

Abstract
As a primary drug for the treatment of acute lymphoblastic leukemia (ALL), encapsulation of L-asparaginase (ASNase) into red blood cells (RBC) has been popular to circumvent immunogenicity from the exogenous protein. Unlike existing methods that perturbs RBC membranes, we introduce a novel method of RBC-incorporation of proteins using the membrane-traslocating low molecular weight protamine (LMWP). Confocal study of fluorescence-labeled LMWP-ovalbumin, as a model protein conjugate, has shown significant fluorescence inside RBCs. Surface morphology by scanning electron microscopy of the RBCs loaded with LMWP-ASNase was indistinguishable with normal RBCs. These drug loaded RBCs also closely resembled the profile of the native erythrocytes in terms of osmotic fragility, oxygen dissociation and hematological parameters. The in vivo half-life of enzyme activity after administering 8 units of RBC/LMWP-ASNase in DBA/2 mice was prolonged to 4.5±0.5 days whereas that of RBCs loaded with ASNase via a hypotonic method was 2.4±0.7 days. Furthermore, the mean survival time of DBA/2 mice bearing mouse lymphoma cell L5178Y was improved by ∼44% compared to the saline control group after treatment with the RBC loaded enzymes. From these data, an innovative, novel method for encapsulating proteins into intact and fully functional erythrocytes was established for potential treatment of ALL.
Keywords: Red blood cell (RBC) carriers, RBC encapsulation, asparaginase therapy, protein transduction domain (PTD) peptide, acute lymphoblastic leukemia (ALL)
Introduction
Acute lymphoblastic leukemia (ALL) is cancer of the white blood cells, the cells that normally fight infections. In patients with ALL, the bone marrow produces excess immature white blood cells, called lymphoblasts, which are unable to help the body fight infections. Normal hematopoiesis is also inhibited resulting in a deficiency of normal white blood cells, red blood cells, and platelets. As a consequence ALL patients often suffer infections, anemia, and easy bleeding. Almost 4,000 cases of ALL are diagnosed annually in the United States alone, approximately two thirds of which are in adolescent children, making ALL the most common cancer in this age group. Indeed, ALL represents 23% of the cancer diagnoses among children younger than 15 years of age, occurring at an annual rate of 30 to 40 per million population [1]. While a cure rate of ∼80% was estimated for childhood ALL, the experience with adult ALL was far less rewarding, as the reported cure rate seldom exceeded 40% [2].
One of the primary drugs used in the treatment of ALL is L-asparaginase (ASNase) from E. coli, which has been in clinical use since 1967 [3]. ASNase is an enzyme which hydrolyzes amino acid L-asparagine (ASN) to L-aspartic acid and ammonia. Most human tissues can self-synthesize ASN from L-glutamine by the action of asparagine synthetase (AS). Certain neoplastic tissues, including ALL cells, however, express a significantly lower level of AS, and thus have to rely solely on extracellular source of ASN to maintain protein synthesis. Systemic depletion of ASN by ASNase would therefore impair protein biosynthesis [4] in these cells, leading to their deaths through cellular dysfunction [5].
Due to the bacterial origin of ASNase, immunogenicity is a major problem with high frequency of hypersensitivity reactions. Most reactions occur within one to several hours after administration, include signs and symptoms typical of anaphylaxis, and are sometimes fatal. So far, remedies for this problem include a change in the route of administration from intravenous to intramuscular or subcutaneous injection, alter source of enzyme from E. coli to Erwinia, or use of pegylated ASNase [6]. Presently referred to as pegaspargase, pegylated ASNase was developed in the 1970s with first clinical trials in 1980s. Attaching polyethylene glycol (PEG) chains increases mass of the enzyme and shields ASNase from proteolytic enzymes, improving pharmacokinetics of ASNase [7]. This pegylated form is better tolerated than the free form, especially when given intramuscularly, and is specifically recommended for treatment in patients who are sensitive to ASNase. According to the review of clinical studies, pegaspargase reduces frequency of ASNase administration from 6-10 to 1-2 times and can be administered to selected ALL patients with hypersensitivity to the E. coli drug for re-induction therapy. However, pegaspargase has not demonstrated superiority over E. coli ASNase for the first remission of ALL [8]. Aside from allergic reactions, the use of ASNase can result in liver dysfunction. Abnormal bilirubin and alkaline phosphatase levels and depression in albumin and lipoprotein levels are often observed [9]. Other toxicities include coagulation abnormalities, pancreatitis, cerebral dysfunction, parotitis, and immune suppression [9].
Another means to overcome toxicity issues related to use of free ASNase was use of erythrocytes as the drug carrier, advantages of which have been thoroughly reviewed by Hamidi and Tajerzadeh [10] In addition to their abundant supply in blood, erythrocytes would protect the loaded ASNase from inactivation by proteolytic degradation and immune surveillance such as destruction by reticuloendothelial system (RES). Previous study by Ataullakhanov and co-workers showed that ASN is able to diffuse freely into human erythrocytes from an external medium [11]. Therefore, ASNase-encapsulated erythrocytes could act as a circulating bioreactor, converting incoming ASN to aspartic acid.
Erythrocytes have an unmatched life-span in circulation when compared with any existing synthetic carriers. In humans, normal erythrocytes have average life-span of 120 days. This means if physical and biological properties of an erythrocyte could be maintained, the encapsulated ASNase would inherit a life-span similar to that of the RBCs. As noted, half-life of free ASNase was about 26 hrs [12] whereas that of pegaspargase was only extended to approximately 15 days in humans [13]. Hence, RBC encapsulation also means that a significantly reduced dosing frequency would be required to maintain the same effective level of ASNase for ALL treatment.
Several methods including drug-induced endocytosis [14], electroporation [15], and hypo-osmotic methods [16-18] have been established to encapsulate drugs into RBCs. Some of these methods involve disruption of the plasma membranes of erythrocytes. With the creation of large pores or perturbations on the cell membrane, a number of impermeable protein drugs including acetaldyhyde dehydrogenase [15], alcohol dehydrogenase [15], ASNase [18], and erythropoietin [19] have been loaded into RBCs. Despite reasonable success, these methods maybe beset by two bottlenecks. First, the disruption of cell membrane often may result in alterations in morphology and surface structure of the erythrocyte, rendering it more susceptible to opsonization and RES clearance. As a consequence a significantly shortened plasma half-life was observed for the processed erythrocytes in a number of cases. Secondly, these membrane pore-opening methods would, both in principle and practice, result in loss of hemoglobin and other important cytosolic constituents of the erythrocyte, thereby impairing its biological functionality in oxygen transport as well as hemodynamics (especially in RBC ghosts created by osmotic rupture-resealing method). Therefore, the quest for a method that would allow for encapsulation of bioactive protein therapeutics into physically and functionally intact erythrocytes continues.
