

Abstract
Background
Three-fourths of cardiac arrest survivors die prior to hospital discharge or suffer significant neurological injury. Excepting therapeutic hypothermia and revascularization, no novel therapies have been developed that improve survival or cardiac and neurological function after resuscitation. Nitrite (NO2−) increases cellular resilience to focal ischemia-reperfusion injury in multiple organs. We hypothesized that nitrite therapy may improve outcomes after the unique global ischemia-reperfusion insult of cardiopulmonary arrest.
Methods and Results
We developed a mouse model of cardiac arrest characterized by 12-minutes of normothermic asystole and a high cardiopulmonary resuscitation (CPR) rate. In this model, global ischemia and CPR was associated with blood and organ nitrite depletion, reversible myocardial dysfunction, impaired alveolar gas exchange, neurological injury and an approximate 50% mortality. A single low dose of intravenous nitrite (50 nmol=1.85 μmol/kg=0.13 mg/kg) compared to blinded saline placebo given at CPR initiation with epinephrine improved cardiac function, survival and neurological outcomes. From a mechanistic standpoint, nitrite treatment restored intracardiac nitrite and increased S-nitrosothiol levels, decreased pathological cardiac mitochondrial oxygen consumption due to reactive oxygen species formation and prevented oxidative enzymatic injury via reversible specific inhibition of respiratory chain complex I.
Conclusion
Nitrite therapy after resuscitation from 12-minutes of asystole rapidly and reversibly modulated mitochondrial reactive oxygen species generation during early reperfusion, limiting acute cardiac dysfunction and death, as well as neurological impairment in survivors.
Keywords: cardiopulmonary resuscitation, heart arrest, ischemia, nitric oxide, reperfusion
Introduction
Nitrite (NO2−), historically considered inert, functions as a reservoir for nitric oxide (NO) 1. During physiological hypoxia and pathological ischemia, nitrite is reduced to NO regulating hypoxic vasodilation, cellular respiration, mitochondrial reactive oxygen species (ROS) generation, angiogenesis2, and cellular death programs3. Nitrite in human plasma exists at concentrations of 100–300 nM3–5 and may be reduced to NO by iron-containing enzymes3, 6 including hemoglobin, myoglobin, neuroglobin, xanthine oxidoreductase, endothelial NO synthase, mitochondrial electron transport chain proteins and the hepatic cytochrome P450 system. The rate and extent of nitrite reduction is coupled to deoxygenation and proton generation. Thus NO generation is coupled to oxygen and pH gradients3, 7 and maximized in ischemic tissues.
Nitrite therapy limits cellular injury and apoptosis after ischemia and reperfusion6. Nitrite therapy is cytoprotective in numerous animal models of focal ischemia-reperfusion injury6 including rodent heart8, brain9, liver8 and kidney, canine heart10 and primate brain11. Systemic nitrite reduction by ceruloplasmin knockout12 or dietary nitrate/nitrite elimination13 increased infarction volume in the liver and heart after experimental ischemia. These studies indicate that physiological systemic nitrite levels modulate host resilience to ischemia. The established safety of human and animal nitrite dosing14 and its potent effects in limiting major organ injury suggest that nitrite represents an ideal therapy for cardiac arrest.
Cardiac arrest results in global multi-organ ischemic injury associated with significant morbidity and mortality15, 16. Over 70% of those resuscitated die prior to hospital discharge15, 16. Excepting the selective application of hypothermia and revascularization, no novel post-resuscitation therapies have been developed that improve survival or cardiac and neurological function15. We explored nitrite therapy in a mouse model of cardiac arrest. We show evidence that nitrite therapy improves cardiac function, survival and neurological function in survivors. Mechanistically, we show that nitrite specifically and reversibly inhibits cardiac complex I, limiting oxidative reperfusion injury. Nitrite’s ease of delivery, established human safety and efficacy in murine cardiac arrest suggest its promise as a novel therapy after cardiac arrest.
