


Abstract
Nucleolin, a multi-domain protein involved in ribosome biogenesis, has been shown to bind the consensus sequence (U/G)CCCG(A/G) in the context of a hairpin loop structure (nucleolin recognition element; NRE). Previous studies have shown that the first two RNA-binding domains in nucleolin (RBD12) are responsible for the interaction with the in vitro selected NRE (sNRE). We have previously reported the structures of nucleolin RBD12, sNRE and nucleolin RBD12–sNRE complex. A comparison of free and bound sNRE shows that the NRE loop becomes structured upon binding. From this observation, we hypothesized that the disordered hairpin loop of sNRE facilitates conformational rearrangements when the protein binds. Here, we show that nucleolin RBD12 is also sufficient for sequence- specific binding of two NRE sequences found in pre-rRNA, b1NRE and b2NRE. Structural investigations of the free NREs using NMR spectroscopy show that the b1NRE loop is conformationally heterogeneous, while the b2NRE loop is structured. The b2NRE forms a hairpin capped by a YNMG-like tetraloop. Comparison of the chemical shifts of sNRE and b2NRE in complex with nucleolin RBD12 suggests that the NRE consensus nucleotides adopt a similar conformation. These results show that a disordered NRE consensus sequence is not a prerequisite for nucleolin RBD12 binding.
INTRODUCTION
Nucleolin is a highly conserved multi-domain protein involved in cell growth and proliferation (1). Although nucleolin functions in a number of important cellular processes, it is most widely known for its participation in steps throughout ribosome biogenesis including rDNA transcription, pre-rRNA processing, ribosome assembly and nucleo-cytoplasmic transport (2). Attempts to characterize how nucleolin participates in ribosome biogenesis have revealed that different domains or combinations of domains of nucleolin perform varied functions. Studies have shown that the N-terminal domain is possibly involved in regulating rDNA transcription (3), the RGG domain interacts with several important ribosomal proteins (4), and the four RNA-binding domains (RBDs) bind to nascent rRNA transcripts (5,6). The RBDs of nucleolin have βαββαβ folds and contain conserved aromatic, hydrophobic and basic amino acids (RNP motifs) that are commonly used for sequence-specific RNA recognition (7). Nucleolin binds specifically to at least two different pre-rRNA sequences using different combinations of its four RBD domains. One, an 11 nt sequence found in the 5′ external transcribed spacer (ETS) of mouse pre-rRNA (nt +655 to +666), which is called the evolutionary conserved motif (ECM), requires all four RBDs of nucleolin for binding (8). Interaction of nucleolin with the ECM, which is conserved from mice to humans, is required for the first U3 snoRNP-mediated cleavage step of pre-rRNA in vitro (9). Another family of recognition sequences was identified by in vitro selection and binding experiments with RNA fragments derived from the 5′ ETS of mouse pre-rRNA. This family of recognition sequences was shown to contain the consensus sequence (U/G)CCCG(A/G) in the context of a hairpin loop structure termed the nucleolin recognition element (NRE) (5). A subsequent study showed that only the first two N-terminal RBDs of nucleolin are necessary for sequence-specific binding of a 68 nt RNA containing the in vitro selected NRE sequence (10). Putative NRE sites have been identified in mouse and human pre-rRNA and are found to cluster within the external and internal transcribed spacers of pre-rRNA near sites of processing, thereby suggesting a role for the NRE in pre-rRNA processing (11).
Our laboratory has previously determined the structure of the first two RBDs of nucleolin (RBD12) in complex with a 22 nt RNA containing the in vitro selected consensus (sNRE) using NMR spectroscopy (12). The structure of the complex reveals that both RBD domains, as well as the 12 residue linker, interact extensively with the NRE consensus sequence. The structure of the complex also shows that nucleolin RBD1 and linker residues contact nucleotides in the loop E (or S-turn) motif (13–17) found in the upper part of the stem of both bound and free sNRE (18). The structure of the nucleolin RBD12–sNRE complex was the first to show two RBDs interacting with a hairpin. This observation and others led to the hypothesis that nucleolin is a pre-rRNA chaperone (12).
RNAs and proteins that interact commonly have disordered regions, which become structured upon complex formation. The presence of disordered regions in a protein, RNA or both is thought to be important for complex formation, since most binding events require enough flexibility in the RNA or protein to facilitate conformational rearrangements necessary to create intricate RNA–protein binding interfaces (19,20). Comparison of the free and bound sNRE structures shows that the flexible NRE loop becomes ordered when the protein binds (18). Therefore, we hypothesized that the loop of sNRE is disordered to allow for conformational rearrangements upon protein binding. Here, we have studied the free RNA structures of two NREs found in the mouse pre-rRNA 5′ ETS, b1NRE (B1: mouse 5′ ETS 515–532) and b2NRE (B2: mouse 5′ ETS 562–578) (EMBL sequence database accession No. M20154) (11,21), using multi-dimensional NMR spectroscopy and compared them with the sNRE structure, which has been re-determined under new solution conditions. Since previous in vitro binding studies used longer oligonucleotides (70–71 nt) containing B1 and B2 NREs and full-length nucleolin (5,21), we first show that, like sNRE, shorter b1NRE and b2NRE constructs (21–24 nt) are sequence specifically recognized by nucleolin RBD12 alone, albeit with weaker affinity than sNRE. Our studies show that while the predicted b1NRE loop structure is not well defined, the predicted b2NRE loop is structured. The b2NRE forms a hairpin with a tetraloop, which is similar to the YNMG tetraloop family (22). In addition, we present evidence that the sNRE and b2NRE sequences are recognized similarly by nucleolin RBD12. Therefore, a disordered NRE loop is not required for nucleolin RBD12 binding.
MATERIALS AND METHODS
RNA synthesis and purification
Unlabeled and uniformly 13C/15N-labeled RNA oligonucleotides were prepared by in vitro transcription and purified as previously described (23). RNAs were annealed at dilute concentrations (1–10 µM) in water and adjusted to the desired salt conditions by the addition of the appropriate stock solution. sNRE buffer consisted of 10 mM potassium phosphate pH 7, 100 mM KCl, 50 µM EDTA, 0.02 % NaN3, and b2NRE buffer consisted of 5 mM potassium phosphate pH 7, 50 µM EDTA, 0.02 % NaN3. All RNAs were subsequently concentrated by ultrafiltration to ∼1.0 mM. Exchange of samples from H2O to 2H2O was achieved through repeated lyophilization. Sample p2H was monitored using a pH meter and corrected by adding 0.4 to the reading (24). Samples were adjusted with 2HCl or NaO2H.
Protein expression and purification
BL21-Codon Plus®-RIL (Stratagene) was transformed with a recombinant pProEX® HT (Life Technologies) plasmid containing an insert encoding the hamster nucleolin (His)6-RBD12 subdomain. Cells grown at 37°C in LB medium containing 100 µg/ml amplicillin and 35 µg/ml chloramphenicol were induced with 1 mM isopropyl-β-d-thiogalactopyranoside at OD600 = 0.6–0.8 for 3 h. Harvested cells were resuspended in buffer A (20 mM sodium phosphate pH 7.4, 500 mM NaCl) with 0.1% Triton® X-100 and 100 µl of protease cocktail inhibitor (Sigma) per liter of culture, and lysed by three freeze–thaw cycles and sonication. After centrifugation, the supernatant was adjusted to 1 M NaCl and treated with 0.1% polyethylenimine (mol. wt 8000) for 10 min to precipitate nucleic acid contaminants. The precipitate was removed by centrifugation for 15 min at 10 000 g. The supernatant was then adjusted to 80% (NH4)2SO4 saturation by the addition of solid (NH4)2SO4 and centrifuged at 10 000 g. The resulting pellet was resuspended in buffer A and dialyzed against 4 l (two exchanges) of buffer A. The dialysate was then applied to a Ni2+ HiTrap Chelating HP column (Pharmacia) and washed with buffer A containing 10 mM imidazole. The bound protein was eluted from the column with buffer A containing 200 mM imidazole. Fractions containing (His)6-RBD12 were pooled and dialyzed against buffer B [20 mM Tris–HCl pH 8.0, 25 mM NaCl, 0.5 mM EDTA, 1 mM dithiothreitol (DTT)]. (His)6-RBD12 was then treated with TEV protease (Life Technologies) for 12 h to cleave the (His)6 from the RBD12 subdomain. The protein mixture was then applied to a Resource Q® (Pharmacia) column and eluted with a NaCl gradient. Fractions containing RBD12 were pooled and concentrated by ultrafiltration to 1 ml. RBD12 was further purified and exchanged into buffer C (50 mM potassium phosphate, 200 mM KCl, 50 µM EDTA, 0.02% NaN3) using a Superdex® 75 (Pharmacia) column.