Recently, a family of small but extraordinarily potent membrane-permeable peptides, classified as “PTD” (protein transduction domain) peptides that include TAT [20], ANTP [21], VP22 [22], poly(arginine) peptides [23], and the non-toxic, naturally occurring low molecular weight protamine (LMWP) developed in our laboratory [24,25] have been discovered. Both in vitro and in vivo studies revealed that, by covalently linking PTD to almost any type of molecular species including proteins (MW>150 kDa; more than 60 different proteins have already been tested [26]) and nano-carriers [27], PTD was able to ferry the attached species across cell membrane of all organ types including the brain [20]. More importantly, it was documented that PTD was neither toxic nor immunogenic [25], and the PTD-mediated cell internalization did not induce any perturbation or alteration of the cell membrane [25]. Since intracellular protein uptake mediated by this PTD peptide was receptor- and transporter-independent, in principle, all cell types including erythrocytes should be transducible. These findings fostered the conceptual framework of the current innovative technology, by utilizing PTD, specifically the non-toxic LMWP, as the tool to achieve a non-invasive encapsulation of ASNase into intact and fully functional erythrocytes, as depicted in Figure 1. In this paper, we present in vitro and preliminary in vivo results to demonstrate the feasibility and potential of this approach in enhancing ASNase therapy for ALL.
Figure 1.
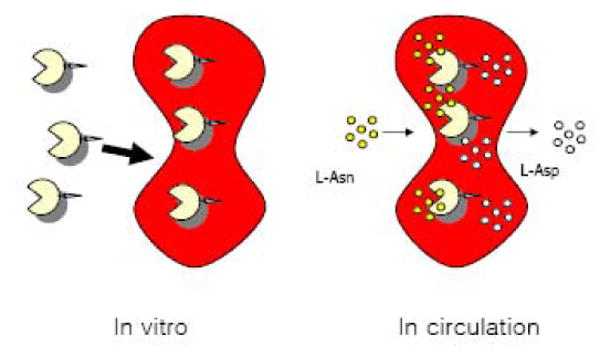
Schematic for PTD-mediated enzyme loading into intact erythrocytes
Materials and Methods
Preparation of LMWP-ASNase Conjugates
Low molecular weight protamine (LMWP) was prepared according to previously described procedures [24]. Activated LWMP was further created by thiolation using the heterobifunctional cross-linker 3-(2-pyridyldithio)propionic acid N-hydroxysuccinimide (SPDP, Sigma-Aldrich, St. Louis, MO). Briefly, 5 mg/ml of LMWP in 50 mM HEPES buffer (pH 8) was mixed with SPDP (1:10 molar ratio) in DMSO and shaken for 1 h at room temperature. The reaction mixture was then treated with 50 mM dithiothreitol (DTT, Sigma-Aldrich) and the thiolated LMWP was purified by HPLC on a heparin affinity column. The product was collected by ultrafiltration, lyophilized, and stored at -20°C until further use.
For conjugation, 5 mg/ml ASNase (Elspar, Ovation, Deerfield, IL, 238 IU/mg) was mixed with SPDP (40 μl of 0.1 M SPDP in ethanol to 1 ml protein solution) in phosphate buffer, and stirred at room temperature for 1 h. Unreacted SPDP was removed by rapid desalting and buffer exchange by FPLC with 0.1 M phosphate buffer (pH 7.4). Activated ASNase was then conjugated with a 10-fold molar excess of the above-prepared LMWP-SH for 24 h at 4°C. The LMWP-ASNase conjugates were isolated by ion-exchange chromatography using a heparin affinity column followed by five rounds of centrifugal filtration (molecular weight cut-off: 5,000 Da). Pooled LMWP-ASNase conjugates were concentrated, and the degree of conjugation was determined by MALDI-TOF mass spectroscopy.
Erythrocyte Uptake of Fluorescence-Labeled LMWP-Ovalbumin Conjugates
Commercial Alexa Fluor-488-labeled ovalbumin (Invitrogen, Carlsbad, CA) was activated using SPDP and then conjugated to LMWP. For uptake experiments, fresh sheep erythrocytes (MP Biomedicals, Solon, OH) were suspended in Hank's balanced salt solution (HBSS) at a density of 5×108 cells/ml, and were then incubated with a 0.5 mg/ml solution of the LMWP-ovalbumin conjugates for 30 min at room temperature under gentle shaking. RBCs were then washed with HBSS, fixed with 2% paraformaldehyde for 20 min, mounted on glass chamber slides, and uptake was examined, using a confocal laser scanning microscopy (LSM 510 META, Carl Zeiss, Jena, Germany).
Enzymatic Activity Assays
ASNase activity was determined by direct nesslerization of produced ammonia, according to previously described procedures [28]. Enzymatic activity units were defined as μmol ammonia per minute. Native ASNase possessed a specific activity of approximately 225-230 units/mg of protein.
Erythrocyte Encapsulation of ASNase
Sheep erythrocytes were suspended in HBSS at a density of 5×108 cells/ml and centrifuged. To the same volume of the RBC suspension, 250 μg/ml of LMWP-ASNase in HBSS was added. The cells were incubated at 37°C for 1 hr, washed with Alsever's solution, and then collected by centrifugation at 800 × g for 3 min. To remove surface bound LMWP-ASNase conjugates, cells were treated with trypsin-EDTA for 5 min at 37°C, followed by washing twice with Alsever's solution. The degree of ASNase loading was then determined by measuring the enzyme activity.
Scanning Electron Microscopy
One hundred microliters of a packed RBC suspension was placed in 6-well plates containing glass coverslips and incubated in 1% glutaraldehyde for 15 min at room temperature. The coverslips containing the fixed RBCs were then air-dried, followed by gold-sputtering for 1 min and examined by scanning electron microscopy (1910 Field Emission Scanning Electron Microscope, Amray) at an accelerating voltage of 15-20 kV.