Methods
Mouse Cardiac Arrest Model
In initial model building we adapted mouse cardiac arrest models from prior reports17, 18 with the goal of nearly 100% resuscitation and 50% 24 hour mortality. To modulate mortality we extended asystolic time from 8 to 12 minutes and added epinephrine to increase return of spontaneous circulation (ROSC). Seven to twelve week old (24–30g) C57BL/6 male mice were used as approved by the NIH IACUC. Anesthesia intraperitoneal ketamine/xylazine (100/15 mg/kg respectively) preceded surgical preparation consisting of orotracheal intubation and placement of right carotid and jugular catheters. Ventilation (ASV, Harvard Instruments, Cambridge, MA) for 10 minutes (minimum) at a rate of 110 bpm and a volume of 15 ml/kg preceded cardiac arrest and produced normal blood gases (Table 1). EKG, arterial blood pressure, exhaled carbon dioxide and rectal temperature were recorded at baseline and during 60 minutes post-resuscitation (Powerlab, ADInstruments, Colorado Springs, CO). Asystole was induced by 100 μl potassium chloride (0.09 g/kg; minimum 2.5% [w/v]) IV bolus and temperature maintained at 36.5°C for 12 minutes. Cardiopulmonary resuscitation (CPR) was performed with rapid (375–425) finger compressions, resumption of mechanical ventilation (25% tidal volume increase) and 10 μg IV epinephrine (500 μl) prior to CPR and 2 μg (100 μl) 1 minute post-CPR. Animals received either 50 nmol (1.85μmol/kg=0.13 mg/kg,) sodium nitrite (in 0.9% saline) or saline placebo IV in randomized, blinded fashion at CPR initiation. This dose and timing of delivery were based on prior work8.
Table 1.
Arterial blood gas results obtained from sham mice or five minutes after CPR from mice randomized to placebo or nitrite
| Post-Arrest Treatment | |||
|---|---|---|---|
| Characteristic | Sham | Placebo | Nitrite |
| pH | 7.35±0.03 | 6.61±0.04 * | 6.73±0.05 |
| PaCO2 (mm Hg) | 38.4±2.7 | 108.3±16.0 * | 66.2±16.2 ** |
| PaO2 (mm Hg) | 253.7±17.6 | 128.2±23.8 * | 210.7±25.7 ** |
| Bicarbonate (mg/dL) | 20.9±0.8 | 9.9±0.9 * | 8.4±0.9 |
| Lactate (mg/dL) | 0.9±0.1 | 16.5±0.5 * | 15.6±1.2 |
Key: All data means±SEM (n=5 per group) analyzed by ANOVA;
, p<0.01 sham vs. post-arrest placebo;
, p<0.05 post-arrest placebo vs. nitrite.
To minimize variability we paired inbred animals to nitrite or placebo based on similar weight, sex, age, delivery date and where possible holding cage. Pairing appeared effective (Table 2). Post-resuscitation care was uniform between groups. A model summary is provided as Figure 1. Sham control animals received anesthesia, surgery and ventilation but no cardiac arrest and the same post-resuscitation care until the similarly timed endpoint. Shams were drawn from the same batch as experimental animals.
Table 2.
Baseline and CPR characteristics of randomized animals
| Post-Arrest Treatment | ||
|---|---|---|
| Characteristic | Placebo | Nitrite |
| Age at Experiment (weeks) | 10±1.8 | 10±1.8 |
| Weight (g) | 27.0±2.3 | 26.5±2.3 |
| Ischemic Time (min) | 12.0±0.01 | 12.0±0.03 |
| DBP 15 sec prior to CPR | 4.3±0.4 | 4.8±0.2 |
| DBP during 15 sec after CPR start | 27.4±2.3 | 27.9±3.5 |
| Successful Resuscitation | 25/28 (89%) | 25/27 (93%) |
| Time to ROSC (sec) | 54.8±30.1 | 43.0±26.3* |
Key:
, p=0.034; DBP=diastolic blood pressure; ROSC=return of spontaneous circulation; n=21 per group
Figure 1. Cardiac arrest experiment, endpoints and cardiovascular effects.

