


Abstract
Bacterial single-stranded DNA-binding proteins (SSBs) are required for DNA replication and repair. We have over-expressed and purified the native form and two His-tagged fusions of the SSB from Thermus thermophilus (TthSSB). The three proteins were found as dimers in solution. They bound in vitro to single-stranded DNA specifically over a temperature range of 4–80°C, and the wild-type protein could withstand incubation at 94°C for 2 min. Addition of TthSSB to PCR halved the elongation time required for the DNA polymerases of T.thermophilus (Tth) and Pyrococcus furiosus (Pfu) to synthesise DNA fragments in PCRs. The presence of TthSSB increased the fidelity of the proof- reading-free DNA polymerase of T.thermophilus. TthSSB was also able to bind single-stranded RNA, allowing a dramatic enhancement of the reverse transcription activity of its cognate Tth DNA polymerase during cDNA synthesis.
INTRODUCTION
Single-stranded DNA-binding proteins (SSBs) are required by all cellular life forms and most viruses. They bind and protect DNA whose double-stranded structure has been disrupted. Such DNA disruptions occur during vital information processing pathways such as replication, recombination and repair (1,2). Most SSBs bind non-specifically to single-stranded DNA (ssDNA), conferring a regular structure upon it, which is recognised and exploited by a variety of enzymes involved in the essential biological processes mentioned above. SSBs are usually required in stoichiometric quantities with respect to the ssDNA substrate, and protect the transiently formed ssDNA against nuclease attack, also preventing the formation of secondary structures [for review see Chase and Williams (3)].
In their soluble form, SSBs are found in different oligomeric states. For example, the SSB of phage T4 and that of Methanococcus jannaschii are monomers in solution; those of filamentous bacteriophages are dimers; in eukaryotes and in Archaea such as Pyrococcus furiosus, they are found as heterotrimers; and most eubacteria and mitochondria form homotetramers (4–9).
Recently, SSB-encoding genes from three different Thermus spp. have been determined. These Thermus spp. SSBs constitute the largest bacterial SSBs so far described (10). They have low similarity to SSBs from proteobacteria, though some conserved regions were identified that correspond to two ssDNA-binding motifs and to the C-terminal end of the protein. Two of these proteins, those from Thermus aquaticus and Thermus thermophilus, have been subjected to a preliminary biochemical characterisation, and have been shown to function as dimers (10), contrary to the homotetrameric forms so far found in bacteria. The presence of two putative ssDNA-binding sites in each monomer led these authors to speculate that the homodimeric structure of the thermophilic SSBs resembles that of the tetrameric form found in mesophilic counterparts and that this configuration could be related to its thermal stability. The sequence similarities between the two ssDNA-binding sites suggest a gene duplication as the origin of the thermophilic protein.
Based on its characteristics, the presence of SSBs during DNA replication is likely to improve the efficiency of the PCR. SSBs of thermophilic origin would be ideal candidates for such an application due to their presumed higher thermostability (11–13).
Here, we describe the purification and characterisation of an SSB from T.thermophilus strain HB8 (TthSSB) and demonstrate that it stimulates the mean rate of DNA synthesis by both bacterial and archaeal DNA polymerases, and the fidelity of the proof-reading-free DNA polymerase from T.thermophilus. We also demonstrate that TthSSB interacts efficiently with RNA, allowing a dramatic increase in the size of the cDNA synthesised by the reverse transcriptase activity of the DNA polymerase of T.thermophilus.
MATERIALS AND METHODS
Bacterial strains
Thermus thermophilus HB8 was obtained from the American Type Culture Collection (Rockville, MD). The Escherichia coli strains DH5αF′ [F′ (lacIq, lacZ+, proAB+), supE44, Δ(lacZYA-argF)U169, (Φ80 lacZΔM15), hsdR17, recA1, endA1, gyrA96, thi1, relA1] (Bethesda Research Laboratories, Gaithersburg, MD) and BL21(DE3) [hsdS, gal λ(lacIts857, ind1, sma7, nin5, lacUV5-T7gene 1)/pLysS (cmr)] (Novagen) were used for genetic constructions and protein expression, respectively. Escherichia coli was grown in LB at 37°C. Ampicillin (100 mg/l), kanamycin (30 mg/l) and/or chloramphenicol (20 mg/l) were added to plates or liquid media when needed.
Cloning of the Thermus thermophilus ssb gene
The open reading frame encoding the SSB was amplified by PCR using T.thermophilus HB8 chromosomal DNA as template. The Tth DNA polymerase was used in PCRs as described by the manufacturer (BIOTOOLS B & M, Madrid, Spain). Primers complementary to the 5′ (5SSB) and 3′ (3SSB or 3HISSSB) ends of the ssb gene described with the accession no. AF079160 were used (Table 1). Purified PCR products were cloned directly into the pCR2.1 vector (TA cloning Kit, Invitrogen). Both strands of the cloned ssb gene were sequenced. The sequence was submitted to the EMBL GenBank (accession no. AJ564626). Next, the amplified DNA fragments were digested either with NdeI and HindIII or with NdeI and EcoRI, and cloned into pET22b or pET28b (Novagen), leading to plasmids pET22-SSBHis, pET22-SSB and pET28-HisSSB. These constructions were used to transform competent BL21(DE3)/pLysS cells.