Electrophoretic mobility shift assay
Internally labeled RNA constructs were prepared using the Riboprobe® in vitro Transcription Systems (Promega) and [α-32P]rCTP (800 Ci/mmol) (ICN). Labeled RNAs were purified by PAGE and quantitated by scintillation counting. The specified RNA (10 fmol) was incubated in TMK buffer [100 mM Tris–HCl pH 7.2, 200 mM KCl, 4 mM MgCl2, 1 mM DTT, 20% glycerol, 0.02% NaN3, 1 U/µl Superasin®, 5 µg/ml bovine serum albumin (BSA), 0.5 mg/ml tRNA] with the indicated amount of nucleolin subdomain for 30 min on ice. The reactions were then loaded onto an 8% polyacrylamide gel (acrylamide:bis-acrylamide 60:1) containing 5% glycerol and 0.5× TGE buffer (25 mM Tris–HCl pH 8.5, 0.2 M glycine, 1 mM EDTA) and run for 1.5 h at 4°C. The gels were dried and exposed to phosphoimager plates.
RNA substrates
5′-Fluorescein-labeled RNAs with sequences corresponding to those shown in Figure 1 were obtained from Dharmacon Research (Boulder, CO), purified, deprotected and desalted. 5′-Fluorescein–NRE substrates were diluted in TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA) and quantified at A260 as described (25).
Figure 1.

Secondary structure representations (left) and nucleolin RBD12 binding isotherms (right) for (A) sNRE26, (B) b1NRE (nucleotides U3–G21 correspond to mouse 5′ ETS: 515–532) and (C) b2NRE (nucleotides A4–U20 correspond to mouse 5′ ETS: 562–578) (EMBL database accession No. M20154) (11). Nucleotides added for the purpose of transcription and stem stability are shown in boxes. The numbering of sNRE26 and b2NRE has been adjusted to correspond to the original sNRE construct (18). Binding isotherms were generated with 2 nM 5′-fluorescein-labeled NRE and varying nucleolin RBD12 concentrations. Each point is the average of five measurements, and error bars represent the standard deviation of those measurements. Data were fit to equation 2, and all R2 values are >95%. (D) Competition experiments of nucleolin RBD12 binding to b2NRE. Millipolarization (mP) values of free 5′-fluorescein-labeled b2NRE (Flb2) (white), Flb2 in the presence of 10 µM nucleolin RBD12 (black), and Flb2 in the presence of 10 µM nucleolin RBD12 and 20 µM (10 000× probe concentration) specified competitor RNA (gray).
Fluorescence anisotropy assays
Fluorescence anisotropy was measured using the Beacon 2000 Variable Temperature Fluorescence Polarization System (Panvera, Madison, WI) with fixed excitation (490 nM) and emission (535 nM). Equilibrium binding assays were performed with a range of protein concentrations and 2 nM 5′-fluorescein-labeled RNA in a final volume of 120 µl containing 50 mM potassium phosphate pH 7.5, 200 mM KCl, 0.1 mM EDTA, 0.02% NaN3, 0.1 µg/µl ultrapure BSA (Panvera), 0.2 µg/µl heparin and 10% glycerol. Samples were read as a blank prior to the addition of probe. After probe addition, samples were incubated for 60 min at 4°C. Each anisotropy measurement is an average of 5–10 polarization measurements taken at 4°C with 20 read cycles. Polarization values were converted to anisotropy values when necessary using equation 1.
A = 2P/3 – P 1
Data are expressed as average mA, where mA is the milli-anisotropy at the indicated amount of protein. Dissociation constants (KD) were calculated by fitting data to equation 2 using KaleidaGraph (25,26).
![]()
Samples for competition experiments were prepared by adding 10 µM nucleolin RBD12 in binding buffer to 20 µM competitor RNA. 5′-Fluorecscein-labeled b2NRE was then added to the sample to a final concentration of 2 nM. Samples were incubated for 1 h and then read as described above.
NMR spectroscopy
All NMR spectra were recorded on Bruker (Billerica, MA) DRX 500 and 600 MHz spectrometers. Assignment of non-exchangeable protons, attached carbons and proximal nitrogens was achieved using a series of two-dimensional experiments [NOESY (27), DQF-COSY (28), MLEV-17 TOCSY (29), CT-HSQC, MQ-H(CNC)H (30) and long-range 1H–15N HSQC (31)] and three-dimensional experiments [3D-HCCH-COSY (32), 3D-HCCH-TOCSY (33) and 3D-NOESY-1H–13C-HMQC (34)] acquired at 278 or 298 K in 100% 2H2O. Exchangeable protons and nitrogens were assigned using NOESY, 1H–15N HMQC, 2D HCCNH-TOCSY (35), 15N-correlated-CPMG-NOESY(36), HCN-TOCSY-CH (37) and H5(C5C4N)H (38) spectra which were acquired in 95% H2O/5% 2H2O at 278 K. Hydrogen bonding patterns were confirmed using JNN-HNN-COSY (39,40). 3JH3′P and 3JCP were measured using 31P spin echo difference CT-HSQC to determine β and ε torsion angles (41). Residual dipolar couplings were measured for 1JHC in C12E6/hexanol at natural abundance using a modified 1H–13C HSQC. Spectra were processed and analyzed using Bruker XWINNMR 2.6 and AURELIA 3.108.
Structure calculations
Inter-proton distances from 2D NOESY experiments at various mixing times (τm = 50, 100, 150, 200 and 300 ms) and 3D NOESY-HMQC (τm = 300 ms) experiments were generated as previously described (41) except for the classification of semi-quantitative NOEs which were as follows: strong (1.8–3.0 Å), medium (2.5–4.5 Å), weak (3.5–5.5 Å) and very weak (4.5–7.0 Å). Ribose conformations were analyzed using the TOCSY experiment as previously described (41). 31P spin echo difference experiments for the determination of β and ε torsion angles were analyzed as described (42). Global χ angle restraints were determined by analysis of the di(6/8,1′) in a short mixing time NOESY (50 ms) (43). γ torsion angles were included for nucleotides with NOE patterns consistent with A-form helical geometry. Hydrogen bonding distance restraints that were either consistent with experimental data or inferred based on initial structure calculation without explicit hydrogen bond restraints showing several of the lowest energy structures with the correct distance and geometry were used.
Structures were calculated using the standard simulated annealing protocols (44) within NIH-XPLOR (45). Starting folds were generated from randomized extended templates. In the next step, converged structures satisfying all NMR restraints were subjected to NOE and torsion angle based refinement followed by refinement against residual dipolar couplings (RDCs). The axial and rhombic components of the alignment tensor were determined by a grid search procedure (46). Selection of converged structures relied on total energy and r.m.s.d. values. Helical parameters of RNA structures were analyzed using 3DNA version 1.4.1 (47).
Coordinate deposition
The coordinates for the 17 and 16 lowest energy structures of sNRE26 and b2NRE, respectively, have been deposited in the Protein Data Bank with accession codes 1QWB and 1QWA.
RESULTS
Studies of nucleolin RBD12 binding to natural NREs
Full-length nucleolin has been shown to interact with in vitro selected NREs and two pre-rRNA NREs (B1 and B2) derived from the mouse 5′ ETS (5). Since previous experiments showed that sequence-specific interaction with an in vitro selected NRE requires only the first two of the four RBDs of nucleolin (10), binding of nucleolin to b1NRE and b2NRE was also expected to only require nucleolin RBD12. To test this, binding of nucleolin RBD12 to oligonucleotides containing the B1 and B2 sequences (b1NRE and b2NRE) (Fig. 1B and C) was assayed using electrophoretic mobility shift assays (EMSAs). Initial gel shifts with nucleolin RBD12 using the procedure previously described (10) resulted in a gel shift for sNRE26 (Fig. 1A), but not for the natural NREs (data not shown). However, a gel shift for the natural NREs was observable in the presence of varying concentrations of nucleolin RBD12 when the running buffer was changed from 0.5× TBE to 0.5× TGE. In addition, better band shifts were obtained by running the gels at lower temperatures. Analysis of the gel shifts run at 4°C using 0.5× TGE showed that nucleolin RBD12 binds sNRE26 and natural NREs with KDs ranging from 5 to 25 nM and 300 to 1000 nM, respectively (data not shown). Since the gel shift results were difficult to quantitate, fluorescence anisotropy assays were used in order to determine the KDs more accurately. Fluorescence anisotropy assays are true equilibrium binding measurements since the assay does not require separation of free and bound RNA, thereby avoiding inaccurate KD determination due to disassociation of the protein–RNA complex during electrophoresis (25). Binding isotherms for sNRE26, b1NRE and b2NRE with nucleolin RBD12 are shown in Figure 1. The KDs determined by fluorescence anisotropy at 4°C for nucleolin RBD12 binding to sNRE, b1NRE and b2NRE are 3 ± 1, 770 ± 210 and 310 ± 110 nM, respectively, and are in good agreement with the EMSA data.