(A) Experimental outline with sample blood pressure and EKG pre-arrest, during arrest, cardiopulmonary resuscitation (CPR), return of spontaneous circulation (ROSC) and recovery phases with endpoints. (B) Physiological data from one experiment depicting heart rate (HR), mean arterial blood pressure (MAP) and exhaled carbon dioxide (CO2). The ischemic period is shaded. (C–E) Post-arrest placebo treated mice are compared to non-arrested shams. (C) Mean HR increases and MAP decreases 60 min post-CPR vs. 1 min pre-arrest (n=21). (D) Cardiac arrest results in significant reductions in left ventricular fractional shortening (FS) and ejection fraction (EF) by echocardiography 75–90 minutes post-CPR (n=6). (E) Cardiac arrest results in diminished right ventricular ejection fraction (RVEF) and increased dilation (RVEDV: RV end-diastolic volume; n=5). Values denoted as means±SEM analyzed by paired t-test; *, p<0.01; †, p=0.038.
Blood Gases and Blood/Tissue Nitrite Levels
Carotid arterial blood was obtained in a heparinized syringe either 55 minutes after anesthesia (sham) or 5 minutes after CPR. Blood was utilized for blood gas analysis (Nova Biomedical, Waltham, MA) or mixed 4:1 with nitrite preservation solution4 or centrifuged to isolate plasma for nitrite measurements. Animals were perfused19 with tissue nitrite preservation solution (1 mM KCN, 0.2% NP-40, 0.8 mM ferricyanide, 0.5 mM NEM, 100 μM DTPA) and brains homogenized for nitrite measurements. Snap frozen hearts obtained 15 minutes after CPR were sectioned at −20°C, placed into ice cold nitrite preservation solution and homogenized for nitrite measurements. All nitrite measurements were determined by tri-iodide based gas-phase reductive chemiluminescence using an NO analyzer (GE Analytic, Boulder, CO) as described previously20, 21 and tissue levels normalized to protein content (BCA Protein Assay, Pierce, Rockford, IL).
Left ventricular echocardiography
Animals had lines removed, wound closure and chest hair removal before transportation for echocardiogram 60 minutes after CPR or sham surgery. Animals were secured to the Vevo 770 (VisualSonics, Toronto, ON, Canada) platform and temperature/EKG were monitored (temperature maintained at >35°C, heart rate >300 beats per minute, supplemental oxygen delivered via nose cone). Parasternal long-axis 2D images of the left ventricle (LV) were obtained using the RMV707B scanhead 75–90 minutes post-CPR. M-mode images were used to measure end-systolic and –diastolic ventricular size and fractional shortening and ejection fraction (EF) calculated with the manufacturer’s software (v.2.3.0).
Cardiac MRI
Anesthetized animals (1–1.5% isoflurane) were MRI imaged 24 hours post-CPR or sham surgery. MRI experiments were carried out in a 7.0T, 16-cm horizontal Bruker MR imaging system (Bruker, Billerica, MA) with Bruker ParaVision 3.0.2 software. Magnevist (Bayer HealthCare, Montville, NJ) diluted 1:10 with sterile 0.9% saline, was administered subcutaneously at a dose of 0.3 mmol/kg. Six short axis slices were used to determine EF and ventricular volumes using CAAS-MRV-FARM software (Pie Medical Imaging, Netherlands).
Histology
Mice were transcardiac perfused with saline followed by 10% buffered formalin19 and brains removed, further fixed, paraffin-embedded, sectioned and stained with hematoxylin and eosin. Seven serial high powered (400×) fields of bilateral CA1 at Bregma -1.5 mm were examined and live and dead cells counted and normalized to hippocampal length as described elsewhere17.
Mitochondria Isolation
Heart mitochondrial isolation was performed by differential centrifugation as described elsewhere22 and protein concentration determined. Fresh mitochondria were used for respirometry, ROS and ATP generation assays and aliquots stored at −80°C for subsequent Complex I activity assays as described previously23.
Aconitase Activity
Hearts snap frozen 15 minutes after CPR or sham surgery were homogenized in the manufacturer’s commercial buffer and lysates prepared by three cycles of freeze/thaw. Aconitase activity was determined spectrophotometrically (340 nm) monitoring NADPH formation using the Bioxytech Aconitase-340 kit (Oxis Research).