Table 1. Oligonucleotides used in this study.
| Name | Sequence (5′–3′) |
|---|---|
| 5SSB | catatggctcgaggcctgaa |
| 3SSB | tcgtgctcaaaacggcaaa |
| 3HISSSB | aagcttcaaatcctcctccg |
| O27-10 | gctcgtagtagtcgtcc |
| O27-27 | cgagcagaccgacgtcc |
| O27-54 | ccaccctcctccttctc |
| O27-55 | atgggcacctccttttg |
| ferred.nde1 | ggtgacatatgccgcacgt |
| ferred.rev | ctactcctccagcacgt |
| orf F.dir | ggaggacatatgagggtggt |
| orf N.rev | ctagtcgggcaggacccgc |
| promUP.ext | cctcctggcaactccggtcct |
| PUC1 | tatctgcgcctctgctgaa |
| PUC2 | ccaaatactgttcttctagtg |
Expression and purification of wild-type and His-tagged SSBs
BL21(DE3)/pLysS cells transformed with pET22-SSB, pET22-SSBHis or pET28-HisSSB were grown to an OD550 of 0.5 before the addition of 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) (14). Cells were harvested 4 h later, resuspended in 20 mM Tris–HCl pH 8.0, and disrupted by sonication. Insoluble debris was removed by centrifugation (15 000 g). The supernatant was heated for 30 min at 70°C to denature mesophilic proteins, which were subsequently discarded by centrifugation (10 000 g for 15 min). Recombinant His-tagged SSBs were further purified from the supernatant by affinity chromatography on Ni-NTA resin (Qiagen). The proteins were eluted with an imidazole gradient (0.05–0.2 M). The SSB-containing fractions, detected by SDS–PAGE (15), were pooled and dialysed against 20 mM Tris–HCl pH 8.0. Protein concentrations were determined by the BCA assay (Pierce). After the addition of glycerol [50% (v/v)], aliquots of the purified proteins were stored at –20°C.
Gel electrophoresis
Proteins were electrophoresed on 13% polyacrylamide gels containing 0.1% SDS under reducing conditions (15) and visualised by staining with Coomassie brilliant blue R-250. Non-denaturing gel electrophoresis (native PAGE) was performed in 4–20% (w/v) gradient polyacrylamide gels. Loading buffer for native gels contained 60 mM Tris–HCl pH 6.8, 10% (v/v) glycerol, 5 mM EDTA and 0.01% bromophenol blue. The samples and the native protein markers (Amersham Pharmacia Biotech) were run for 48 h on native PAGE to ensure that all proteins had achieved mobility according to their respective size.
DNA methods
All DNA manipulations were carried out according to Sambrook et al. (16). Restriction enzymes were used as indicated by the suppliers. [α-32P]dATP and [γ-32P]ATP (3000 Ci/mmol) were obtained from Amersham Pharmacia Biotech. DNA labelling was carried out with [α-32P]dATP (20 µCi) in a final volume of 80 µl containing 5 µg of HindIII-digested bacteriophage φ29 DNA, 100 mM each of dCTP, dTTP and dGTP, and 10 U of the Klenow DNA polymerase (New England Biolabs). After 20 min incubation at room temperature, the reaction was stopped by adding EDTA up to 6 mM, and the enzyme was heat-inactivated (65°C, 10 min). After filtration through Sephadex G-50 to eliminate non-incorporated nucleotides, the 273 bp φ29 HindIII DNA fragment was purified from agarose gels using the Qiagen gel extraction kit.
RNA isolation and in vitro synthesis of RNAs
RNA was purified from exponential cultures of T.thermophilus HB8 that had been subjected to a 4 h incubation at 70°C under unstirred conditions in the presence of 40 mM KNO3. This treatment induces the transcription of two operons encoding a nitrate reductase (17) and a specific electron transport chain (unpublished) required for nitrate respiration in this organism (17). The mRNA of these operons was used as template in our RT–PCR assays using specific oligonucleotides as primers. After cell harvesting, total RNA was purified using the Tri reagent ls kit (Molecular Research Center, Inc., Cincinatti, OH).
In vitro transcription was carried out with T7 RNA polymerase according to the manufacturer’s instructions (Promega) using pET22-SSB plasmid as template. Transcription reactions were digested with DNase RQ1 (RNase-free DNase I, Promega) and RNAs were analysed by agarose gel electrophoresis.
Gel mobility shift assays
Reactions with DNA were carried out in a final volume of 20 µl of binding buffer [50 mM Tris–HCl pH 7.5, 4% (v/v) glycerol], containing a mixture of 200 pg of double-stranded and 200 pg of heat-denatured single-stranded α-32P-labelled HindIII fragment (273 bp) from φ29 DNA (see above), and different amounts of the SSBs. After incubation for 5 min at the indicated temperatures, samples were immediately loaded onto 4% polyacrylamide/bis-acrylamide (80:1) non-denaturing gels containing 12 mM Tris-acetate (pH 7.5) and 1 mM EDTA. Gels were run for 2 h (8 V/cm) at 4 or 25°C. Gels were then dried and subjected to autoradiography.