The KD values determined here are only 10-fold lower than previously reported for non-specific controls (10). Therefore, in order to confirm that the nucleolin RBD12–natural NRE interaction was really sequence specific, a competition experiment was conducted for one of the natural NREs, b2NRE. The results of a competitive binding assay in which a mutant b2NRE sequence is tested for the ability to remove a 5′-fluorescein-labeled b2NRE (Flb2) from the binding site of nucleolin RBD12 are shown in Figure 1D. A decrease in polarization indicates that the unlabeled, competitor RNA is capable of interacting with nucleolin RBD12. For example, addition of 20 µM (10 000× probe concentration) unlabeled b2NRE to the 10 µM RBD12–2 nM Flb2 mixture decreases the observed polarization value since the unlabeled b2NRE is competing with Flb2 for the same binding site of nucleolin RBD12. Likewise, mutation of non-consensus nucleotides (b2G15A and b2U16C) does not prevent the competitor RNA from being bound by nucleolin RBD12, as can be seen by the decrease in polarization to levels within experimental error of the unlabeled b2NRE control. However, mutation of the CCCG sequence of the NRE (b2C10U, b2C11U/C12U and b2G13A) completely abolishes the ability to bind competitively, which is expected since the consensus sequence does not vary at these positions. Mutation of U9 or A14 (b2U9C and b2A14G) results in a 25–50% decrease in polarization, indicating that mutations in these positions are tolerated but decrease the affinity. Interestingly, deletion of U16, a non-consensus nucleotide in the b2NRE loop, results in the inability to bind competitively (Fig. 1D). The reason for the lack of competitive binding by b2ΔU16 is discussed below. Taken together, these results show that nucleolin RBD12 sequence specifically recognizes natural NREs with nanomolar affinity.
We note that the KDs observed for the nucleolin RBD12 and natural NRE interactions are higher than those previously determined by filter binding assay using full-length nucleolin and B1 and B2 constructs of ∼70 nt in length (5). Our previous structural work with the nucleolin RBD12–sNRE complex (12) and ongoing structural investigations of the nucleolin RBD12–b2NRE complex (C.Johansson et al., in preparation) suggest that the remaining RBDs of nucleolin would not be able to bind the b2NRE loop since an additional domain would sterically clash with another RBD and/or RNA stem. Therefore, the higher affinities nucleolin displays for the longer B1 and B2 constructs are probably due to non-specific binding of the remaining two RBDs and/or the RGG domain to other regions of B1 and B2 rRNA.
NMR spectroscopy of the sNRE
We previously reported the structure of the free sNRE and showed that it consisted of a stem with seven base pairs, the top three of which are non-Watson–Crick base pairs that form a loop E motif (13–17), and a disordered hairpin loop. This structure was determined at low salt conditions (2–5 mM added salt) because at higher salt concentrations some duplex formation occurred (18). Since nucleic acid structure can be greatly affected by the concentrations of counterions (48,49), the possibility remained that the disordered loop of sNRE could be due to the low ionic strength of the buffer. In order to study sNRE under higher salt conditions (100 mM KCl) while preventing the formation of duplex, an sNRE construct with a more stable, longer stem, sNRE26, was designed (Fig. 1A) and a folding protocol that traps the hairpin conformation was used (see Materials and Methods). Correct folding of sNRE26 was assayed by NMR spectroscopy and showed that the expected base pairs in the stem and loop E motif of the hairpin were present. No imino proton signals arising from the loop were observed at pH 7. The addition of either more KCl to 200 mM or multivalent ions [1–10 mM Mg2+, Ca2+ or Co(NH3)63+], which have been shown to be important for some RNA structures (49,50), did not change the 1D NMR spectrum (data not shown). Since the absence of imino proton resonances is sometimes due to exchange broadening, we assayed the effect of pH by NMR spectroscopy (Fig. 2). As the pH is lowered, all imino proton resonances are broadened, especially those of the terminal G and iminos arising from the loop E motif. The effect of pH on the iminos of sNRE26 is unexpected since lowering the pH of a sample usually decreases the linewidth of imino proton resonances (51). A DNA hairpin with a similar loop sequence also displays this unusual pH dependence of the imino proton resonances, and has been proposed to have a pH-dependent dimerization (52,53).
Figure 2.

One-dimensional spectra (500 MHz) of the imino proton resonances of sNRE26 at 278 K as a function of pH. Resonance assignments are indicated.
Assignments of the proton and carbon resonances of sNRE26 were obtained using homonuclear and heteronuclear two- and three-dimensional experiments as previously described (18,23). The base to H1′/H5 region of a 200 ms NOESY spectrum of sNRE26 at 100 mM KCl, pH 7 is shown in Figure 3A. Analysis of the NOESY and other spectra showed that nucleotides G –1 to A8 and A15 to C24 form an A-form stem and a loop E motif, as previously seen (18). However, significant changes are observed for the loop resonances. Compared with the NOESY and 1H–13C HSQC spectra of the sNRE taken at low salt and pH (18), the proton and carbon dispersion is significantly better for nucleotides C10–G13. With the improved chemical shift dispersion, sequential base–H1′ connectivities can be observed from U9 to C12, suggesting that there is stacking of these nucleotides. However, the intensities of these peaks in NOESY and HSQC spectra are more than twice the intensities of those in the loop E motif and stem, suggesting that U9, C10, C11 and C12 are conformationally dynamic. The largest difference observed under the new conditions is for G13. At low salt and pH 6, the G13H8 proton has a strong NOE to both the G13H1′ and G13H3′, indicating that it is switching between the syn and anti conformations (18). At pH 7 and 100 mM KCl, the H8–H1′ NOE is twice as intense as the H8–H2′, peak and a H8–H3′ intranucleotide NOE is not observable in a 50 ms NOESY. This shows that at higher salt concentrations and pH, G13 is exclusively in the syn conformer.
Figure 3.

NOESY spectra of the base proton–H1′ region of sNRE26 and b2NRE. (A) The 500 MHz NOESY spectrum of sNRE26 at 298 K and τm = 200 ms. Sequential connectivities from G –1 to A8, C10 to C12 and G13 to C24 are shown in red, black and blue, respectively. The base–H1′ sequential connectivities for G19 are not shown since it resonates upfield at 3.82 p.p.m. There is no base–H1′ sequential connectivity between A15 and G16; however, NOEs from A15–U17 are observed. In addition, NOEs from U17 to A15 indicate that the sugar of A15 is flipped. (B) The 600 MHz NOESY spectrum of b2NRE at 298 K and τm = 300 ms. Base–H1′ sequential connectivities from G2 to C11 and G13 to C22 are indicated by red and blue lines, respectively. A break in the sequential connectivities is observed between A14 and G15, which is due to the fact that the A13H1′ resonance is broadened to baseline at 298 K. In both spectra, the intranucleotide cross-peaks are labeled. In addition, peaks arising from AH2 resonances are indicated with green lines and labeled accordingly.
Solution structure of sNRE26
Structures of sNRE26 were initially calculated from random starting structures using NOEs and dihedral angle restraints (54). Nucleotides –1 to 8 and 15 to 24 form an A-form stem and loop E motif, as expected. In addition, A9 stacks on A8, as seen in both the free and bound sNRE (12,18). However, even with 19 additional NOEs and dihedral angle restraints for the loop, nucleotides 9–14 did not converge into a single structure. Furthermore, incorporation of RDCs neither helped to define the loop structure nor improved the long-range order of the stem and loop E motif. Since incorporation of RDCs from the disordered loop would adversely affect RDC refinement of the stem and loop E motif, loop RDCs were removed and only the loop E motif and stem were refined using RDCs. This resulted in an improved long-range order for the stem and loop E motif nucleotides. Figure 4A shows the superposition from nucleotides –1 to 8 and 15 to 24 of the 17 lowest energy structures after RDC refinement (Table 1).
Figure 4.