Statistical Analysis
Data appears as means±standard error (SEM) with analysis performed using GraphPad Prism 5 (La Jolla, CA). Continuous data were compared between three groups using one-way ANOVA with post-hoc Bonferroni adjustment, between two groups using paired Student’s t-test for variables that are normally distributed and Wilcoxon for variables that are not normally distributed and for variables measured at multiple time points from same subjects using repeated measures ANOVA. Mitochondrial experiments were performed as multiple paired experiments at discrete times and therefore analyzed at each time utilizing a paired t-test. Mortality was assessed by Kaplan-Meier survival analysis (log rank test). A two-tailed p<0.05 was considered statistically significant
Statement of Responsibility
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Cardiac arrest physiological effects
Cardiac arrest resulted in metabolic (lactic) and respiratory acidosis and oxygen depletion (Table 1). Ischemic times and resuscitation rates were similar between groups (Table 2). Cardiac arrest led to transient hyperemia soon after resuscitation (Figure 1B) as described by others17. One hour after resuscitation, placebo treated mice exhibited signs of myocardial depression based on decreased blood pressure, increased tachycardia (Figure 1C) and decreased ejection fractions (Figure 1D) compared to pre-arrest baseline. Twenty four hours later LVEF had normalized but right ventricular (RV) failure remained based on RV dilation and diminished RVEF (Figure 1E). Since right sided pressures were not assessed, it’s unclear whether this was due to pulmonary hypertension or loss of RV contractility.
Cardiac arrest depletes systemic nitrite which is reversed by IV therapy
Nitrite is reduced to NO and NO-modified proteins during focal ischemia in rodents13, 24. We tested the hypothesis that basal systemic levels of nitrite would be reduced by ischemic consumption during cardiac arrest and IV nitrite could restore levels (Figure 2A). In placebo treated mice, ischemia depleted whole blood nitrite levels (0.64±0.05μM) and nitrite therapy repleted these levels (1.01±0.06 μM) to near baseline (1.15±0.10μM). These findings were mirrored in plasma (Figure 2B), heart (Figure 2E/F) and a similar trend noted in brain (Figure 2C). S-nitrosation of the cardiomyocyte L-type calcium channel25 and mitochondrial complex I23, 25 are protective post-translational modifications resulting from nitrite therapy or ischemic preconditioning. We found that total S-nitrosothiol modified protein concentration (Figure 2F) did not change with ischemia but nitrite therapy significantly increased these levels (p<0.05).
Figure 2. Systemic nitrite depletion after global ischemia and therapeutic repletion.

Whole blood (A) and plasma (B) nitrite levels (n=6) measured after 12 minutes of global ischemia and 5 minutes reperfusion are depleted in placebo vs. sham, and nitrite therapy increases levels with similar trends in brain (C). (D) A sample reductive chemiluminescence tracing measuring nitrite in heart 15 min after CPR. Peaks represent total tissue nitrite (first peak), S-nitrosothiols (second peak) and the mercury stable fraction (third peak; not visible). (D) Ischemia depletes heart nitrite in placebos and nitrite therapy significantly increases total levels and (E) S-nitrosothiols (n=7). Values denoted as means±SEM analyzed by ANOVA; *, p<0.01 (placebo vs. sham); †, p<0.01 and ‡, p<0.05 (nitrite vs. placebo).
Nitrite repletion improves cardiac function
Increased systemic nitrite in treated mice was associated with improved cardiac function. Just before CPR, animals exhibited similar vascular loading based on identical asystolic pressures (Table 2). Nitrite didn’t decrease diastolic blood pressure, a coronary perfusion pressure surrogate, during CPR and ROSC occurred sooner (p=0.034) than placebo treatment. Consistent with improved cardiac function, nitrite-treated mice exhibited trends towards less tachycardia than placebo-treated controls (68.7±12.1 vs. 94.7±17.8 beat per minute increase) and less hypotension (9.1 ± 3.2 vs. 12.6±4.2 mm Hg decrease).