Reactions with RNA were carried out in a final volume of 30 µl of binding buffer (100 mM Tris–HCl pH 8, 0.1 mM EDTA) containing 10 µg of in vitro-transcribed RNA, and different amounts of the SSBs. After 5 min incubation at 70°C, samples were analysed by agarose gel electrophoresis, and RNA was further detected by ethidium bromide staining.
PCR amplifications
Primers used for PCR were O27-27 and O27-10 (Table 1). PCRs were performed with DNA polymerases from T.thermophilus or P.furiosus according to the manufacturer’s instructions (BIOTOOLS B & M). We used 50 pmol of each primer and 30 or 100 pg of plasmid pNIT7 (18) and chromosomal DNA as templates, respectively. The PCR conditions used were: 1 min at 96°C for denaturation, followed by 25 cycles of: 1 min at 94°C, 1 min at 56°C and various elongation times at 72°C. A final elongation step was performed for 10 min at 72°C.
RT–PCR amplifications
RT–PCR amplifications were performed using the Retrotools kit (BIOTOOLS B & M). Reverse transcription was performed in a 20 µl reaction volume containing 10 pmol of a specific primer (Table 1), 1 µg of T.thermophilus total RNA and 1 U of Tth DNA polymerase. The reaction mixtures were incubated for 5 min at 75°C to denature the RNA, 1 min at 50°C for primer hybridisation and 45 min at 70°C for cDNA synthesis. Then, the reaction volume was increased up to 50 µl by adding 6 µl of standard DNA synthesis buffer (BIOTOOLS B & M) and 10 pmol of the appropriate upstream primer. PCR amplification was performed as described above.
Primer extension
Oligonucleotide PROMUP.ext (25 pmol) was labelled with [γ-32P]ATP using T4 polynucleotide kinase (New England Biolabs). After 1 h incubation at 37°C, the reaction was stopped by heating the sample (10 min at 65°C). For primer extension, 25 pmol of the labelled oligonucleotide were incubated (5 min at 75°C, 1 min at 60°C, 45 min at 70°C) with 1 µg of RNA from anaerobically induced cultures of T.thermophilus, and 1 U of Tth DNA polymerase (BIOTOOLS B & M). Samples were then loaded onto 6% polyacrylamide gels containing 8 M urea, subjected to electrophoresis, and detected by autoradiography.
PCR fidelity assays
The 2686 bp plasmid pUC19 was amplified by PCR with the divergent oligonucleotides PUC1 and PUC2 (Table 1). The PCR program was 1 min at 94°C, 1 min at 56°C and 3 min at 72°C. The amplified fragments were purified, treated with the Klenow fragment of DNA polymerase (30 min, 30°C), and phosphorylated with the phage T4 polynucleotide kinase (New England Biolabs). After ligation, the PCR-amplified pUC19 DNA was used to transform competent E.coli DH5αF′ cells. Transformants were selected on LB containing ampicillin and X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactoside). Mutation frequencies were calculated from the ratio of white to blue colonies, bearing in mind that the target DNA, the α-lacZ gene fragment, is 340 bp long.
RESULTS
Expression and purification of SSBs
The ssb genes of Thermus spp. of references AF079160, AF276705 and AF146075 encode proteins of 263, 264 and 266 amino acids, respectively. Based on these sequences, we designed suitable oligonucleotides to amplify the ssb gene from our T.thermophilus strain. The cloned gene encodes a 263 amino acid protein identical in sequence to that published by Dabrowski et al. (10) except for two conservative amino acid changes at positions 93 (R to K) and 241 (V to A).
The gene was cloned in E.coli vectors, in such a way that either wild-type SSB, and a N- or a C-terminal His-tagged derivates were over-expressed from a promoter dependent on T7 phage RNA polymerase (see Materials and Methods). The wild-type SSB and its N- and C-terminal His-tagged variants were named TthSSB, HisSSB and SSBHis, respectively.
The results, presented in Figure 1A (lanes 1–3), show that TthSSB and its His-tagged variants appear soluble, and no or very small amounts were detected in the particulate fraction (not shown). The mobility of the purified proteins in SDS–PAGE (Fig. 1A, lanes 4–6) was consistent with expectations based on their theoretical molecular weight, except for SSBHis (lane 6), which appeared as a larger protein than HisSSB (lane 5) despite having nine fewer amino acids.
Figure 1.

Purification of T.thermophilus SSBs and oligomerisation status. SSBs were produced in E.coli BL21(DE3)/pLysS and were purified as described in Materials and Methods. (A) Analysis of proteins by 13% SDS–PAGE. Approximately 5 μg of protein was separated and stained with Coomassie brilliant blue R-250. Lane M, molecular size markers; lanes 1–3, cell lysates from E.coli BL21(DE3)/pLysS containing pET22-SSB, pET28-HisSSB or pET22-SSBHis, respectively; lanes 4–6, purified T.thermophilus SSB, HisSSB and SSBHis, respectively. (B) Analysis of proteins by 4–20% native PAGE. Approximately 10 μg of protein was separated and stained with Coomassie brilliant blue R-250. Lane M, native molecular size markers; lanes 1–3, purified T.thermophilus SSB, HisSSB and SSBHis, respectively. The electrophoretic mobility of the dimers (d) and monomers (m) is indicated. Numbers on the left correspond to the size (kDa) of molecular mass markers.