Osmotic Fragility Measurements
Normal or LMWP--ASNase loaded erythrocytes were suspended at concentrations of 50% hematocrit, and 20 μl of the suspended cells were then added to 1.0 ml solutions of hypotonic saline with increasing osmolality ranging from 0 to 300 mOsm/kg. The solutions were incubated at 37°C for 30 min, centrifuged, and the absorbance of each supernatant was measured at 540 nm. The absorbance for 0 mOsm/kg solution was taken as 100% hemolysis. The osmolality of each solution was measured using a vapor pressure osmometer (Wescor, Logan, UT).
Determination of Hematological Parameters
A 10% solution of ASNase loaded erythrocytes in HBSS was washed three times and lysed with distilled water. The resulting hemolysate was centrifuged and the supernatant was diluted to 5 ml with a 1:1 mixture of HBSS and distilled water. Dissolved oxygen was measured by using a Clark electrode (World Precision Instruments, Sarasota, FL) connected to a data acquisition system. Oxyhemoglobin was measured using a 37°C thermostated spectrophotometer (UV 2501PC, Shimadzu, Columbia, MD) at 540 nm, and changes in oxyhemoglobin content of the hemolysate due to decreases in oxygen concentration were monitored.
To determine the mean corpuscular volume, mean corpuscular hemoglobin, and mean corpuscular hemoglobin content, erythrocytes were analyzed using a commercially available veterinary hematology system (Drew Scientific, Dallas, TX).
Pharmacokinetics of ASNase-Loaded Erythrocytes
All animal experiments were performed in accordance with the Guide for Laboratory Animal Facilities and Care (NIH publication 85-23, revised 1985) and the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the University of Utah.
Erythrocytes collected from DBA/2 mice (6 weeks old, Harlan, Indianapolis, IN) were treated with LMWP-ASNase conjugates using the previously described procedures. The plasma half-life based on enzymatic activity was calculated after intravenous injection of the ASNase-loaded RBCs into the DBA/2 mice. As a control, ASNase was encapsulated into red blood cell carriers using the conventional hypotonic rupture/resealing, according to a previously established procedure [18]. In both groups, each individual mouse was given 8 units of RBC-loaded ASNase (n=4 each). Total ASNase activity from recipient blood was measured by Nesslerization.
Survival of Tumor-Bearing Mice after administration of LMWP-ASNase-Loaded Erythrocytes
L5178Y cells (American Type Culture Collection (ATCC), Manassas, VA) were cultured in RPMI 1640 (Gibco, Grand Island, NY) supplemented with 10% FBS at 37°C in a humidified atmosphere of 5% CO2 environment. DBA/2 mice were then given intraperitonial injections of 7 × 105 cells in 0.1 ml HBSS. Five days after tumor implantation, mice with narrow bodyweight range were selected and divided into two groups: 1) control groups given saline only; and 2) erythrocytes loaded with LMWP-ASNase (n=5 each). Each individual mouse received intravenous injection of 8 units of erythrocytes loaded with LMWP-ASNase, and the survival of the tumor-bearing mice was monitored.
Statistical Analysis
All data are expressed as mean ± S.D. A two-tailed t-test was used in assessing the hematological parameters and the log-rank test was used in the survival study of the tumor-bearing mice. A p-value of less than 0.05 was considered significant.
Results
LMWP-Mediated Encapsulation of Proteins into Erythrocyte Carriers
To provide physical evidence of the ability of LMWP to deliver proteins into erythrocytes, we first activated LMWP by introducing a thiol moiety to the N-terminus, and then conjugating it to a commercial ovalbumin that was already labeled with the fluorescent dye, Alex Fluor 488. While control native RBCs showed weak autofluorescence from hemoglobin excitation (Figure 2A), RBCs incubated with the Alexa Fluor 488-labeled ovalbumin also displayed only weak fluorescence on the cell surface, with no observable uptake of the labeled protein within the interior of the RBCs (Figure 2B). However, after conjugation of ovalbumin with LMWP, significant intracellular fluorescence was detected within the RBC carriers (Figure 2C).
Figure 2.
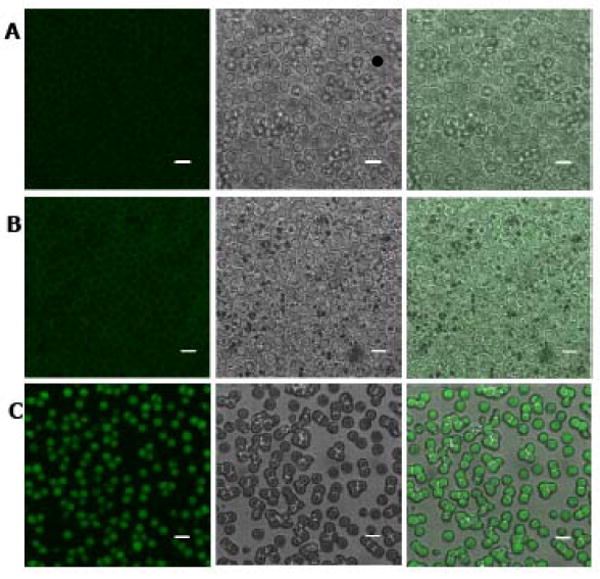
Confocal microscopy images of control RBCs (row A), RBCs incubated with Alexa Fluor 488-labeled ovalbumin (OVA-488; row B), and LMWP-ovalbumin (LMWP-OVA-488; row C). First column: fluorescence mode; second column: DIC mode; third column: superimposition. Scale bar = 5 μm. (Microscope: Carl Zeiss, Jena Germany, LSM 510 META. Objective: 63X, 1.2N. A., Water immersion, Zeiss cat. no. 440668. Temperature: 20°C. Mounting medium: ProLong Gold, Invitrogen, Carlsbad, CA. Acquisition software: LSM 510 Release Version 4.2 Service Pack 1).
Stability of RBC-Encapsulated ASNase
Results in Figure 2 already provided physical evidence that LMWP was able to transport protein molecules into erythrocytes. By following the same protocol, we conjugated ASNase with LMWP via disulfide linkages. MALDI-TOF mass spectra indicated that between 1 and 4 LMWP peptides were conjugated to each ASNase monomer (data not shown). Importantly, the specific activity of ASNase was found to be ∼150 U/mg, which is approximately 60% of the original enzyme, after conjugation.