Nitrite significantly improved post-arrest LVEF (54.4±2.4%; Figure 3A/C) compared to placebo-treated mice (43.5±2.9%; p=0.007). Heart rate and blood pressure were similar between treatment groups throughout post-ROSC monitoring (data not shown, p>0.2). Given similar baseline volume loading pre-CPR and similar mean blood pressures prior to imaging, the LVEF improvements likely indicate improved LV contractility. RVEF measured by MRI (Figure 3B/D) 24 hours post-CPR was significantly better in nitrite treated mice (54.7±1.3%) than placebo (42.8±1.7%; p< 0.001). Consistent with improved pulmonary perfusion, gas exchange 5 minutes post-CPR was improved with nitrite therapy compared to placebo (Table 1).
Figure 3. Improved cardiac function after nitrite therapy.

(A) Sample M-mode echocardiogram images obtained 75–90 minutes post-CPR from a pair of mice randomized to placebo (n=6) or nitrite therapy (n=8). (C) Left ventricular ejection fraction (LVEF) was significantly improved with nitrite therapy. (B) Sample MRI images 24 hours after CPR demonstrate normal LVEF but a dilated right ventricle with improved right ventricular ejection fraction (RVEF) in nitrite-treated vs. placebo animals (n=5). Values denoted as means±SEM analyzed by paired t-test; *, p<0.01.
Nitrite repletion improves survival and survivor neurological function
Death after ROSC occurred 1–6 hours post-CPR after which all animals survived until the subsequent day (Figure 4). We observed bradycardia and hypotension progressing to asystole in mice dying early (while monitored) consistent with death from post-ischemic myocardial stunning and heart failure. Nitrite therapy significantly improved survival (19/25 [76%]) compared to placebo (12/25 [48%]; hazard ratio 2.72 [95% confidence interval 1.1–6.7]; p=0.033).
Figure 4. Nitrite therapy improves survival.

After resuscitation, animals died 1–6 hours after CPR. Nitrite improved 22 hour survival compared to placebo (*, p=0.033; n=28/27 for placebo/nitrite groups).
Since a third of cardiac arrest survivors have neurological disability, we blindly assigned neurological scores18 to all 1 day surviving mouse pairs (n=11) (Figure 5A). The median score for nitrite-treated mice (11) was significantly better than placebo (9; p=0.020). In all pairs, placebo-treated mice exhibited equal or more severe impairment than nitrite-treated mice. We measured rectal temperature 22 hours post-resuscitation (Figure 5B) since rodent hypothermia correlates with severity of injury26. Nitrite-treated mice had better thermoregulation (34.9±0.4°C) than placebo-treated mice (32.4±0.9°C; p = 0.013). Sham surgery didn’t impair neurological scores (all 12) or thermoregulation (36.5±0.5°C). Hippocampal CA1, know to be selectively vulnerable to global ischemia, showed no histological injury at 24 hours consistent with prior observations27. Seventy-two hour survival experiments were therefore performed (n=3) demonstrating increased CA1 neuronal injury in placebo- compared to nitrite-treated mice (Figure 5C, D).
Figure 5. Nitrite neuroprotection.

Neurological function in paired 22 hour survivors (n=11) was improved with nitrite based on (A) neurological scores and (B) thermoregulation (rectal temperature). (C) At 72 hours post-CPR, moderate to severe hippocampal CA1 cell death was noted in placebo-treated brains while lesser injury was noted in nitrite-treated survivors. Hematoxylin and eosin staining, bar indicates 40 μm. (D) Summary of live and dead cells (per millimeter of CA1) in serial high-powered fields. Data presented as median (A) or means±SEM (B,D); *, p=0.016; †, p=0.013; ‡, p<0.01; #, p<0.05 comparing nitrite to placebo.