Equilibrium non-denaturing gel electrophoresis was used to study the oligomerisation state of the purified proteins. To this end, protein samples were heated for 10 min at 70°C before being loaded onto the gel. The results presented in Figure 1B show that the wild-type protein migrated to a position above that of bovine serum albumin (66 kDa protein). Bearing in mind the calculated molecular weight of TthSSB (29.8 kDa) and that the migration position of proteins in these gels may deviate slightly from their expected position because of differences in charge distribution or shape, these results are likely to imply that TthSSB forms dimers in solution. Similarly, both His-tagged proteins migrated as dimers. A minor protein fraction of TthSSB and SSBHis migrated to positions corresponding to their respective monomers.
DNA–SSB interaction assays
When different concentrations of each of the three proteins were incubated at 70°C in the presence of a mixture of labelled ssDNA (∼200 pg) and dsDNA (∼200 pg), a specific ssDNA bandshift was observed. This bandshift was specific to ssDNA molecules even at the highest SSB concentration used (Fig. 2A). However, comparison of the amount of retarded ssDNA for identical protein concentrations revealed that their ssDNA binding activity was not identical. Whereas HisSSB behaved similarly to the wild-type protein, SSBHis showed a lower binding capacity at the same concentrations. In the latter case, free ssDNA was still detected even at the highest protein concentration assayed. Similar results were observed when the bandshift experiments were performed at 4°C (data not shown).
Figure 2.

Single-stranded DNA binding activity of SSBs and their ssDNA binding activity at different temperatures. (A) Bandshift assays were performed with purified T.thermophilus SSBs and a mixture of 5′-end-labelled ssDNA (200 pg) and dsDNA (200 pg) molecules corresponding to the 273 bp fragment of the φ-29 genome. The assays were carried out in the presence of the indicated amounts of proteins. (B) Bandshift assays were performed with purified TthSSB (25 ng) after heating for 5 min at the indicated temperatures. (C) Bandshift assays were performed with purified TthSSB (25 ng) after heating at 94°C for the indicated times. The electrophoretic mobilities of unbound DNA and SSB–DNA complexes are indicated. SSB, wild-type; H-SSB, HisSSB; SSB-H, SSBHis.
In order to analyse the effect of temperature on ssDNA binding activity, TthSSB was incubated at different temperatures with the labelled mixture of ssDNA and dsDNA. Figure 2B shows that TthSSB was able to bind to ssDNA over a wide temperature range (between 4 and 80°C). While native TthSSB was able to bind all the ssDNA at 80°C, either binding activity or protein stability was affected at higher temperatures. In fact, only small amounts of ssDNA were partially retarded at 90°C. In these experiments, the dsDNA melted above 70°C, leading to an increase in the total amount of ssDNA in the assay.
The thermostability of TthSSB was also studied. To this end, TthSSB was pre-incubated at 94°C for different times, cooled down to 70°C, and immediately subjected to bandshift assays. As shown in Figure 2C, TthSSB could withstand up to 2 min incubation at 94°C, without any detectable loss of its ssDNA-specific binding activity.
Effect of SSBs on PCR-based DNA amplification
In order to test whether the presence of TthSSB could improve the activity of its cognate thermostable DNA polymerase, PCR assays were carried out under suboptimal conditions (short elongation times) in the presence or absence of TthSSB. As shown in Figure 3, the DNA polymerase from T.thermophilus was unable to amplify a 3.2 kb DNA fragment from chromosomal DNA under these suboptimal conditions. In contrast, the presence of TthSSB had a dramatic effect on this reaction even at low concentrations (Fig. 3A). Both His-tagged derivatives had a similarly striking effect, but SSBHis was slightly less efficient (Fig. 3B).
Figure 3.

Effect of T.thermophilus SSBs on PCR assays. (A) PCR amplifications were carried out in the absence or presence of the indicated amount of TthSSB (ng) using T.thermophilus HB8 chromosomal DNA as template and Tth DNA polymerase. (B) PCR amplifications were carried out in the absence or presence of 2 ng of TthSSB, HisSSB and SSBHis using T.thermophilus HB8 chromosomal DNA as template and Tth DNA polymerase. (C) PCR amplifications were carried out in the absence or presence of 2 ng of native TthSSB using Tth and Pfu DNA polymerases for the elongation times, as indicated in the figure, and pNIT7 plasmid as template. The oligonucleotides used for the PCR assay were 027-10 and 027-27 (Table 1). Lane M: molecular weight markers (4370, 2899, 2498, 2201 and 1933 bp). SSB, wild-type; H-SSB, HisSSB; SSB-H, SSBHis.