When RBCs were incubated with the LMWP-ASNase conjugates at a total enzyme concentration of 100 IU/ml, a loading efficiency of 4% was observed, with a loading capacity of 8 IU ASNase per 100 μl packed erythrocytes. No hemolysis or loss of hemoglobin was detected during the loading process. In addition, no leaching of ASNase activity from the loaded RBCs was detected during the first 14 days of incubation in isotonic buffer at 4°C. After this incubation period, both control and ASNase-loaded RBCs began to show signs of disintegration. When the loaded RBCs were lysed after 14 days of incubation, greater than 70 % of the ASNase activity were recovered. Since the RBCs were treated with trypsin and washed with Alsever's solution after enzyme loading, the recovered enzymatic activity was clearly from the intracellularly entrapped ASNase.
Erythrocyte Functionality after Encapsulation of LMWP-ASNase
Since preservation of surface morphology and shape of the RBC after drug loading are important, we examined the surface morphology of the ASNase-loaded RBCs obtained by using the LMWP-mediated cell internalization process. While the ASNase-loaded RBCs created using the conventional osmotic rupture-resealing technique showed distinctive changes in surface morphology, RBCs treated with LMWP-ASNase displayed morphology identical to that of the control erythrocytes, with full preservation of the customary biconcave shape and no observable deformities (Figure 3A). Also of great significance was that when RBCs loaded with LMWP-ASNase were incubated in solutions of decreasing salt concentration, the displayed osmotic fragility curve was virtually identical to the result of control erythrocytes. As revealed in Figure 3B, the onset of RBC rupture in both groups occurred to superimpose at approximately 150 mOsm.
Figure 3.
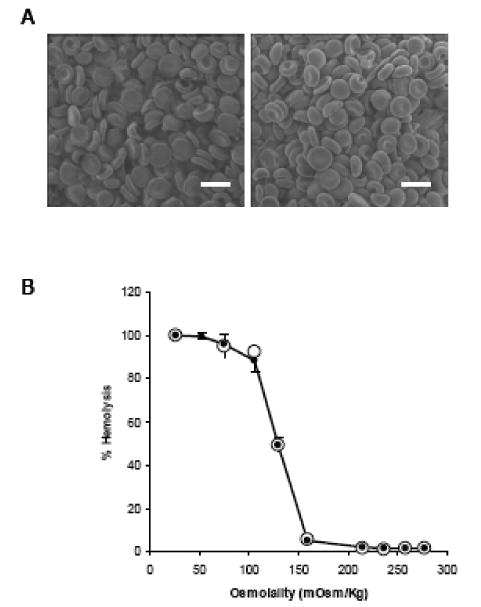
(A) Scanning electron microscopy (SEM) images of control, normal RBCs (left) and LMWP-ASNase-loaded RBCs (right). Cells were fixed with glutaraldehyde. Scale bar = 5 μm. (B) Osmotic fragility curve for control RBCs (●) and LMWP-ASNase-loaded RBCs (○).
In addition to confirming the structural integrity of the LMWP-ASNase loaded RBCs, we also examined their functional hematological properties. As noted, with conventional methods for protein encapsulation, the loss of intracellular components during pore formation or osmotic swelling could result in changes to the normal oxygen-binding capabilities of the drug-loaded erythrocytes. When subjected to varying oxygen concentrations, we found that the hemoglobin of the LMWP-ASNase loaded RBCs exhibited the characteristic sigmoidal profile indicative of cooperative binding. The Hill coefficient and pO50 values for the LMWP-ASNase loaded RBCs were equivalent to those of the control erythrocytes or the previously established values forsheep RBCs (Table 1), confirming the presence of the same level of fully functional hemoglobin. Additionally, the mean corpuscular volume (MCV), mean cell hemoglobin (MCH), and mean corpuscular hemoglobin content (MCHC) were all equivalent between normal erythrocytes and the LMWP-ASNase loaded cells (Table 1). These data demonstrated that cells loaded with ASNase using LMWP were virtually identical to normal erythrocytes with regard to both structural and functional properties.
Table 1.
Oxygen dissociation and hematological parameters of untreated (i.e., Control) and LMWP/ASNase-treated (i.e., Loaded) erythrocytes.
| Oxygen Dissociation Parameters | |||
|---|---|---|---|
| Control | Loaded | Ref. [40] | |
| Hill Coefficient | 3.15 ± 0.32 | 3.07 ± 0.34 | 3.14 ± 0.12 |
| pO50 (mmHg) | 37.44 ± 0.03 | 38.64 ± 0.02 | 40.0 ± 1.0 |
| Hematological Parameters | |||
| Control | Loaded | ||
| Hb (g/dL) | 15.47 ± 0.06 | 15.93 ± 1.10 | |
| HCT (%) | 40.80 ± 1.87 | 42.37 ± 2.18 | |
| MCV (fL) | 32.63 ± 0.06 | 32.53 ± 0.06 | |
| MCH (pg) | 12.37 ± 0.50 | 12.23 ± 0.45 | |
| MCHC(g/dL) | 37.97 ± 1.62 | 37.60 ± 1.45 | |
| RDW (%) | 22.37 ± 0.23 | 22.23 ± 0.40 | |
Hb = hemoglobin; HCT = hematocrit; MCV = mean corpuscular volume; MCH = mean corpuscular hemoglobin; MCHC = mean corpuscular hemoglobin concentration; and RDW = red cell distribution width.
Prolonged Circulation Time of RBC-Encapsulated ASNase
Since RBCs loaded with LMWP-ASNase closely resembled normal erythrocytes, we hypothesized that encapsulated ASNase would inherit the same, extended circulation half-life of normal erythrocytes. To test this hypothesis, we injected LWMP-ASNase loaded RBCs into the tail vein of DBA/2 mice, and the circulation half-life of these cells were determined via the linear portion of the elimination phase. For comparison, we also prepared ASNase-loaded RBCs with equivalent enzyme concentrations using the conventional hypoosmotic rupture/resealing technique. While both types of RBCs showed a biphasic disappearance of enzymatic activity over time, the LMWP-ASNase loaded RBCs displayed a significantly longer circulation half-life (4.5±0.5 days) than that of RBCs prepared using the hypoosmotic rupture/resealing encapsulation method (2.4±0.7 days), as depicted in Figure 4.