Nitrite specifically and reversibly inhibits complex I
Based on prior observations23, 28 we hypothesized that nitrite therapy in vivo would transiently S-nitrosate and inhibit mitochondrial complex I resulting in a decrease in reperfusion ROS. Heart mitochondria from mice treated with nitrite isolated 5, 15 and 60 minutes post-CPR exhibited reduced state 3 respiration using the complex I substrate pyruvate (Figure 6A) at 5 and 15 minutes but not one hour compared to placebo. Nitrite therapy did not reduce electron transport efficiency based on respiratory control ratio (5min: 9.5±1.7; 60 min: 13.9±4.4) which was similar to the placebo-treated group (5min: 7.5±1.0; 60min: 10.4±4.6). Complex II (succinate) dependent state 3 respiration was similar at all times (Figure 6B) and did not change with rotenone inhibition (data not shown) indicating nitrite’s complex I specificity. Complex I activity measured by NADH oxidation at 5 and 60 minutes confirmed the respirometry findings (Figure 6C). Using pyruvate as substrate, we consistently found increased oxygen consumption by placebo-treated post-arrest mitochondria compared to pre-arrest despite reduced complex I (NADH consumption). This suggests pathological oxygen consumption to form ROS rather than for energy production. Complex I inhibition was reversed 60 minutes post-CPR as measured by respirometry (Figure 6A) and NADH oxidation (Figure 6C). ATP generation was similar between 60 minute post-arrest nitrite (58.1±8.1 nmol/min/mg protein) and placebo (57.1±8.7 nmol/min/mg) treated groups and pre-arrest mice (65.1±4.3 nmol/min/mg) indicating no persistent functional complex I inhibition.
Figure 6. Nitrite therapy effects on mitochondrial function.

Heart mitochondrial respiration time course where zero represents CPR start and shaded area indicates arrest. (A) Inhibition of pyruvate (complex I) mediated respiration was present at 5 (n=8) and 15 minutes (n=4) post-CPR but reversed by 1 hour. (B) Succinate (complex II) mediated respiration rates did not differ (n ≥ 4 for each time). Inset: sample traces of mitochondrial oxygen consumption 5 minutes post-CPR. (C) Complex I activity measured in sub-mitochondrial particles by NADH oxidation. Compared to placebo, nitrite therapy significantly reduced complex I activity 5 minutes post-CPR (n=7) which was reversed by 60 minutes. Inset: sample tracing 5 minutes post-CPR. Values denoted as means±SEM, analyzed at each time by paired t-test; *, p<0.01; †, p=0.021; ‡, p=0.014.
Nitrite limits reperfusion ROS generation
Consistent with pathological oxygen conversion to ROS, peroxide generation by respiring mitochondria (pyruvate substrate) 5 and 15 minutes post-CPR exceeded that of pre-arrest mice but normalized by 1 hour (Figure 7A). Cardiac mitochondria from nitrite-treated mice had significantly less peak (5 minutes post-CPR) ROS production (14.0±3.2 pmol/min/mg protein) compared to placebo-treated mice (26.6±4.4 pmol/min/mg; p<0.01). The abundant mitochondrial enzyme aconitase is susceptible to oxidative modification decreasing its activity and thus useful as an indicator of oxidative damage. Cardiac arrest significantly decreased aconitase activity (Figure 7B). Compared to placebo (14.8±8.4 mU/mg protein), nitrite therapy attenuated this loss of aconitase function (61.6±11.4 mU/mg protein; p=0.05).
Figure 7. Nitrite therapy effects on mitochondrial oxidative burst and injury.
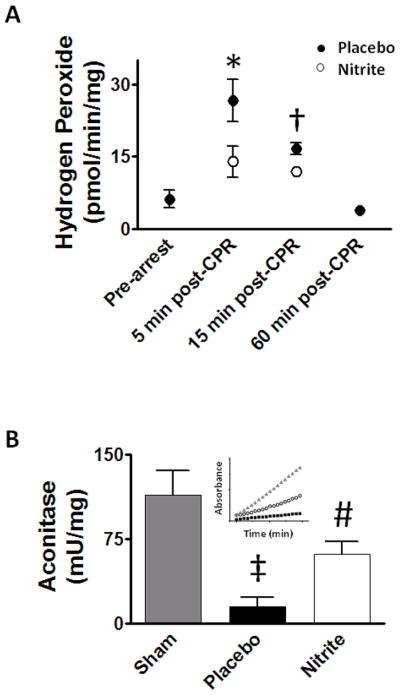
(A) Reactive oxygen species (ROS) generation was measured as mitochondrial peroxide production using the Amplex Red assay. Cardiac arrest increased ROS at 5 and 15 minutes which resolved by 60 minutes. Compared to placebo, nitrite reduced ROS production 5 minutes (n=7) after CPR with a similar trend at 15 minutes (n=4). (B) Cardiac arrest resulted in a significant loss of heart aconitase activity which improves with nitrite therapy. Inset: sample trace from each group. Values denoted as means±SEM, analyzed by paired t-test (ROS) or ANOVA (aconitase); *, p<0.01; †, p=0.071; ‡, p<0.01 sham (n=11) vs. placebo(n=8); #, p=0.05 nitrite (n=8) vs. placebo.