As the above effects on DNA synthesis were observed at short elongation times, we decided to check whether the SSBs affected the mean rate of DNA synthesis of the thermostable DNA polymerase. As expected from the manufacturer’s declared rate of DNA synthesis (1000 nt/min), the DNA polymerase from T.thermophilus was able to amplify the target 3.2 kb DNA fragment when elongation times longer than 2 min were used, but not at shorter elongation times. However, the fragment was detected when TthSSB was present even with elongation times of 1 min. With a 30 s elongation time, the DNA fragment was not amplified even in the presence of TthSSB.
These results support the idea that the Tth DNA polymerase approximately doubled the mean rate of DNA synthesis in the presence of TthSSB. Interestingly, this stimulatory effect of TthSSB was also observed with the heterologous DNA polymerase from the Archaea P.furiosus (Fig. 3C, Pfu). In this case, the low rate of DNA synthesis of this enzyme (500 nt/min) did not allow amplification of the target DNA even at the longer elongation time used (3 min), unless TthSSB was present.
TthSSB increases the fidelity of the proof-reading-deficient DNA polymerase of T.thermophilus
A possible effect of TthSSB on the fidelity of the proof-reading-deficient Tth DNA polymerase was investigated (Materials and Methods). In brief, plasmid pUC19 was amplified by PCR in the presence or absence of TthSSB using divergent oligonucleotides, and the protocol described in Materials and Methods was followed for PCR product religation before transformation in E.coli cells. The use of a chromogenic substrate (X-gal) on selection plates made it possible to distinguish between plasmid with a non-mutated α-lacZ gene fragment and those in which mutations inactivated this gene. As shown in Table 2, the number of white colonies (LacZ defective) decreased significantly in the presence of TthSSB. Therefore, TthSSB increases the fidelity of this DNA polymerase. Despite this, the fidelity of the Tth DNA polymerase in the presence of TthSSB in these assays was an order of magnitude lower than that obtained with the proof-reading-proficient Pfu DNA polymerase in the absence of TthSSB (0.15%).
Table 2. Effect of TthSSB on the fidelity of Tth DNA polymerase during PCR.
| PCR no. | Without SSB | With SSB | ||
|---|---|---|---|---|
| White (%) | Total | White (%) | Total | |
| 1 | 2.38 | 5544 | 1.6 | 3301 |
| 2 | 2.44 | 7725 | 1.97 | 4958 |
| 3 | 2.88 | 7463 | 1.69 | 2767 |
| 4 | 2.11 | 7282 | 1.73 | 7555 |
| Average (%) | 2.45 ± 0.2 | 1.74 ± 0.1 | ||
Four parallel amplifications were developed under identical conditions. After re-ligation of the amplified fragment, the percentage of white colonies was determined.
Effect of SSBs on reverse transcription
The DNA polymerase from T.thermophilus displays reverse transcriptase (RT) activity in the presence of MnCl2 (19). Thus, the synthesis of cDNA and subsequent amplification of this DNA by PCR can be achieved in a single assay. Since the presence of TthSSB stimulated DNA synthesis by the DNA polymerase of T.thermophilus, we wondered whether it could also stimulate the reverse transcriptase activity of this enzyme.
As shown in Figure 4A, the amount of a 264 bp cDNA fragment amplified by RT–PCR from total RNA of T.thermophilus increased in the presence of TthSSB (lane 1+). For longer fragments, the effect of TthSSB was very pronounced. Whereas standard RT–PCR assays failed to detect products of 1085, 1932 and 3285 bp (lanes 2, 3 and 4, respectively), the presence of TthSSB had a dramatic effect, allowing the amplification of these cDNA (lanes +). For the largest cDNA amplified, unspecific products were also detected (lane 4+), most probably because of the low annealing temperature used during the first cycles of the PCR to allow the hybridisation of a non-perfect match forward primer (ferred.nde1).
Figure 4.