Figure 4.
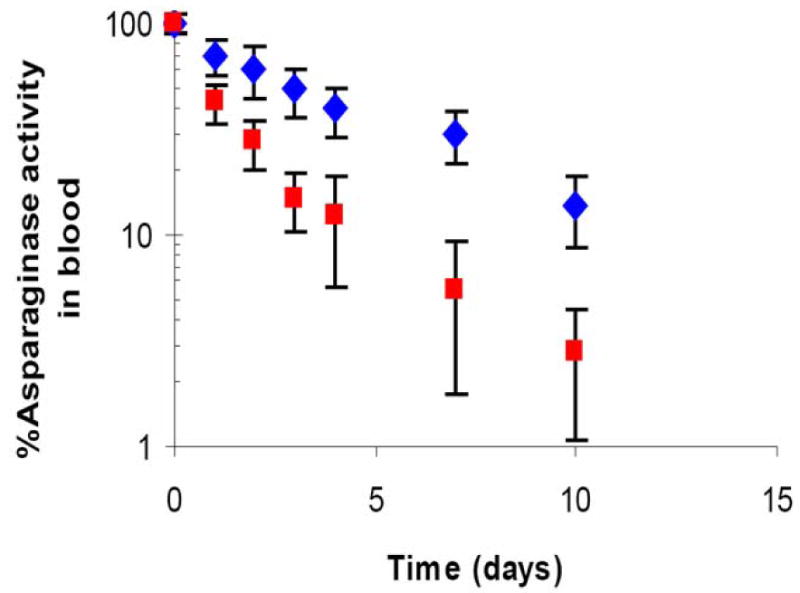
Time profile of ASNase activity in blood in DBA/2 mice (n=4). LMWP-ASNase loaded RBC (8 IU ASNase per mouse; blue diamond) and ASNase-loaded RBC ghosts (8 IU ASNase per mouse; red square) were given via intravenous injection through tail vein. ASNase activities in whole blood specimens were measured by nesslerization.
Therapeutic Efficacy of RBC-Encapsulated ASNase
Lastly, we injected RBCs loaded with LMWP-ASNase into L5178Y lymphoma-bearing DBA/2 mice to examine the therapeutic efficacy of the encapsulated enzyme. On day five after tumor injection and when symptoms became apparent, 8 IU of RBC-loaded ASNase were intravenously injected. Compared to control animals which received saline injections, the animals treated with LMWP-ASNase loaded RBCs showed significant increase in mean survival time, almost 44%, from 10.0±1.4 days to 14.4 ±2.3 days, as shown in Figure 5.
Figure 5.
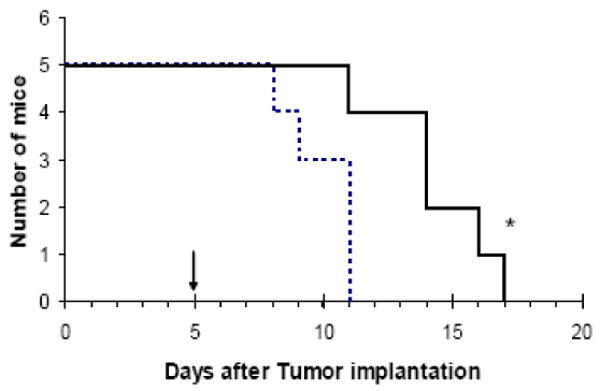
Kaplan-Meier survival curve for DBA/2 mice bearing L5178Y lymphoma cells. RBC-loaded LMWP-ASNase (8 IU ASNase per mouse; solid line) or saline (dotted line) were given on Day 5, when symptom from tumor burden was apparent (n=5, *p<0.05).
Discussion
Specific substrate selectivity and unparalleled reaction efficiency bestow proteins with the promise of being potent therapeutic agents. Yet, clinical applications of protein drugs face two major hurdles; one is their premature degradation and inactivation by endogenous proteases as well as elimination by the reticuloendothelial system (RES), and the other is the manifestation of immunological responses and toxic side effects by the host immune system towards foreign protein compounds. To overcome such problems, the most widely employed strategy is encapsulation of the protein into a soluble and biocompatible drug carrier; which can either be a synthetic polymer (e.g. PLGA) or a multi-component particulate structure such as liposomes or cells. Among all of these carrier systems, erythrocytes feature a number of distinctive advantages. Not only are erythrocytes the most abundant cells in human body and thus affordable for use in drug encapsulation, but they are also completely biocompatible and biodegradable, particularly when autologus RBCs are used. In addition, the biconcave disk shape of erythrocytes endows them with the highest surface to volume ratio (1.9×104 cm/g) usable for drug encapsulation. Most importantly, erythrocytes possess a lifespan in circulation of approximately 120 days, which is significantly longer than any of the currently existing carriers.
ASNase represents a typical example of these protein therapeutics. Despite its wide use in treating ALL, clinical application of ASNase is hindered by the short plasma half-life and high potential in inducing allergic responses. Hence, ASNase has been attempted in almost every method involved in RBC encapsulation. We therefore selected ASNase to examine the PTD-mediated RBC encapsulation technology, not only because of the clinical significance of ASNase, but also a direct comparison of the benefits of the new method over existing techniques could be readily attained and justified. We also chose the LMWP peptide developed in our laboratory as the representative PTD, simply because it possessed similar membrane-translocating activity to the most widely studied TAT and yet was proven in animal studies to be neither toxic or immunogenic [25]. Furthermore, to provide a direct physical evidence of the utility of the new method, we decided to study the LMWP-mediated cell uptake examined via confocal fluorescence microscopy, by utilizing the Alexa 488 fluorescent dye-labeled ovalbumin as the protein marker. As demonstrated in Figure 5, despite a significant overlap between the emission wavelengths of Alexa 488 and absorption wavelengths of hemoglobin, the green fluorescence from the LMWP-ovalbumin conjugates inside the cells was nevertheless quite evident. The horizontal sections along the z-axis (z sections) were also taken to ensure that the conjugates were indeed inside the RBCs (data not shown). In contrast, without the aid of LMWP, the dye-labeled ovalbumin could not enter the cells, and only weak fluorescence was observed on the surfaces of RBCs. Based on these findings, we were convinced that LMWP would be able to translocate other attached proteins such as ASNase into living RBCs.