Structure of sNRE26. (A) Stereoview of the 17 lowest energy structures. The heavy atom superposition is from –1 to 8 and 15 to 24 of sNRE26. Only the heavy atoms are shown, and the bases of the loop (9–14) have been removed to show only the backbone. (B) Lowest energy structure of sNRE26 with G13 and A14 bases in the loop shown. G13 is syn in all structures and partially stacks on A14 in four of the 17 lowest energy structures. (C) Schematic representation of the sNRE26 structure. Nucleotides are colored red (U), green (C), orange (A) and cyan (G).
Table 1. Structural statistics for sNRE26 and b2NRE.
| Experimental data used for structure calculations |
sNRE26 |
b2NRE |
| NOE-derived distance restraints | 605 (average: 23/nt) | 490 (average: 23/nt) |
| Intra-nucleotide NOEs | 308 | 268 |
| Inter-nucleotide NOEs | 297 | 222 |
| Hydrogen bond for paired residues | 47 | 46 |
| Dihedral restraints | 102 | 91 |
| Residual dipolar couplings | ||
| 1DC-H (Hz) | 9 | 19 |
| Additional restraints | ||
| Base pair planarity restraints | 10 | 9 |
| R.m.s.d. from experimental restraintsa | ||
| Distance restraints (Å)b | 0.01 ± 0.001 | 0.04 ± 0.0005 |
| Dihedral restraints (°)c | 0.075 ± 0.029 | 0.16 ± 0.035 |
| Dipolar couplings (Hz) | 0.044 ± 0.047 | 0.35 ± 0.08 |
| Deviations from idealized geometrya | ||
| Bonds (Å) | 0.0042 ± 0.00005 | 0.0048 ± 0.00005 |
| Angles (°) | 1.03 ± 0.005 | 1.01 ± 0.003 |
| Impropers (°) | 0.35 ± 0.003 | 0.40 ± 0.007 |
| Overall r.m.s.d. (Å) (heavy atoms) | (residues –1–8, 15–24)a | (residues 2–15, 17–22)a |
| From mean structure | 1.10 ± 0.36 | 1.02 ± 0.18 |
| Mean pair-wise | 1.68 ± 0.48 | 1.31 ± 0.64 |
aAveraged over the accepted structures.
bNo violations >0.2 Å.
cNo violations >5°.
Even at higher pH and salt concentrations, the loop of sNRE is disordered. This conclusion is supported by the observation that the proton and carbon chemical shifts of U9–C12 are close to the values of the free nucleotides. Although the loop is disordered, differences between the sNRE structures determined under the different conditions are seen on the 3′ side of the loop. G13 is syn rather than alternating between the syn and anti conformers as seen previously (18). In addition, NOEs for G13 suggest that it is stacked on A14 and some of the structures have it stacked at least partially on A14 (Fig. 4B). NOEs from A14H8 and A14H2 to A15 sugar protons and A8H2, respectively, suggest that A14 stacks on A15 and A8 in a zipper-like fashion seen in the bound form of the sNRE–nucleolin RBD12 complex (12). However, A14H2 also has an NOE to A7H2, which causes A14 to be positioned almost perpendicular to A8 and A15 (Fig. 4B). This is a physically unreasonable location that did not violate the NOE, dihedral angle or RDC restraints, suggesting that more than one conformation is present in fast exchange. Taken together, these results show that loop nucleotides U9–C12 of the sNRE are disordered and that loop nucleotide A14 exists in at least two conformations.
NMR spectroscopy of b1NRE
The folding procedure and buffer conditions used for sNRE26 were used for b1NRE to ensure hairpin formation. Correct folding of b1NRE was assayed by 1D and 2D NMR spectroscopy of the imino proton resonances (data not shown), and indicated that b1NRE folds into the predicted hairpin loop structure (Fig. 1B). No iminos from the loop were detected, and lowering of the pH resulted in broadening of all imino proton resonances as previously seen for the sNRE26. Assignment of the base and H1′ region of a NOESY spectrum show NOEs consistent with an A-form helix from G1 to G8 and C17 to C24. NOEs from G8 to C10 suggest that these residues are stacked; however, no NOEs are observed for the C12, G13 or C16 in the base–H1′ region (data not shown). NOEs for A14 and A15 were not assigned due to the fact that the resonances arising from these residues were extremely broad. Neither increasing the ionic strength of the buffer with either KCl (200 mM) or divalent ions, nor lowering of the ionic strength of the buffer improved the spectra to allow assignment of NOEs to A14 or A15. Since conditions for high resolution structure determination of b1NRE were not found, no further study of b1NRE was conducted.
NMR spectroscopy of b2NRE
The folding procedure and salt conditions used for sNRE26 were initially used for b2NRE; however, dilution experiments showed that 100 mM KCl and the high RNA concentration used for NMR resulted in some dimer formation. In order to study b2NRE at concentrations required for NMR, the ionic strength of the sample buffer was lowered to 5 mM potassium phosphate. Under these conditions, NMR spectra show that the predominant form was the hairpin, with six iminos observed from stem Watson–Crick base pairs (Fig. 1C) and three broad imino proton resonances from the loop nucleotides. Lowering the pH resulted in line broadening of all imino proton resonances, as previously seen for sNRE26. NOESY and UH3/AH2 dual detected JNN-COSY (39) experiments identified two of the three loop imino resonances as Watson–Crick base pairs formed by C8·G15 and U9·A14. The last imino resonance was identified as G13 since the imino resonance is still observable in 1D NMR and 2D NOESY spectra of a b2NRE mutant in which U16 is deleted (b2ΔU16).
Assignment of the proton and carbon resonances was accomplished through a series of 2D and 3D experiments similar to those applied to sNRE26 (23). The base–H1′/H5 region of a NOESY spectrum of b2NRE is shown in Figure 3B. Sequential NOEs between C7 and C8 and weak non-sequential NOEs between G15 and G17 indicate that the 6 bp stem and two additional base pairs of the b2NRE loop are stacked to form a continuous helix, with U16 bulged out of the helix. The loop nucleotides C10–G13 have unusual sequential and long-range NOEs, which indicates that these nucleotides are structured. Loop nucleotides C11 and C12 also have the only C2′-endo sugar puckers in b2NRE. NOESY spectra (50 ms) show that the G13H8–H1′ NOE is twice as intense as the H8–H2′ NOE. The G13H8–H1′ NOE intensity and downfield proton (G13H3′ and H2′ at 5.42 and 4.81 p.p.m., respectively) and carbon (G13C8 at 141.44 p.p.m.) chemical shifts indicate that G13 is syn (43,55).
Solution structure of b2NRE
The solution structure for b2NRE was initially calculated as described for sNRE26. From the lowest energy structures satisfying all experimental restraints, 10 structures were selected as the starting ensemble for further refinement. Since two distinct folds were present in the starting ensemble of structures, a modified refinement strategy was utilized (56). Briefly, the refinement protocol was run 10 times for each starting structure using significantly different random starting velocities to obtain an ensemble of 100 refined structures. The 50 lowest energy structures were subsequently sorted into two distinct families, which were then refined further with RDCs using DA and R values that were separately optimized by grid search for each fold. The RDC refinement resulted in two structurally and energetically different families of structures. The family of structures with the lower overall energy and back-calculated RDCs in better agreement with experimental RDCs was chosen as the final ensemble (Fig. 5A). The 16 lowest energy structures have an r.m.s.d. to the mean of 1.02 Å (not including U16), with no NOE violations >0.2 Å or dihedral angles violations >5° (Table 1).
Figure 5.

Structure of b2NRE. (A) Stereoview of the superposition of the 16 lowest energy structures. Only the heavy atoms are shown. (B) Major groove view of the lowest energy structure of b2NRE. (C) Stereoview of the superposition of nucleotides 8–15 highlighting the tetraloop structure. (D) Schematic representation of the b2NRE structure.
b2NRE forms a hairpin that is separated into two domains by a bulged uridine (Fig. 5A and D). The first domain is a 6 bp stem (nucleotides 2–7 and 17–22) consistent with A-form RNA. The second part is comprised of a 2 bp stem capped by a tetraloop structure (nucleotides 8–15). The two domains stack on one another to form a quasi-continuous helix, with U16 positioned outside the helix in the minor groove (Fig. 5A). U16 does not significantly affect local structure of the surrounding nucleotides, except G15, which has a sugar pucker in equilibrium between C2′- and C3′-endo conformations. However, the bulged residue causes distortions in the stacking of the C7·G17 and C8·G15 base pairs as reflected in changes in the roll parameter. The tetraloop capping the two Watson–Crick base pairs in the upper domain of b2NRE begins with a non-canonical base pair formed between the anti C10 and syn G13, which has the C10 O2-carbonyl and the G13 amino and imino protons hydrogen bonded. The second nucleotide of the tetraloop (C11) is in the minor groove of the stem, with the H6–H5 edge of the base pointing towards the C10 sugar moiety, and the Watson–Crick face and amino protons exposed to solvent. The third nucleotide of the loop, C12, is positioned in the major groove and is partially stacked on the non-Watson–Crick CG base pair. The H6–H5 edge of the C12 base points towards the C11 sugar moiety, while the Watson–Crick face is exposed to solution (Fig. 5C). C11 and C12 also have ε torsion angles that deviate from the usual trans to gauche–, while G13 and A14 have γ torsion angles that deviate from the usual gauche+ to trans. The turn in the backbone occurs after the third nucleotide in the tetraloop. Comparison with known loop structures reveals that the CCCG tetraloop of b2NRE is similar to the UUCG tetraloop structure (57).