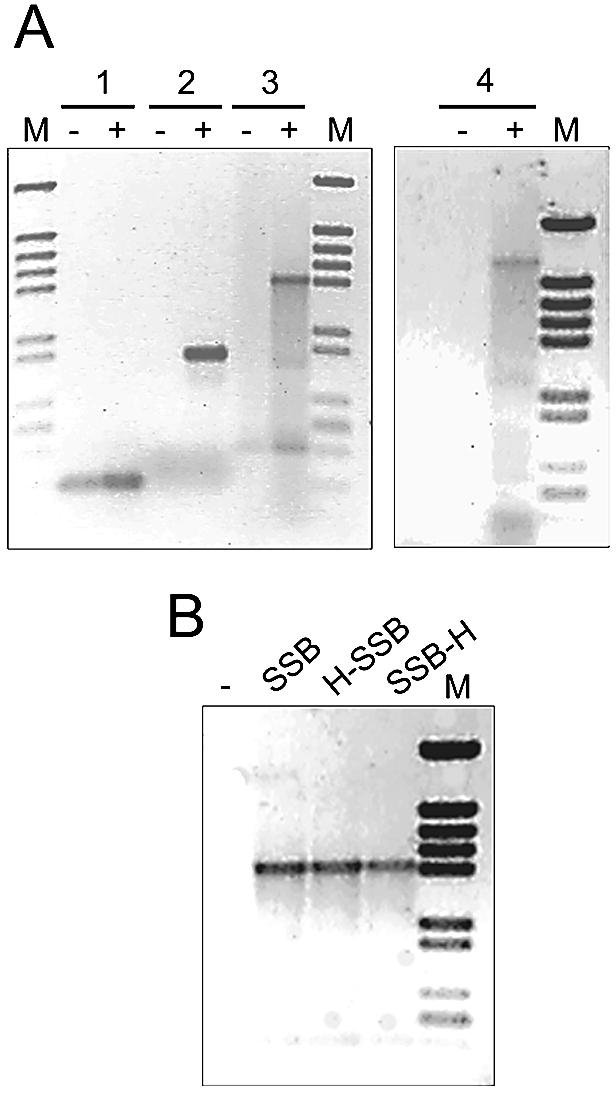
Effect of T.thermophilus SSBs on RT–PCR assays. (A) RT–PCR amplifications were carried out on 2 µg samples of total RNA from nitrate anoxia-induced cultures of T.thermophilus HB8, in either the absence (lanes –) or presence (lanes +) of 2 ng of native SSB. The oligonucleotide pairs used for reverse transcription (RT) and subsequent amplification (PCR) steps were: (1) ferred.rev (RT) and ferred.nde1 (PCR); (2) 027-55 (RT) and 027-54 (PCR); (3) orfN.rev (RT) and orfF.dir (PCR); and (4) orfN.rev (RT) and ferred.nde1 (PCR). (B) RT–PCR amplifications were carried out in the absence (–) or presence of 2 ng of TthSSB, HisSSB and SSBHis using the oligonucleotides orfN.rev (RT) and orfF.dir (PCR). Lane M: molecular weight markers (4370, 2899, 2498, 2201, 1933, 1331, 1150, 759, 611, 453 and 273 bp); SSB, wild-type; H-SSB, HisSSB; SSB-H, SSBHis.
The His-tagged derivates also had a similarly large effect on the detection of the 1932 bp cDNA fragment (Fig. 4B). The elongation times used for the PCR step of these experiments were long enough to allow the amplification of the expected fragments in the absence of TthSSB when DNA was used as template instead of RNA (data not shown). Therefore, TthSSB apparently acted directly on the RT step and not on the subsequent PCR.
This assumption was further supported by primer extension reactions carried out in the absence or presence of TthSSB. For this purpose, we used RNA preparations of nitrate anoxia-induced cultures of T.thermophilus, and a primer (PROMUP.ext) which hybridises near to the expected 5′ termini of the mRNA. The results of Figure 5A show that the amount of cDNA products synthesised in the presence of TthSSB was higher than in its absence. The presence of small cDNA products was most probably the consequence of the heterogeneity in the 5′ termini of the mRNA template used, as they were also detected when the AMV viral reverse transcriptase (Promega) was used in the RT reaction (not shown). These data support the view that SSB would improve the reverse transcriptase activity of the Tth DNA polymerase.
Figure 5.

Effect of TthSSB on RT assay and ssRNA binding activity. (A) RT assays were carried out in the absence or presence of 2 ng of TthSSB, using the oligonucleotide PROMUP.ext. Thermus thermophilus HB8 total RNA (4 µg) was used as template. (B) Bandshift assays were performed with TthSSB and in vitro transcribed RNA (10 µg) using 1% agarose gels. Lanes 1–8 correspond to 0, 1.5, 3, 6, 12, 24, 48 and 96 µg of TthSSB, respectively. Lane M: molecular weight markers (4370, 2899, 2498, 2201, 1933, 1331, 1150, 759 and 611 bp).
In order to check whether the effect on RT was a consequence of a direct interaction of TthSSB with the template mRNA, we used mRNA retarding assays. To this end, purified TthSSB was incubated at 70°C for 10 min with a 792 nt mRNA. TthSSB–ssRNA complexes were then identified by agarose gel electrophoresis. As shown in Figure 5B, TthSSB was able to bind the mRNA and produce complexes, which migrated more slowly than free RNA (Fig. 5B). Interestingly, an inverse relationship between the amount of TthSSB used and the rate of RNA migration was observed.
DISCUSSION
A thermophilic SSB from T.thermophilus HB8 was purified and its effects on the activity of thermophilic DNA polymerases assayed. The sequence of the ssb gene cloned encodes a protein whose sequence differs by two amino acids from that previously published for T.thermophilus (20).
TthSSB and its His-tagged derivatives were shown to be homodimers in solution, suggesting that neither the C- nor the N-terminus is critically involved in protein dimerisation. In addition, HisSSB displayed identical ssDNA binding activity to that of the wild type, implying that the four ssDNA-binding sites of each dimer are fully accessible, and that the overall protein architecture is conserved. In contrast, SSBHis showed decreased ssDNA binding activity. This defect could be related to a specific role in ssDNA binding for the C-terminal sequence of this protein, which is highly conserved among many SSBs, including that of E.coli (21,22). In the latter protein, it has been suggested that the C-terminal sequence moderates the affinity for ssDNA and interacts with components of the replication machinery (22,23). It is therefore possible that the observed lower ssDNA binding activity of SSBHis is caused by steric constraints imposed by the histidine tag in the binding of ssDNA. In addition, the C-terminal His-tag could also add steric constraints to the interaction between SSB and DNA polymerases, affecting the stimulatory effect of SSB-His on PCR and RT–PCR (see below).