Indeed, by utilizing an optimized encapsulation protocol, our results showed that a reasonable ASNase loading efficiency (∼4%) and RBC loading capacity (8 IU of ASNase per 100 μL of packed RBCs) was achieved. Clinically, the dose regimen for ASNase as a sole induction agent in the treatment of ALL is about 200 IU/kg body weight [29]. Hence, even based on our currently established loading protocol, this clinical dosing regimen, which can be translated into a dose of 3 mL of ASNase-loaded RBCs per kg of body weight, is obviously quite achievable.
In vitro characterization showed there was no leaching or activity decay of the RBC-encapsulated ASNase over a 3-day incubation period; under which the erythrocytes remained visibly intact. Since LMWP was linked to ASNase via disulfide linkages, it was speculated that detachment of LMWP from ASNase via degradation of such bonds by the elevated glutathione activity in the cytosol caused the membrane-impermeable ASNase to be trapped inside of the erythrocyte. In addition, it has so far not been established that the PTD-mediated cell entry is a reversible process. Hence, the permanent entrapment of the protein drug inside RBCs could also result from this irreversible translocation mechanism. Overall, the absence of leaching and activity decay of the entrapped ASNase fulfills one of the essential requirements for the ASNase-loaded RBCs to be eventually used clinically.
Evidences gathered from our experimental results all point towards the same direction; i.e. erythrocytes after processing by the new encapsulation method remain both structurally and functionally intact. The morphology of treated RBCs were indistinctive from the untreated cells (see Figure 3A), whereas all of the important cellular parameters including MCV, MCH, and MCHC were all statistically indistinguishable from literature or experimental results obtained for untreated RBCs (see Table 1). The most convincing evidence came from the oxygen transport capability of the treated erythrocytes, as ASNase-loaded RBCs displayed virtually statistically indistinguishable oxygen transport characteristics, such as the measured Hill coefficients and pO50 values, from the untreated, normal RBCs (see Table 1).
It should be noted that erythrocytes treated by any of the existing encapsulation methods, either by electroporation or hypotonic dilution, may result in a loss of cellular constituents such as hemoglobin. Several investigators reported that under a carefully managed process of hypotonic dialysis and with the aid of rejuvenating agent during the resealing procedure, drug-loaded erythrocytes could reserve an intact structure and initial chemical balance similar to those of native RBCs [30,31]. Nevertheless, in this hypo-osmotic drug loading method, a pore-opening and a resealing step, both involving dialysis, were required. Thus far, the largest protein being encapsulated in RBCs by using this method was alcohol oxidase from Pichea pastoria [32], which had a molecular weight (675 KDa) 10-fold larger than that of hemoglobin (65 KDa), the major component of an erythrocyte. Since dialysis is an equilibrium process and with such large pores being created on the cell membrane, in theory and practice, it is inevitable that some constituents in the cytosol such as hemoglobin, glutathione, or cytoskeleton would be leaked out of the erythrocyte. Indeed, loss of hemoglobin was observed in RBCs treated with the hypo-osmotic dialysis method, as evidenced by a decrease in MCH after resealing [30]. As known, aside from the principal activity in oxygen transport, RBCs also carry out other important biological functions including energy (ATP)-involved metabolic processes as well as scavenging of oxidative stress [33]. Hence, a loss of hemoglobin would not only impair the oxygen transport functions of RBCs, but also affect their ability in managing oxidative stress. On the other hand, a loss of the cytoskeletal constituents would compromise the structural integrity of erythrocytes, rendering them prone to destruction or recognition by cells in the phagocytic system.
Osmotic fragility of the treated RBCs has been widely adopted as a measure or revealing sign of the membrane integrity after undergoing drug loading process. Numerous reports have been found in the literature to indicate changes in osmotic fragility curves after loading erythrocytes with the hypotonic methods [30,34]. Chiarantini and co-workers reported that after hypotonic dialysis followed by hypotonic or isotonic washing cells exhibited distinctly different osmotic fragility profiles as well as earlier onset of rupture compared to normal erythrocytes [34]. These findings signified the assumption that the erythrocyte membranes were considerably weakened during these loading processes. In sharp contrast, our results demonstrated that erythrocytes being processed through the LMWP-mediated encapsulation method displayed a nearly superimposed osmotic fragility profile to that of normal erythrocytes, with both samples showing an identical onset of rupture osmolarity at about 150 mOsm (see Figure 4B). These findings further confirm our hypothesis that the PTD-mediated protein translocation is a rather non-invasive process that apparently does not compromise the erythrocyte membrane with weakened structural integrity.
One of the proofs of the benefits of the LMWP-mediated cell encapsulation method stemmed from the in vivo pharmacokinetic study of the plasma half-life of the ASNase-loaded erythrocytes. For comparison, erythrocytes loaded with ASNase via a hypotonic method were used as a control. Consistent with the findings by other investigators [16,19,30], the hypotonic method resulted in changes in morphology and surface structures of many of the treated erythrocytes and, as a consequence, significantly shortened the circulating half-lives of such cells. An overall half-life of approximately 2.4 days was found for erythrocytes treated with the hypotonic method whereas, in contrast, erythrocytes treated with the LMWP-mediated method exhibited significantly prolonged plasma half-life of 4.5 days; almost doubled the value for the hypotonic treated erythrocytes (see Figure 4). However, we believe that there is a room for improvement just because biological carriers like RBCs require more strict measures of handling precautions [15], as evidenced by the fact that the reported half-lives of manipulated RBC in rodents show variable results [17,18,30,35]. Therefore, upon optimization of RBC encapsulation method via PTD peptide, it is expected that the half life of enzyme loaded RBCs will increase compared to the results we have obtained from the preliminary study.