Nucleolin RBD12 binds sNRE and b2NRE similarly
The structures of free b2NRE and sNRE show two major differences. First, the NRE loop of b2NRE is structured while the NRE loop of sNRE is disordered. Secondly, the upper part of the sNRE stem contains a eukaryotic loop E motif, which is not present in b2NRE. In order to determine if either of these structural differences affects nucleolin RBD12–NRE recognition, a comparison of b2NRE and sNRE in complex with nucleolin RBD12 was made. Figure 6A and B shows 1H–13C-HSQC spectra of 13C/15N-labeled sNRE and b2NRE in 1:1 complexes with nucleolin RBD12. The nucleotides of the NRE consensus sequence of bound b2NRE and sNRE have similar chemical shifts, suggesting that they have the same conformation and molecular environment in both complexes. Comparison of the 1H–15N HSQCs of nucleolin RBD12 in complex with sNRE and b2NRE indicates that the structure of the protein is also similar in the two complexes (Fig. 7A). The residues involved in binding the NRE consensus sequence in the sNRE complex, most of which are part of the conserved RNP motifs of the RBDs, have very similar chemical shifts in the b2NRE complex, suggesting that the NRE is recognized in the same way. However, small chemical shift differences do exist for certain residues involved in NRE recognition, e.g. residues K94 and F56, which suggest slight differences between the two complexes (Fig. 7A). In contrast, larger chemical shift differences are observed for residues in RBD1, which contact the loop E motif in the sNRE complex, e.g. residues T52 and V27 (Fig. 7A and B). This is to be expected since b2NRE does not have a loop E motif. Therefore, the NRE loop consensus nucleotides of nucleolin RBD12–sNRE and nucleolin RBD12–b2NRE complexes probably adopt similar structures, whereas differences in the complex structures are likely to be present where nucleolin RBD12 contacts the upper part of the stem in the sNRE complex.
Figure 6.
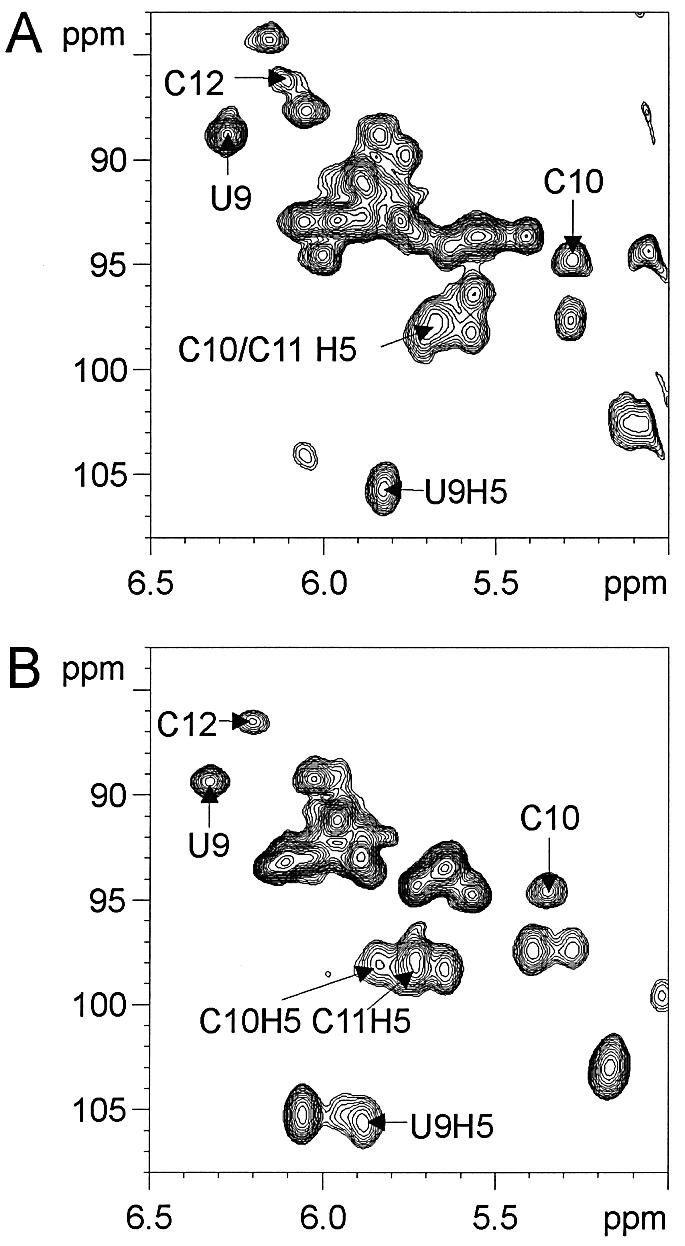
Consensus loop NRE nucleotides of sNRE and b2NRE have similar chemical shifts. 1H–13C HSQC of the H1′–C1′ and H5–C5 region of 13C/15N-labeled (A) sNRE26 and (B) b2NRE in a 1:1 complex with nucleolin RBD12 at 310 K. Assignments of some of the well dispersed peaks are shown (12). Samples were between 0.5 and 1 mM in complex.
Figure 7.

Comparison of nucleolin RBD12 in complex with sNRE and b2NRE. (A) 1H–15N HSQC spectra of nucleolin RBD12 in a 1:1 complex with sNRE (purple) and b2NRE (red). Some of the amino acids are labeled (12). Gray labels indicate no or very little chemical shift difference (|ΔN|+|ΔH|<65 Hz); yellow labels indicate small chemical shift changes (|ΔN|+|ΔH|<75 ≥65Hz); orange labels indicate moderate chemical shift differences (|ΔN|+|ΔH|<90 ≥75Hz); and red labels indicate large chemical shift differences (|ΔN|+|ΔH|≥90 Hz). (B) Chemical shift differences observed in (A) illustrated on the lowest energy structure of the nucleolin RBD12–sNRE (12). The largest chemical shift differences are clustered where nucleolin RBD12 contacts the S-shaped backbone of sNRE.
DISCUSSION
The loop of sNRE is disordered
The original structure determination of sNRE at low ionic strength and pH 6 showed that the NRE loop was disordered and suggested that the loop switches between two conformations depending on the glycosidic torsion angle of G13 (18). RNA structure is often dependent on concentrations and types of counterions (48,49). For instance, the L3 hairpin loop of the hepatitis delta virus ribozyme is structured at 100 mM NaCl, but has multiple conformations at lower salt concentrations (58). Although it has been suggested that the disordered sNRE loop facilitates conformational rearrangements upon protein binding (18), it was also possible that the loop structure was disordered due to the low ionic strength and/or pH of the buffer. Here, we re-examine the structure of the sNRE under salt conditions identical to those used in the study of the nucleolin RBD12–sNRE complex and at higher pH. The re-examination shows that the loop remains disordered under the new conditions. However, the data for G13 and A14 suggest that these residues have some structural differences compared with the previously determined loop. First, we see no evidence for the syn–anti equilibrium reported previously for the G13 nucleotide at low pH and ionic strength. Instead, G13 is exclusively in the syn conformer. Other RNAs have also been observed to have nucleotides with glycosidic torsion angles strongly dependent on ionic strength and/or pH (58–62). Secondly, the syn G13 shows some evidence of stacking on A14. Thirdly, A14 has NOEs that cannot be satisfied by one structure, indicating exchange between at least two conformations.