The inactivation assays demonstrated that TthSSB can withstand short heating periods at 94°C, suggesting that it could be used to stimulate the activity of thermostable DNA polymerases at high temperatures. In fact, the presence of TthSSB shortened the elongation time required to synthesise a specific DNA fragment by the Tth DNA polymerase. Such stimulation could be the result of direct interactions between TthSSB and its cognate DNA polymerase. However, as an increase in DNA synthesis was also observed when an archaeal DNA polymerase was used, it follows that the effect of TthSSB was not due to specific interactions with its cognate DNA polymerase, but was most probably an indirect consequence of its binding to ssDNA.
There are several putative explanations for such an indirect effect on DNA synthesis stimulation. For example, the TthSSB–ssDNA complexes could avoid the formation of folded ssDNA structures, which are generally believed to block the progression of the DNA polymerase. On the other hand, the TthSSB could prevent non-productive binding of the DNA polymerase to ssDNA, directing it to the specific dsDNA regions where the primers hybridise, thus allowing a more effective re-usage of the template. In any of these contexts, the nature of the DNA polymerase will not be relevant.
The intrinsic properties of thermostable DNA polymerases, which contribute to variation in fidelity during synthesis, are not fully understood. A number of factors are thought to contribute to the overall fidelity of a DNA polymerase. These include the frequency at which a DNA polymerase incorporates incorrect nucleotides during synthesis, and the presence of integral 3′–5′ exonuclease activity that can remove mispaired bases (proof-reading activity). In general, enzymes that possess associated 3′–5′ exonuclease-dependent proof-reading activity exhibit greater replication fidelity than non-proof-reading DNA polymerases (24), such as that of Thermus spp. Therefore, the increase in fidelity of the Tth DNA polymerase observed in the presence of TthSSB could be associated with a higher rigidity of the DNA template, which promotes greater steric hindrance between template and incorrect nucleotides, as postulated for the E.coli SSB (25,26). Alternatively, the effect of TthSSB on ssDNA conformation could result in a better identification of non-Watson–Crick pairs by the polymerase, and the concomitant decrease in the frequency of incorporation of incorrect nucleotides during synthesis.
The effect of TthSSB on the efficiency of reverse transcription is one of its most surprising effects. Our results clearly show that TthSSB interacts with RNA. Thus, with the exception of poly(dC), the affinity of gp32, an SSB from phage T4, for the corresponding ribo-homopolynucleotide is about one order of magnitude lower than that for a similar deoxyribo-homopolynucleotide (27). Much lower affinities for ribonucleotides have been reported for bacterial SSB, except for specific binding of the E.coli SSB to its own mRNA. In the case of TthSSB, our data suggest that there is an efficient and unspecific interaction between TthSSB and RNA under the conditions used to promote the reverse transcriptase activity of the Tth DNA polymerase. In this context, and as discussed above in relation to DNA synthesis on DNA templates, the stimulatory effect on cDNA synthesis could be associated with the unfolding effect of TthSSB on the template RNA, and not with specific interactions between this protein and its cognate DNA polymerase. Direct applications of TthSSB in RNA detection are clearly derived from these results.
Acknowledgments
ACKNOWLEDGEMENTS
This work has been supported by project grants from the Comunidad Autónoma de Madrid (07M-0096-2000) and the Ministerio de Ciencia y Tecnología (BIO2001-1267), Spain. An institutional grant from the Fundación Ramón Areces is also acknowledged. C.P. was a postdoctoral fellow of the Comunidad Autónoma de Madrid, and F.C. was a postgraduate fellow of the Spanish Ministerio de Educación, Cultura y Deporte. ‘Ramón y Cajal’ program of the Spanish Ministry of Science and Technology supported W.J.J.M.