For the ASNase-loaded erythrocytes to function desirably, another essential requirement is that the entrapped drug must be able to retain its original therapeutic capability. To validate this criterion, we tested the therapeutic functions of the ASNase-loaded erythrocytes against a L5178Y lymphoma tumor-bearing DBA/2 mouse model. As can be seen in Figure 5, administration of ASNase-loaded erythrocytes was able to considerably increase the median survival time of the mice (14.4 days), when compared to the median survival time of 10 days observed from the control, the saline-injected animal group; a remarkable enhancement of the survival time by 44%. It should be quite easy to assess the prowess of the proposed RBC-encapsulation technology in ASNase therapy, after comparing our results with findings by others under similar in vivo conditions. As reported, a merely 16.7% enhancement in survival time over the control was observed by other investigators following intravenous injection of 8 IU of free ASNase to L5178Y tumor-bearing mice [36]. Previously, TAT-ASNase was investigated for potential application for targeted therapy of ALL [37, 38]. Same tumor model (L5178Y/DBA/2) showed that the mean survival time of L5178Y cell implanted DBA/2 mice treated with free ASNase showed no improvement with respect to untreated control group [38]. It is also noteworthy that Alpar and Lewis [39] investigated ASNase-loaded in RBCs, which reports impressively longer survival in treated animals. However, in that work, different tumor cell lines and animal models (6C3HED cell lines, C3H mice) was used. The median survival time of the mice in the untreated control group was about 18 days whereas L5178Y implanted DBA/2 mice in our study as well as previous studies [38] showed only 10 days, suggesting the tumor burden in DBA/2 strikes animals more quickly and severely than the C3H mice. The difference is further evidenced by in vitro doubling time of the two cell lines – L5178Y divides more rapidly than 6C3HED cells (data not shown). A more extensive animal investigation designed to further demonstrate the long-term benefits of this new approach in ASNase therapy, such as the alleviation of ASNase-induced toxic and immunologic responses, is currently in progress in our laboratory.
In summary, an innovative method for encapsulation of therapeutically active ASNase into functionally intact erythrocytes was developed toward enhanced ASNase therapy for ALL. Because of the non-invasive nature of PTD-mediated cell entry, our results showed that the structural and functional integrity of both the loaded ASNase and processed erythrocytes were completely reserved. Erythrocytes treated by this encapsulation method not only exhibited a long plasma half-life similar to that of untreated RBCs, but also displayed a enhanced therapeutic effects of the entrapped protein drug, presumably via protection of the drug by erythrocyte from possible proteolytic degradation and phagocytic clearance. It should be noted that a full preservation of both structure and functionality of the treated erythrocytes is of great clinical significance because theoretically this would provide the flexibility of replacing an unrestrictive amount of blood (or RBCs) from the patient with drug-loaded erythrocytes, should situations warrant such a clinical management. Based on the extraordinary simplicity of this encapsulation method, it is envisioned that a plasmapheresis-type of blood auto-transfusion device could be designed to allow RBCs being separated from the drawn blood of a patient, processed with LMWP-ASNase in a bioreactor for cell encapsulation, ASNase-loaded RBCs are then merged back with the other blood components, and finally returned to the patient for in situ drug therapy for ALL. Overall, this universal method of encapsulation may also be applied to several other protein drugs. Applications of this technology on the treatment of cocaine overdose, oxidative stress, and various types of cancers are currently being pursued in our laboratory.
Acknowledgments
The authors would like to thank Professor Bruce Palfey (Department of Biological Chemistry, University of Michigan) and Professor Mark E. Meyerhoff (Department of Chemistry, University of Michigan), and Dr. Joseph Yang (School of Medicine, Wayne State University) for valuable technical assistance and advice. Professor Victor C. Yang is currently a recipient of the Cheung Kong Scholar at the School of Chemical Engineering, Tianjin University, Tianjin, China. This study was supported by NIH grants HL55461, HL59705, and CA114612.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.http://www.cancer.gov/
- 2.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:66–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 3.Hill JJ, Roberts J, Loeb E, Khan A, MacLellan A, Hill AW. L-Asparaginase therapy for leukemia and other malignant neoplasms. Remission in human leukemia. JAMA. 1967;202:882–888. [PubMed] [Google Scholar]
- 4.Cooney DA, Handschumacher RE. L-Asparaginase and L-asparagine metabolism. Annu Rev Pharmacol. 1970;10:421–440. doi: 10.1146/annurev.pa.10.040170.002225. [DOI] [PubMed] [Google Scholar]
- 5.Becker FF, Broome JD. L-Asparaginase: inhibition of early mitosis in regenerating rat liver. Science. 1967;156:1602–1603. doi: 10.1126/science.156.3782.1602. [DOI] [PubMed] [Google Scholar]
- 6.Shepherd GM. Hypersensitivity reactions to chemotherapeutic drugs. Clin Rev Allergy Immunol. 2003;24:253–262. doi: 10.1385/CRIAI:24:3:253. [DOI] [PubMed] [Google Scholar]
- 7.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2(3):214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 8.Graham M. Pegaspargase: a review of clinical studies. Adv Drug Deliv Rev. 2003;55:293–1302. doi: 10.1016/s0169-409x(03)00110-8. [DOI] [PubMed] [Google Scholar]
- 9.Oettgen HF, Tallal L, Tan CC, Murphy ML, et al. In: Experimental and Clinical Effects of L-Asparaginase. Grundman E, Oettgen HF, editors. Springer; New York: 1997. pp. 219–243. [Google Scholar]