The sNRE consensus was identified by in vitro selection using nucleolin as a ligand. The solution conditions used in the in vitro selection and binding reactions ranged from 150 to 200 mM NaCl or KCl, from 1 to 4 mM MgCl2, from 50 to 100 mM Tris and pH from 7.4 to 7.5 (5). The pH and salt concentrations used in this structural study of sNRE26 are more similar to those used in the in vitro selection and binding assays than those of the previous structural study (18). Therefore, the re-examined structure better reflects what was selected for using in vitro evolution. In vitro selection can generate a high-affinity RNA target to a protein in two ways. (i) The RNA target can be selected for the ability to form a rigid structure that is similar to the bound conformation (i.e. lock-and-key binding mechanism). (ii) An RNA target with a disordered binding region can be selected, so that the RNA has enough flexibility for conformational rearrangements to occur when the protein or ligand binds. Although the first mechanism theoretically avoids energetic penalties associated with binding, structured RNAs tend not to have functional groups important for specific protein–RNA interactions exposed to solution. The second mechanism is more commonly seen in protein–RNA recognition, since it allows for the creation of intimate protein–RNA interfaces, with the only cost being entropic penalties associated with the ordering of flexible elements (19,20). The nucleolin RBD12–sNRE complex shows that the NRE consensus nucleotides are in an extended conformation across the surface of the β-sheets of the RBDs, and that the S-shaped backbone of the loop E motif is contacted by RBD1 and the linker residues (12). While the loop E motif was most probably selected to stabilize the protein–RNA complex by providing additional contacts between the protein and RNA (18), the disordered loop of sNRE was probably selected to allow enough flexibility in the loop to facilitate creation of the intricate protein–RNA interface found in the nucleolin RBD12–sNRE complex. The loop of b1NRE, one of the two natural NRE sequences studied here, is also conformationally heterogeneous.
b2NRE contains a bulged uridine
The b2NRE RNA forms a hairpin with an 8 bp stem capped by a tetraloop, in contrast to the predicted 6 bp stem and eight nucleotides in the loop (Fig. 1C). The 8 bp stem is interrupted between the sixth and seventh base pairs by a bulged uridine, which lies in the minor groove. Bulged uridines in the context of G·C/C·G and G·C/G·C stacks have been observed in both solution and crystal structures. In the crystal structure of the RNA duplex r(gugucgcac)2 (63) and in the solution structure of the P4 element from bacterial RNase P (64), the helices, which are interrupted by a 5′-bulged uridine in the context of a G·C/C·G stack, are basically straight. The solution structure of the P5.1 hairpin of Bacillus RNase P shows that the 5′-bulged uridine in the context of a G·C/G·C stack was observed to induce a significant kink in the helix (∼100°) (65). In contrast, the 3′-bulged uridine surrounded by an analogous C·G/C·G stack in b2NRE has no significant effect on the global geometry of the helix, which is essentially straight.
Structure calculations of b2NRE without RDCs resulted in two families of structures. One family had an ∼90° bend at the bulged uridine similar to that observed by Leeper et al. (65), while the other family did not have a bent helix. Neither ensemble of structures had NOE or dihedral angle violations. Therefore, both families of structures were refined against RDCs with independently determined DA and R values of the alignment tensor. Since the RDC refined structures maintained all features of the starting ensembles, selection of one of the two families as the final ensemble was based on overall lower energy and agreement of back-calculated RDCs with experimental values. These selection criteria identified the b2NRE family with the straight helix as the final ensemble. The observation of a structural family with a bent helix similar to that seen by Leeper et al. in our calculations that is notably higher in overall energy and has back-calculated RDCs that agree less with experimental values than the ensemble of structures with the straight helix, suggests that the large kink seen in the P5.1 helix may need re-evaluation (65).
The b2NRE forms a YNMG tetraloop with a closing U·A base pair
The CCCG tetraloop of b2NRE is structurally similar to the UNCG family of tetraloops, and therefore can be considered as a member of the newly described YNMG tetraloop family where Y is either a C or U, N is any nucleotide, and M is either an A or C (22). Most members of the YNMG family that have been characterized structurally and/or thermodynamically have a C·G closing base pair. In vitro selection using temperature gradient gel electrophoresis has shown that YNMG tetraloops are most stable with a closing C·G base pair, with the exception of the CUUG tetraloop, which is most stable with a G·C closing base pair (66). Recently, the structure of the CACG tetraloop demonstrated that the general YNMG structure can be maintained even when a U·G reverse wobble closing base pair is present (67). In the case of b2NRE, the tetraloop is closed by a Watson–Crick U·A base pair. Although most of the structural features of the YNMG tetraloops are maintained with a U·A closing base pair, some differences are observed. The base pair made by the first nucleotide (C10) and fourth nucleotide (G13) is similar to that observed for the YNMG family, with the G13 imino and amino protons hydrogen bonding to the O2-carbonyl of C10 (57,67). However, the 2′-hydroxyl proton of C10, which was not observed in NMR spectra, is not within hydrogen bonding distance of the G13 O6-carbonyl. In comparison with the structure of the cUUCGg tetraloop (57), the stacking of G13 shifts slightly from the A13 sugar moiety to stacking more on the A13 base. In other members of the YNMG family, the third loop nucleotide is hydrogen bonded to the pro-R phosphate oxygen of the second loop nucleotide (22). The C12 amino protons in b2NRE are detectable, but do not have NOEs that would suggest a C12 amino/pro-R phosphoryl oxygen hydrogen bond (57). The final b2NRE ensemble does not have the C12 amino protons within hydrogen bonding distance of the C11 pro-R phosphate oxygen in the b2NRE structure. Interestingly, the orientation of the C11 and C12 bases within a YNMG type tetraloop structure has the hydrogen bond donors and acceptors exposed to solvent. This orientation may be important for protein recognition (66,67).
Nucleolin RBD12 can recognize structured and disordered NREs
We have shown that like the sNRE, natural NREs can bind sequence specifically to nucleolin RBD12. Comparison of the chemical shifts of the RNA in the nucleolin RBD12–sNRE and nucleolin RBD12–b2NRE complexes suggests that consensus nucleotides of b2NRE in complex with nucleolin RBD12 are in a conformation similar to that observed in the nucleolin RBD12–sNRE complex (12). Therefore, when the free b2NRE and nucleolin RBD12 form a complex, a large conformational rearrangement in the loop must occur. Conformational rearrangement of a folded ligand can be energetically costly since it requires the unfolding of pre-existing structure (19). Thermodynamic studies of a cCCCGg tetraloop have shown that it is one of the least stable tetraloops of the YNMG family (22). Furthermore, the CCCG tetraloop of b2NRE is on top of a U·A base pair, which results in the tetraloop being even less stable. The energetic penalty associated with conformational rearrangement of the b2NRE NRE consensus is probably proportional to the low stability of the uCCCGa tetraloop sequence; therefore, the structured NRE consensus sequence of b2NRE does not prevent binding. Interestingly, the b2ΔU16 RNA did not competitively bind nucleolin RBD12 in the presence 10 000-fold less b2NRE even though it has all NRE consensus nucleotides and forms an identical hairpin structure (data not shown). Structural studies of the nucleolin RBD12–b2NRE complex show that the bulged U16 does not interact with the protein in the nucleolin RBD12–b2NRE complex (C. Johansson et al., in preparation). UV melting indicates that b2ΔU16 is ∼3.3 kcal/mol more stable than b2NRE (data not shown), consistent with the removal of a bulged nucleotide (68). Therefore, the bulged uridine in b2NRE appears to be present to decrease the stability of the upper domain in order to facilitate conformational transitions. Similarly, the Rev–RRE complex and TAR have extra-helical bases that are not involved in protein–RNA contacts, but are necessary for conformational transitions (19).
Although the nucleolin RBD12 sequence specifically recognizes the natural NREs derived from the 5′ ETS of mouse pre-rRNA, their affinity for nucleolin RBD12 is 100- to 500-fold less than that of the sNRE. The lower affinity of the natural NREs for nucleolin RBD12 is consistent with nucleolin’s transient association with pre-rRNA (5,10). b2NRE is more structured and rigid than the flexible sNRE loop, and this may contribute to the lower affinity observed for b2NRE. However, differences in flexibility cannot fully explain the trend in KDs since b1NRE, the natural NRE which displays conformational heterogeneity, binds more weakly than b2NRE. In addition, a mutant of b2NRE, b2G15A, which is similar to b2NRE except for conformational heterogeneity on the 3′ side of the loop (NMR spectra not shown), binds just as well as b2NRE. Therefore, flexibility of the NRE is probably important for binding, but is not the primary determinant of affinity. Since the largest protein chemical shift differences between the nucleolin RBD12–sNRE and the nucleolin RBD12–b2NRE complexes are found in residues that contact the loop E motif, it is likely that the presence of the loop E motif in the in vitro selected NREs is responsible for the difference in affinity observed between sNRE and the natural NRE targets.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Robert Peterson for assistance with the NMR spectroscopy, Dr Carla Theimer for helpful discussion and critical reading of the manuscript, Dr Philippe Bouvet for helpful discussion, Brittany Yerby for assistance with protein preparation, and Evan Feinstein for figure and manuscript preparation. This research was supported by NIH grant GM37254 (J.F.), a Blanceflor Foundation (Sweden) postdoctoral fellowship to C.J., and an EMBO postdoctoral fellowship to L.T.