Discussion
We examine the effects of nitrite repletion on mitochondrial function, reperfusion ROS generation, organ function and survival in a 12-minute mouse cardiac arrest model. Cardiac arrest results in systemic nitrite depletion and low dose nitrite replacement (therapy with 50 nmol) at CPR initiation repletes these levels to near baseline and increases cardiac S-nitrothiols. Therapeutic nitrite repletion and S-nitrosation in heart is associated with transient, reversible inhibition of complex I reducing mitochondrial reperfusion ROS generation and oxidative injury. Nitrite improved pulmonary gas exchange, cardiac contractility and survival with a suggestion of neuroprotection.
Moderate NO reperfusion therapy is known to be protective29 but NO formation is limited by NO synthase’s dependence on oxygen and reduced substrates30. Nitrite acts as a reservoir for NO during ischemia and nitrite reduction generates NO through NOS-independent pathways 6, 31, 32. We demonstrate that global ischemia depletes nitrite systemically reducing its availability to act as a reperfusion NO source. This profound depletion with brief global ischemia was surprising but not unprecedented13 and explains why our nitrite dose did not achieve the “optimal” plasma levels (11.9 μM) noted after focal ischemia8. Nitrite repletion early in reperfusion provides a NOS-independent source of NO to ischemic tissues.
Mitochondrial complexes I and III are major sources of pathological reperfusion ROS 33 and transient, reversible inhibition of complex I has been proposed as a mechanism to achieve cardioprotection34–36. The protective effects of complex I inhibition have been described for nitrite23, S-nitrosothiol donors28 and amobarbital34 and observed during classical ischemic preconditioning25. Complex I has numerous cysteine residues available for S-nitrosation with resultant inhibition of electron flow37, 38. Nadtochiy and colleagues S-nitrosated complex I in cardiomyocytes and isolated heart using S-nitroso-2-mercaptopropionyl glycine with associated reduction in ROS production and improved cardiac contractility after ischemia-reperfusion28. Similar to our findings, these authors noted reversal of S-nitrosation and complex I inhibition 30 minutes after ischemia. Shiva and colleagues provided the first evidence that nitrite S-nitrosates complex I and reduces ROS production in liver mitochondria after in vitro ischemia-reperfusion23 though inhibition persisted for 5 hours and was bypassed via complex II. Sun and colleagues have shown complex I to be one of several proteins S-nitrosated in cardioprotective ischemic preconditioning 25.
Nitrite therapy is complex I specific based on the lack of effects using succinate. Complex I efficiency is uneffected therefore this isn’t due to complex I damage. Complex I inhibition is reversible based on restored oxygen and NADH consumption and ATP generation by 60 minutes. The increase in complex I oxygen consumption with placebo in the absence of increased NADH oxidation implies pathological oxygen consumption to form ROS rather than ATP which is prevented with nitrite. Based on our ROS and aconitase data, nitrite is an antioxidant. This mechanism complements prior observations of reduced tissue nitrotyrosine staining9, 39, lipid peroxidation9 and superoxide production9 with nitrite therapy.
Cardiac arrest’s poor prognosis is driven primarily by brain and heart injury15. Excepting hypothermia, no beneficial post-resuscitation therapies exist since CPR’s description ~50 years ago. Present post-resuscitation care is largely supportive15. Nitrite’s role as a novel therapeutic would be of great importance in this setting.
Human myocardial dysfunction (stunning) is common cardiac arrest40, 41, ultimately reversible42 and strongly associated with mortality40, 41 The molecular mechanisms of myocardial stunning after cardiac arrest remain unknown but loss of excitation-contraction coupling is believed to result from ROS injury and calcium-mediated proteolysis43. Nitrite, by reducing ROS, may mitigate stunning similar to other antioxidants44. The reduction in myocardial dysfunction likely explains the 50% relative survival advantage we noted. Further work is needed to characterize nitrite’s effects on brain injury but our results are encouraging.