- 10.Hamidi M, Zarrin A, Foroozesh M, Mohammadi-Samani S. Applications of carrier erythrocytes in delivery of biopharmaceuticals. J Control Release. 2007;118(2):145–60. doi: 10.1016/j.jconrel.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 11.Ataullakhanov FI, Vitvitskii VM, Zhabotinskii AM, Pichugin AV. Permeability of human erythrocytes to asparagine. Biokhimiia. 1985;50(10):1733–1737. [PubMed] [Google Scholar]
- 12.Avramis VI, Sencer S, Periclou AP, et al. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood. 2002;99(6):1986–1994. doi: 10.1182/blood.v99.6.1986. [DOI] [PubMed] [Google Scholar]
- 13.Ho DH, Brown NS, Yen A, et al. Clinical pharmacology of polyethylene glycol-L-asparaginase. Drug Metab Dispos. 1986;14:349–352. [PubMed] [Google Scholar]
- 14.Benn-Bassat I, Bensch KG, Schrier SL. Drug-induced erythrocyte membrane internalization. J Clin Invest. 1985;51(7):1833–1844. doi: 10.1172/JCI106985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millan CG, Marinero MLS, Castaneda AZ, Lanao JM. Drug, enzyme, and peptide delivery using erythrocyte as carriers. J Control Rel. 2004;95:27–49. doi: 10.1016/j.jconrel.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 16.Hamidi M, Tajerzadeh H, Dehpour AR, Rouini MR, Ejtemaee-Mehr S. In vitro characterization of human intact erythrocytes loaded by enalaprilat. Drug Deliv. 2001;8:223–230. doi: 10.1080/107175401317245903. [DOI] [PubMed] [Google Scholar]
- 17.Ihler GM, Glew RH, Schnure FW. Enzyme loading of erythrocytes. Proc Natl Acad Sci USA. 1973;70:2663–6. doi: 10.1073/pnas.70.9.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Updike SJ, Wakamiya RT, Lightfoot EN., Jr Asparaginase entrapped in red blood cells: action and survival. Science. 1976;193:681–683. doi: 10.1126/science.821145. [DOI] [PubMed] [Google Scholar]
- 19.Garin MI, Lopez RM, Sanz S, Pinilla M, Luque J. Erythrocytes as carriers for recombinant human erythropoietin. Pharm Res. 1996;13(6):869–874. doi: 10.1023/a:1016049027661. [DOI] [PubMed] [Google Scholar]
- 20.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 21.Joliot A, Pernelle C, Deagostini-Bazin H, Prochiantz A. Antennapedia homeobox peptide regulates neutral morphogenesis. Proc Natl Acad Sci USA. 1991;88:1864–1868. doi: 10.1073/pnas.88.5.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elliott G, O'Hare P. Intercellular trafficking and protein delivery by a hyrpesvirus structural protein. Cell. 1997;88:223–233. doi: 10.1016/s0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 24.Chang LC, Lee HF, Yang Z, Yang VC. Low molecular weight protamine (LMWP) as nontoxic heparin/low molecular weight heparin antidote (I): preparation and characterization. AAPS PharmSci. 2001;3(3):E17. doi: 10.1208/ps030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park YJ, Chang LC, Liang JF, Moon C, Chung CP, Yang VC. Nontoxic membrane translocation peptide from protamine, low molecular weight protamine (LMWP), for enhanced intracellular protein delivery: in vitro and in vivo study. Faseb J. 2005;19(11):1555–1557. doi: 10.1096/fj.04-2322fje. [DOI] [PubMed] [Google Scholar]
- 26.Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27(2):85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Nori A, Jensen KD, Tijerina M, Kopečková P, Kopeček J. Subcellular trafficking of HPMA copolymer–Tat conjugates in human ovarian carcinoma cells. J Control Release. 2003;91:53–59. doi: 10.1016/s0168-3659(03)00213-x. [DOI] [PubMed] [Google Scholar]
- 28.Ho PP, Milikin EB, Bobbitt JL, et al. Crystalline L- asparaginase from Escherichia coli B. I. Purification and chemical characterization. J Biol Chem. 1970;245(14):3708–15. [PubMed] [Google Scholar]
- 29.American Hospital Formulary Service (AHFS) Drug Information: Asparaginase. American Society of Health-System Pharmacists; Bethesda, MD: 2001. [Google Scholar]
- 30.Kravtzoff R, Ropars C, Laguerre M, Muh JP, Chassainge M. Erythrocytes as carriers for L-asparaginase. Methodological and mouse in vivo studies. J Pharm Pharmacol. 1990;42:473–476. doi: 10.1111/j.2042-7158.1990.tb06598.x. [DOI] [PubMed] [Google Scholar]
- 31.Chalmers RA. Comparison and potential of hypo-osmotic and iso-osmotic erythrocyte ghosts and carrier erythrocytes as drug and enzyme carriers. Bibl Haematol. 1985;51:15–24. doi: 10.1159/000410223. [DOI] [PubMed] [Google Scholar]
- 32.Magnani M, Fazi A, Magnani F, Rossi L, Mancini U. Methanol detoxification by enzyme-loaded erythrocytes. Biotechnol Appl Biochem. 1993;18:217–226. [PubMed] [Google Scholar]
- 33.Berg JM, Tymoczko JL, Stryer L. Biochemistry. 6th. Freeman and Company; New York, NY: 1988. [Google Scholar]
- 34.Chiarantini L, Johnson J, Deloach JR. Optimized recirculation survival of mouse carrier erythrocytes. Blood Cells. 1991;17:607–617. [PubMed] [Google Scholar]
- 35.Kravtzoff R, Desbois I, Lamagnere JP, et al. Improved pharmacodynamics of L-asparaginase-loaded in human red blood cells. Eur J Clin Pharmacol. 1996;49:465–470. doi: 10.1007/BF00195932. [DOI] [PubMed] [Google Scholar]
- 36.Kodera Y, Sekine T, Yasukohchi T, et al. Stabilization of L-asparaginase modified with comb-shaped poly(ethylene glycol) derivatives, in vivo and in vitro. Bioconjug Chem. 1994;5:283–6. doi: 10.1021/bc00028a001. [DOI] [PubMed] [Google Scholar]
- 37.Li YT, Kwon YM, Spangrude GJ, Liang JF, Chung H, Park YJ, Yang VC. Preliminary In Vivo Evaluation of the PTD-Modified ATTEMPTS Approach in Enhancing Asparaginase Therapy. J Biomed Mater Res A. doi: 10.1002/jbm.a.32204. accepted. [DOI] [PubMed] [Google Scholar]
- 38.Kwon YM, Li YT, Liang JF, Park YJ, Chang LC, Yang VC. PTD-modified ATTEMPTS system for enhanced asparaginase therapy: A proof-of-concept investigation. J Control Release. 2008;130:252–8. doi: 10.1016/j.jconrel.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alpar HO, Lewis DA. Therapeutic Efficacy of Asparaginase Encapsulated in Intact Erythrocytes. Biochem Pharmacol. 1985;34:257–261. doi: 10.1016/0006-2952(85)90133-9. [DOI] [PubMed] [Google Scholar]
- 40.Moraga F, Monge C, Riquelme R, Llanos AJ. Fetal and Maternal Blood Oxygen Affinity: A Comparative Study in Llamas and Sheep. Comp Biochem Physiol. 1996;115A:111–115. doi: 10.1016/0300-9629(96)00016-3. [DOI] [PubMed] [Google Scholar]