REFERENCES
- 1.Meyer R.R., Glassberg,J. and Kornberg,A. (1979) An Escherichia coli mutant defective in single-strand binding protein is defective in DNA replication. Proc. Natl Acad. Sci. USA, 76, 1702–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wold M.S. (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem., 66, 61–92. [DOI] [PubMed] [Google Scholar]
- 3.Chase J.W. and Williams,K.R. (1986) Single-stranded DNA binding proteins required for DNA replication. Annu. Rev. Biochem., 55, 103–136. [DOI] [PubMed] [Google Scholar]
- 4.Bochkarev A., Pfuetzner,R.A., Edwards,A.M. and Frappier,L. (1997) Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA Nature, 385, 176–181. [DOI] [PubMed] [Google Scholar]
- 5.Kelly T.J., Simancek,P. and Brush,G.S. (1998) Identification and characterization of a single-stranded DNA-binding protein from the archaeon Methanococcus jannaschii.Proc. Natl Acad. Sci. USA, 95, 14634–14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komori K. and Ishino,Y. (2001) Replication protein A in Pyrococcus furiosus is involved in homologous DNA recombination. J. Biol. Chem., 276, 25654–25660. [DOI] [PubMed] [Google Scholar]
- 7.Shamoo Y., Friedman,A.M., Parsons,M.R., Konigsberg,W.H. and Steitz,T.A. (1995) Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature, 376, 362–366. [DOI] [PubMed] [Google Scholar]
- 8.Stassen A.P., Folmer,R.H., Hilbers,C.W. and Konings,R.N. (1994) Single-stranded DNA binding protein encoded by the filamentous bacteriophage M13: structural and functional characteristics. Mol. Biol. Rep., 20, 109–127. [DOI] [PubMed] [Google Scholar]
- 9.Yang C., Curth,U., Urbanke,C. and Kang,C. (1997) Crystal structure of human mitochondrial single-stranded DNA binding protein at 2.4 Å resolution. Nature Struct. Biol., 4, 153–157. [DOI] [PubMed] [Google Scholar]
- 10.Dabrowski S., Olszewski,M., Piatek,R., Brillowska-Dabrowska,A., Konopa,G. and Kur,J. (2002) Identification and characterization of single-stranded-DNA-binding proteins from Thermus thermophilus and Thermus aquaticus—new arrangement of binding domains. Microbiology, 148, 3307–3315. [DOI] [PubMed] [Google Scholar]
- 11.Chakravarty S. and Varadarajan,R. (2002) Elucidation of factors responsible for enhanced thermal stability of proteins: a structural genomics based study. Biochemistry, 41, 8152–8161. [DOI] [PubMed] [Google Scholar]
- 12.Kumar S., Tsai,C.J. and Nussinov,R. (2000) Factors enhancing protein thermostability. Protein Eng., 13, 179–191. [DOI] [PubMed] [Google Scholar]
- 13.Scandurra R., Consalvi,V., Chiaraluce,R., Politi,L. and Engel,P.C. (1998) Protein thermostability in extremophiles. Biochimie, 80, 933–941. [DOI] [PubMed] [Google Scholar]
- 14.Hanahan D. (1983) Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol., 166, 557–580. [DOI] [PubMed] [Google Scholar]
- 15.Laemmli U. and Favre,M. (1973) Maturation of the head of bacteriophage T4.I; DNA packaging events. J. Mol. Biol., 80, 575–599. [DOI] [PubMed] [Google Scholar]
- 16.Sambrook J., Fritsch,E. and Maniatis,T. (1989) Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 17.Ramírez-Arcos S., Fernández-Herrero,L.A. and Berenguer,J. (1998) A thermophilic nitrate reductase is responsible for the strain specific anaerobic growth of Thermus thermophilus HB8. Biochim. Biophys. Acta, 1396, 215–227. [DOI] [PubMed] [Google Scholar]
- 18.Ramírez-Arcos S. (1997) Caracterización y transferencia horizontal de una nitrato reductasa de Thermus thermophilus HB8. Universidad Autónoma de Madrid, Madrid. [Google Scholar]
- 19.Myers T.W. and Gelfand,D.H. (1991) Reverse transcription and DNA amplification by a Thermus thermophilus DNA polymerase. Biochemistry, 30, 7661–7666. [DOI] [PubMed] [Google Scholar]
- 20.Dabrowski S., Olszewski,M., Piatek,R. and Kur,J. (2002) Novel thermostable ssDNA-binding proteins from Thermus thermophilus and T.aquaticus—expression and purification. Protein Expr. Purif., 26, 131–138. [DOI] [PubMed] [Google Scholar]
- 21.Curth U., Genschel,J., Urbanke,C. and Greipel,J. (1996) In vitro and in vivo function of the C-terminus of Escherichia coli single-stranded DNA binding protein. Nucleic Acids Res., 24, 2706–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohman T.M. and Ferrari,M.E. (1994) Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu. Rev. Biochem., 63, 527–570. [DOI] [PubMed] [Google Scholar]
- 23.Kinebuchi T., Shindo,H., Nagai,H., Shimamoto,N. and Shimizu,M. (1997) Functional domains of Escherichia coli single-stranded DNA binding protein as assessed by analyses of the deletion mutants. Biochemistry, 36, 6732–6738. [DOI] [PubMed] [Google Scholar]
- 24.Showalter A.K. and Tsai,M.D. (2002) A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry, 41, 10571–10576. [DOI] [PubMed] [Google Scholar]
- 25.Kunkel T.A., Meyer,R.R. and Loeb,L.A. (1979) Single-strand binding protein enhances fidelity of DNA synthesis in vitro.Proc. Natl Acad. Sci. USA, 76, 6331–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer R.R. and Laine,P.S. (1990) The single-stranded DNA-binding protein of Escherichia coli.Microbiol. Rev., 54, 342–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Hippel P.H., Kowalczykowski,S.C., Lonberg,N., Newport,J.W., Paul,L.S., Stormo,G.D. and Gold,L. (1982) Autoregulation of gene expression. Quantitative evaluation of the expression and function of the bacteriophage T4 gene 32 (single-stranded DNA binding) protein system. J. Mol. Biol., 162, 795–818. [DOI] [PubMed] [Google Scholar]