REFERENCES
- 1.Srivastava M. and Pollard,H.B. (1999) Molecular dissection of nucleolin’s role in growth and cell proliferation: new insights. FASEB J., 13, 1911–1922. [PubMed] [Google Scholar]
- 2.Ginisty H., Sicard,H., Roger,B. and Bouvet,P. (1999) Structure and function of nucleolin. J. Cell Sci., 112, 761–772. [DOI] [PubMed] [Google Scholar]
- 3.Roger B., Moisand,A., Amalric,F. and Bouvet,P. (2002) Repression of RNA polymerase I transcription by nucleolin is independent of the RNA sequence that is transcribed. J. Biol. Chem., 277, 10209–10219. [DOI] [PubMed] [Google Scholar]
- 4.Bouvet P., Diaz,J.-J., Kindbeiter,K., Madjar,J.-J. and Amalric,F. (1998) Nucleolin interacts with several ribosomal proteins through its RGG domain. J. Biol. Chem., 273, 19025–19029. [DOI] [PubMed] [Google Scholar]
- 5.Ghisolfi-Nieto L., Joseph,G., Puvion-Dutilleul,F., Amaric,F. and Bouvet,P. (1996) Nucleolin is a sequence specific RNA-binding protein—characterization of targets on pre-ribosomal RNA. J. Mol. Biol., 260, 34–53. [DOI] [PubMed] [Google Scholar]
- 6.Herrera A.H. and Olson,M.O. (1986) Association of protein C23 with rapidly labeled nucleolar RNA. Biochemistry, 25, 6258–6264. [DOI] [PubMed] [Google Scholar]
- 7.Birney E., Kumar,S. and Krainer,A.R. (1993) Analysis of the RNA-recognition motif and RS and RGG domains: conservation in metazoan pre-mRNA splicing factors. Nucleic Acids Res., 21, 5803–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginisty H., Amalric,F. and Bouvet,P. (2001) Two different combinations of RNA-binding domains determine the RNA binding specificity of nucleolin. J. Biol. Chem., 276, 14338–14343. [DOI] [PubMed] [Google Scholar]
- 9.Ginisty H., Serin,G., Ghisolfi-Nieto,L., Roger,B., Libante,V., Amalric,F. and Bouvet,P. (2000) Interaction of nucleolin with an evolutionarily conserved pre-ribosomal RNA sequence is required for the assembly of the primary processing complex. J. Biol. Chem., 275, 18845–18850. [DOI] [PubMed] [Google Scholar]
- 10.Serin G., Joseph,G., Ghisolfi,L., Bauzan,M., Erard,M., Amalric,F. and Bouvet,P. (1997) Two RNA-binding domains determine the RNA-binding specificity of nucleolin. J. Biol. Chem., 272, 13109–13116. [DOI] [PubMed] [Google Scholar]
- 11.Serin G., Joseph,G., Faucher,C., Ghisolfi,L., Bouche,G., Amalric,F. and Bouvet,P. (1996) Localization of nucleolin binding sites on human and mouse pre-ribosomal RNA. Biochimie, 78, 530–538. [DOI] [PubMed] [Google Scholar]
- 12.Allain F.H.-T., Bouvet,P., Dieckmann,T. and Feigon,J. (2000) Molecular basis of sequence specific recognition of RNA stem–loops by nucleolin. EMBO J., 19, 6870–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leontis N.B. and Westhof,E. (1998) A common motif organizes the structure of multi-helix loops in 16 S and 23 S ribosomal RNAs. J. Mol. Biol., 283, 571–583. [DOI] [PubMed] [Google Scholar]
- 14.Correll C.C., Freeborn,B., Moore,P.B. and Steitz,T.A. (1997) Metals, motifs and recognition in the crystal structure of a 5S rRNA domain. Cell, 91, 705–712. [DOI] [PubMed] [Google Scholar]
- 15.Wimberly B., Varani,G. and Tinoco,I.,Jr (1993) The conformation of loop E of eukaryotic 5S ribosomal RNA. Biochemistry, 32, 1078–1087. [DOI] [PubMed] [Google Scholar]
- 16.Szewczak A.A. and Moore,P.B. (1995) The sarcin/ricin loop, a modular RNA. J. Mol. Biol., 247, 81–98. [DOI] [PubMed] [Google Scholar]
- 17.Correll C.C., Munishkin,A., Chan,Y.L., Ren,Z., Wool,I.G. and Steitz,T.A. (1998) Crystal structure of the ribosomal RNA domain essential for binding elongation factors. Proc. Natl Acad. Sci. USA, 95, 13436–13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouvet P., Allain,F.H.-T., Finger,L.D., Dieckmann,T. and Feigon,J. (2001) Recognition of pre-formed and flexible elements of an RNA stem–loop by nucleolin. J. Mol. Biol., 309, 763–775. [DOI] [PubMed] [Google Scholar]
- 19.Leulliot N. and Varani,G. (2001) Current topics in RNA–protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry, 40, 7947–7956. [DOI] [PubMed] [Google Scholar]
- 20.Williamson J.R. (2000) Induced fit in RNA–protein recognition. Nature Struct. Biol., 7, 834–837. [DOI] [PubMed] [Google Scholar]
- 21.Bourbon H.M., Michot,B., Hassouna,N., Feliu,J. and Bachellerie,J.P. (1988) Sequence and secondary structure of the 5′ external transcribed spacer of mouse pre-RNA. DNA, 7, 181–191. [DOI] [PubMed] [Google Scholar]
- 22.Proctor D.J., Schaak,J.E., Bevilacqua,J.M., Falzone,C.J. and Bevilacqua,P.C. (2002) Isolation and characterization of a family of stable RNA tetraloops with the motif YNMG that participate in tertiary interactions. Biochemistry, 41, 12062–12075. [DOI] [PubMed] [Google Scholar]
- 23.Dieckmann T. and Feigon,J. (1997) Assignment methodology for larger oligonucleotides: application to an ATP-binding RNA aptamer. J. Biomol. NMR, 9, 259–272. [DOI] [PubMed] [Google Scholar]
- 24.Roberts G.C.K. (ed.) (1993) NMR of Macromolecules. Oxford University Press, New York, NY. [Google Scholar]
- 25.Heyduk T., Ma,Y., Tang,H. and Ebright,R.H. (1996) Fluorescence anisotropy: rapid, quantitative assay for protein–DNA and protein–protein interaction. Methods Enzymol., 274, 492–503. [DOI] [PubMed] [Google Scholar]
- 26.Heyduk T. and Lee,L.C. (1990) Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc. Natl Acad. Sci. USA, 87, 1744–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macura S. and Ernst,R.R. (1980) Elucidation of cross relaxation in liquids by two-dimensional NMR spectroscopy. Mol. Phys., 41, 95–117. [Google Scholar]
- 28.Rance M., Sørensen,M., Bodenhausen,G., Wagner,G., Ernst,R.R. and Wüthrich,K. (1983) Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun., 117, 479–485. [DOI] [PubMed] [Google Scholar]
- 29.Griesinger C., Otting,G., Wüthrich,K. and Ernst,R.R. (1988) Clean TOCSY for 1H spin system identification in macromolecules. J. Am. Chem. Soc., 110, 7870–7872. [Google Scholar]
- 30.Fiala R., Czernek,J. and Sklenar,V. (2000) Transverse relaxation optimized triple-resonance NMR experiments for nucleic acids. J. Biomol. NMR, 16, 291–302. [DOI] [PubMed] [Google Scholar]
- 31.Sklenar V., Piotto,M., Leppik,R. and Saudek,V. (1993) Gradient-tailored water suppression for 1H–15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson., 102, 241–245. [Google Scholar]
- 32.Clore G.M., Bax,A., Driscoll,P.C., Wingfield,P.T. and Gronenborn,A.M. (1990) Assignment of the side-chain 1H and 13C resonances of interleukin-1β using double- and triple-resonance heteronuclear three-dimensional NMR spectroscopy. Biochemistry, 29, 8172–8184. [DOI] [PubMed] [Google Scholar]
- 33.Bax A., Clore,G.M. and Gronenborn,A.M. (1990) 1H–1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J. Magn. Reson., 88, 425–431. [Google Scholar]
- 34.Nikonowicz E.P. and Pardi,A. (1993) An efficient procedure for assignment of the proton, carbon and nitrogen resonances in 13C/15N labeled nucleic acids. J. Mol. Biol., 232, 1141–1156. [DOI] [PubMed] [Google Scholar]
