural and immunohistochemical study. J Bone Joint Surg Br. 2007;89:1666–72. doi: 10.1302/0301-620X.89B12.19258. [DOI] [PubMed] [Google Scholar]
- 37.Molina-Holgado E, Ortiz S, Molina-Holgado F, Guaza C. Induction of COX-2 and PGE(2) biosynthesis by IL-1beta is mediated by PKC and mitogen-activated protein kinases in murine astrocytes. Br J Pharmacol. 2000;131:152–9. doi: 10.1038/sj.bjp.0703557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murata Y, Olmarker K, Takahashi I, Takahashi K, Rydevik B. Effects of lumbar sympathectomy on pain behavioral changes caused by nucleus pulposus-induced spinal nerve damage in rats. Eur Spine J. 2006;15:634–40. doi: 10.1007/s00586-005-1020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Obata K, Tsujino H, Yamanaka H, Yi D, Fukuoka T, Hashimoto N, et al. Expression of neurotrophic factors in the dorsal root ganglion in a rat model of lumbar disc herniation. Pain. 2002;99:121–32. doi: 10.1016/s0304-3959(02)00068-4. [DOI] [PubMed] [Google Scholar]
- 40.Obata K, Yamanaka H, Dai Y, Mizushima T, Fukuoka T, Tokunaga A. Activation of extracellular signal-regulated protein kinase in the dorsal root ganglion following inflammation near the nerve cell body. Neuroscience. 2004;126:1011–21. doi: 10.1016/j.neuroscience.2004.04.036. [DOI] [PubMed] [Google Scholar]
- 41.Ohnmeiss DD, Vanharanta H, Ekholm J. Degree of disc disruption and lower extremity pain. Spine. 1997;22:1600–5. doi: 10.1097/00007632-199707150-00015. [DOI] [PubMed] [Google Scholar]
- 42.Peng B, Wu W, Li Z, Guo J, Wang X. Chemical radiculitis. Pain. 2007;127:11–6. doi: 10.1016/j.pain.2006.06.034. [DOI] [PubMed] [Google Scholar]
- 43.Pohjolainen T, Jekunen A, Autio L, Vuorela H. Treatment of acute low back pain with the COX-2-selective anti-inflammatory drug nimesulide: results of a randomized, double-blind comparative trial versus ibuprofen. Spine. 2000;25:1579–85. doi: 10.1097/00007632-200006150-00019. [DOI] [PubMed] [Google Scholar]
- 44.Robinson AP, White TM, Mason DW. Macrophage heterogeneity in the rat as delineated by two monoclonal antibodies MRC OX-41 and MRC OX-42, the latter recognizing complement receptor type 3. Immunology. 1986;57:239–47. [PMC free article] [PubMed] [Google Scholar]
- 45.Saal JS. The role of inflammation in lumbar pain. Spine. 1995;20:1821–7. doi: 10.1097/00007632-199508150-00013. [DOI] [PubMed] [Google Scholar]
- 46.Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–5. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 47.Samad TA, Sapirstein A, Woolf CJ. Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets. Trends Mol Med. 2002;8:390–6. doi: 10.1016/s1471-4914(02)02383-3. [DOI] [PubMed] [Google Scholar]
- 48.Schafer A, Hall T, Briffa K. Classification of low back-related leg pain – a proposed patho-mechanism-based approach. Man Ther. 2007 doi: 10.1016/j.math.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci USA. 1994;91:12013–7. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Selim AJ, Ren XS, Fincke G, Deyo RA, Rogers W, Miller D, et al. The importance of radiating leg pain in assessing health outcomes among patients with low back pain. Results from the Veterans Health Study. Spine. 1998;23:470–4. doi: 10.1097/00007632-199802150-00013. [DOI] [PubMed] [Google Scholar]
- 51.Sun JH, Yang B, Donnelly DF, Ma C, LaMotte RH. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J Neurophysiol. 2006;96:2189–99. doi: 10.1152/jn.00222.2006. [DOI] [PubMed] [Google Scholar]
- 52.Tachihara H, Kikuchi S, Konno S, Sekiguchi M. Does facet joint inflammation induce radiculopathy? An investigation using a rat model of lumbar facet joint inflammation. Spine. 2007;32:406–12. doi: 10.1097/01.brs.0000255094.08805.2f. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi H, Suguro T, Okazima Y, Motegi M, Okada Y, Kakiuchi T. Inflammatory cytokines in the herniated disc of the lumbar spine. Spine. 1996;21:218–24. doi: 10.1097/00007632-199601150-00011. [DOI] [PubMed] [Google Scholar]
- 54.Tegeder I, Niederberger E, Vetter G, Brautigam L, Geisslinger G. Effects of selective COX-1 and -2 inhibition on formalin-evoked nociceptive behaviour and prostaglandin E(2) release in the spinal cord. J Neurochem. 2001;79:777–86. doi: 10.1046/j.1471-4159.2001.00613.x. [DOI] [PubMed] [Google Scholar]
- 55.Thompson SW, Vernallis AB, Heath JK, Priestley JV. Leukaemia inhibitory factor is retrogradely transported by a distinct population of adult rat sensory neurons: co-localization with trkA and other neurochemical markers. Eur J Neurosci. 1997;9:1244–51. doi: 10.1111/j.1460-9568.1997.tb01479.x. [DOI] [PubMed] [Google Scholar]
- 56.van Tulder MW, Scholten RJ, Koes BW, Deyo RA. Nonsteroidal anti-inflammatory drugs for low back pain: a systematic review within the framework of the Cochrane Collaboration Back Review Group. Spine. 2000;25:2501–13. doi: 10.1097/00007632-200010010-00013. [DOI] [PubMed] [Google Scholar]
- 57.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 58.White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, et al. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci USA. 2005;102:14092–7. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winkelstein BA, Weinstein JN, DeLeo JA. The role of mechanical deformation in lumbar radiculopathy: an in vivo model. Spine. 2002;27:27–33. doi: 10.1097/00007632-200201010-00009. [DOI] [PubMed] [Google Scholar]
- 60.Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA. 1999;96:7723–30. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yaksh TL, Dirig DM, Conway CM, Svensson C, Luo ZD, Isakson PC. The acute antihyperalgesic action of nonsteroidal, anti-inflammatory drugs and release of spinal prostaglandin E2 is mediated by the inhibition of constitutive spinal cyclooxygenase-2 (COX-2) but not COX-1. J Neurosci. 2001;21:5847–53. doi: 10.1523/JNEUROSCI.21-16-05847.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoshizawa H, Kobayashi S, Morita T. Chronic nerve root compression. Pathophysiologic mechanism of nerve root dysfunction. Spine. 1995;20:397–407. doi: 10.1097/00007632-199502001-00001. [DOI] [PubMed] [Google Scholar]
- 63.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–10. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]