- 35.Sklenar V., Dieckmann,T., Butcher,S.E. and Feigon,J. (1996) Through-bond correlation of imino and aromatic resonances in 13C,15N-labeled RNA via heteronuclear TOCSY. J. Biomol. NMR, 7, 83–87. [DOI] [PubMed] [Google Scholar]
- 36.Mueller L., Legault,P. and Pardi,A. (1995) Improved RNA structure determination by detection of NOE contacts to exchange-broadened amino protons. J. Am. Chem. Soc., 117, 11043–11048. [Google Scholar]
- 37.Simorre J.P., Zimmermann,G.R., Mueller,L. and Pardi,A. (1996) Triple-resonance experiments for assignment of adenine base resonances in C-13/N-15-labeled RNA. J. Am. Chem. Soc., 118, 5316–5317. [Google Scholar]
- 38.Wöhnert J., Ramachandran,R., Görlach,M. and Brown,L.R. (1999) Triple-resonance experiments for correlation of H5 and exchangeable pyrimidine base hydrogens in 13C, 15N-labeled RNA. J. Magn. Reson., 139, 430–433. [DOI] [PubMed] [Google Scholar]
- 39.Luy B. and Marino,J.P. (2000) Direct evidence for Watson–Crick base pairs in a dynamic region of RNA structure. J. Am. Chem. Soc., 122, 8095–8096. [Google Scholar]
- 40.Hennig M. and Williamson,J.R. (2000) Detection of N–H center·center·center·N hydrogen bonding in RNA via scalar couplings in the absence of observable imino proton resonances. Nucleic Acids Res., 28, 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H., Pok,K., Butcher,S.E., Kang,S., Chanfreau,G. and Feigon,J. (2001) A novel family of RNA tetraloop structure forms the recognition site for S.cerevisiae RNase III. EMBO J., 20, 7240–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marino J.P., Schwalbe,H. and Griesinger,C. (1999) J-coupling restraints in RNA structure determination. Acc. Chem. Res., 32, 614–623. [Google Scholar]
- 43.Wijmenga S.S. and van Buuren,B.N.M. (1998) The use of NMR methods for conformational studies of nucleic acids. Prog. Nucl. Magn. Reson. Spec., 32, 287–387. [Google Scholar]
- 44.Varani G., Aboul-ela,F. and Allain,F.H.-T. (1996) NMR investigation of RNA structure. Prog. Nucl. Magn. Reson. Spec., 29, 51–127. [Google Scholar]
- 45.Schwieters C.D., Kuszewski,J.J., Tjandra,N. and Clore,G.M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson., 160, 65–73. [DOI] [PubMed] [Google Scholar]
- 46.Clore G.M., Gronenborn,A.M. and Tjandra,N. (1998) Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson., 131, 159–162. [DOI] [PubMed] [Google Scholar]
- 47.Lu X.J., Shakked,Z. and Olson,W.K. (2000) A-form conformational motifs in ligand-bound DNA structures. J. Mol. Biol., 300, 819–840. [DOI] [PubMed] [Google Scholar]
- 48.Saenger W. (1984) Principles of Nucleic Acid Structure. Springer-Verlag, New York, NY. [Google Scholar]
- 49.Feigon J., Butcher,S.E., Finger,L.D. and Hud,N.V. (2001) Solution nuclear magnetic resonance probing of cation binding sites on nucleic acids. Methods Enzymol., 338, 400–420. [DOI] [PubMed] [Google Scholar]
- 50.Dallas A. and Moore,P. (1997) The loop E–loop D region of Escherichia coli 5S rRNA: the solution structure reveals an unusual loop that may be important for binding ribosomal proteins. Structure, 5, 1639–1653. [DOI] [PubMed] [Google Scholar]
- 51.Wüthrich K. (1986) NMR of Proteins and Nucleic Acids. John Wiley & Sons, New York, NY. [Google Scholar]
- 52.Avizonis A.Z. and Kearns,D.R. (1995) Structural characterization of d(CAACCCGTTG and d(CAACGGGTTG) mini-hairpin loops by heteronuclear NMR: the effects of purines versus pyrimidines in DNA hairpins. Nucleic Acids Res., 23, 1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Avizonis D.Z. and Kearns,D.R. (1995) Kinetic and thermodynamic characterization of DNA duplex–hairpin interconversion for two DNA decamers—d(CAACGGGTTG) and d(CAACCCGTTG). Biopolymers, 35, 187–200. [DOI] [PubMed] [Google Scholar]
- 54.Brünger A.T. (1992) X-PLOR (Version 3.1) Manual. Yale University Press, New Haven, CT. [Google Scholar]
- 55.Ghose R., Marino,J.P., Wiberg,K.B. and Prestegard,J.H. (1994) Dependence of 13C chemical shifts on glycosidic torsional angles in ribonucleic acids. J. Am. Chem. Soc., 116, 8827–8828. [Google Scholar]
- 56.Allen M., Varani,L. and Varani,G. (2001) Nuclear magnetic resonance methods to study structure and dynamics of RNA–protein complexes. Methods Enzymol., 339, 357–376. [DOI] [PubMed] [Google Scholar]
- 57.Allain F.H.T. and Varani,G. (1995) Structure of the P1 helix from group I self-splicing introns. J. Mol. Biol., 250, 333–353. [DOI] [PubMed] [Google Scholar]
- 58.Lynch S.R. and Tinoco,I. (1998) The structure of the L3 loop from the hepatitis delta virus ribozyme: a syn cytidine. Nucleic Acids Res., 26, 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ippel J.H., Lanzotti,V., Galeone,A., Mayol,L., Vandenboogaart,J.E., Pikkemaat,J.A. and Altona,C. (1995) Slow conformational exchange in DNA minihairpin loops—a conformational study of the circular dumbbell d[pCGC-TT-GCG-TT]. Biopolymers, 36, 681–694. [DOI] [PubMed] [Google Scholar]
- 60.van Dongen M.J., Heus,H.A., Wymenga,S.S., van der Marel,G.A., van Boom,J.H. and Hilbers,C.W. (1996) Unambiguous structure characterization of a DNA–RNA triple helix by 15N- and 13C-filtered NOESY spectroscopy. Biochemistry, 35, 1733–1739. [DOI] [PubMed] [Google Scholar]
- 61.Hall K.B. and Tang,C.G. (1998) C-13 Relaxation and dynamics of the purine bases in the iron responsive element RNA hairpin. Biochemistry, 37, 9323–9332. [DOI] [PubMed] [Google Scholar]
- 62.Addess K.J., Basilion,J.P., Klausner,R.D., Rouault,T.A. and Pardi,A. (1997) Structure and dynamics of the iron responsive element RNA—implications for binding of the RNA by iron regulatory binding proteins. J. Mol. Biol., 274, 72–83. [DOI] [PubMed] [Google Scholar]
- 63.Xiong Y., Deng,J.P., Sudarsanakumar,C. and Sundaralingam,M. (2001) Crystal structure of an RNA duplex r(GUGUCGCAC)2 with uridine bulges. J. Mol. Biol., 313, 573–582. [DOI] [PubMed] [Google Scholar]
- 64.Schmitz M. and Tinoco,I. (2000) Solution structure and metal-ion binding of the P4 element from bacterial RNase P RNA. RNA, 6, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leeper T.C., Martin,M.B., Kim,H., Cox,S., Semenchenko,V., Schmidt,F.J. and Van Doren,S.R. (2002) Structure of the UGAGAU hexaloop that braces Bacillus RNase P for action. Nature Struct. Biol., 9, 397–403. [DOI] [PubMed] [Google Scholar]
- 66.Jucker F.M. and Pardi,A. (1995) Solution structure of the CUUG hairpin loop—a novel RNA tetraloop motif. Biochemistry, 34, 14416–14427. [DOI] [PubMed] [Google Scholar]
- 67.Du Z.H., Yu,J.H. andino,R. and James,T.L. (2003) Extending the family of UNCG-like tetraloop motifs: NMR structure of a CACG tetraloop from coxsackievirus B3. Biochemistry, 42, 4373–4383. [DOI] [PubMed] [Google Scholar]
- 68.Turner D.H., Sugimoto,N. and Freier,S.M. (1988) RNA structure prediction. Annu. Rev. Biophys. Biophys. Chem., 17, 167–192. [DOI] [PubMed] [Google Scholar]