We designed a mouse model of cardiac arrest with prolonged asystole to study the effects of nitrite on heart and brain injury after resuscitation. Our model utilizes hyperkalemia to induce arrest, limiting its clinical relevance and potentially causing artifacts which may be organ protective (eg cardioplegia) or injurious (endothelial damage perhaps causing RV dysfunction). In the context of these limitations, we demonstrate improvements in gas exchange, heart and brain function and survival. We demonstrate that nitrite transiently inhibits complex I resulting in an antioxidant effect. The ease in delivering IV nitrite,, its established human safety14, 31, its reproducible cytoprotective effects in multiple organs and species6 all suggest that nitrite represents a promising post-resuscitation therapy after cardiac arrest.
Acknowledgments
The authors gratefully acknowledge Kenneth Jeffries (NHLBI), Drs. Danhong Zhao (University of South Dakota) and Huashan Wang (University of Chicago) and the NHLBI Laboratory of Animal Medicine and Surgery, Pathology and Microscopy cores for assistance in model development and experimental methodologies.
Funding Sources
This work was funded by and performed in the Division of Intramural Research, National Heart, Lung and Blood Institute, NIH. Dr. Gladwin is supported by the Institute for Transfusion Medicine (ITxM) and the Hemophilia Center of Western Pennsylvania.
Footnotes
Clinical Summary
Cardiac arrest results in significant morbidity and mortality driven mainly by the cardiac and neurological injury resulting from global ischemia and reperfusion injury. Although resuscitation rates can exceed 65% for some rhythms, between 50-75% of these patients will die before hospital discharge and up to a third of survivors will suffer significant brain injury. Only hypothermia has shown clinical benefit as a post-resuscitation therapy in a subset of patients. Clearly additional therapies are needed. The recent finding that nitrite acts as an ischemic reservoir for enzyme independent nitric oxide generation has resulted in numerous animal studies where it has proven beneficial in reducing focal ischemic organ injury. Based on promising results in focal heart and brain ischemia, Dezfulian et al. have adapted a mouse model of cardiac arrest to model the high clinical mortality and myocardial and neurological dysfunction associated with cardiac arrest. Within this model, nitrite therapy given at the start of resuscitation resulted in significant improvements in survival and myocardial and neurological function in survivors. The authors further investigate the potential mechanism for cardioprotection which involves nitrite’s role as a mitochondrial antioxidant early in resuscitation. The significant benefits attributed to nitrite, along with its ease of delivery and known primate and human safety data make this a promising therapy for a condition with few current therapeutic options.
This work was completed within the intramural research program of the Clinical Center and National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD.
Subject Codes: [151] Ischemic biology - basic studies, [91] Oxidant stress, [130] Animal models of human disease, [25] CPR and emergency cardiac care
Disclosures
Dr. Gladwin is co-inventor on NIH/Government patents for the use of nitrite salts for cardiovascular indications and the use of nitrite to detoxify hemoglobin based oxygen carriers.
References
- 1.Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, Patel RP. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. American journal of physiology. 2006;291:H2026–2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- 2.Kumar D, Branch BG, Pattillo CB, Hood J, Thoma S, Simpson S, Illum S, Arora N, Chidlow JH, Langston W, Teng X, Lefer DJ, Patel RP, Kevil CG. Chronic sodium nitrite therapy augments ischemia-induced angiogenesis and arteriogenesis. Proceedings of the National Academy of Sciences. 2008;105:7540–7545. doi: 10.1073/pnas.0711480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 4.Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S, Power GG, Kelm M, Gladwin MT, Schechter AN. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood. 2005;106:734–739. doi: 10.1182/blood-2005-02-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleinbongard P, Dejam A, Lauer T, Jax T, Kerber S, Gharini P, Balzer J, Zotz RB, Scharf RE, Willers R, Schechter AN, Feelisch M, Kelm M. Plasma nitrite concentrations reflect the degree of endothelial dysfunction in humans. Free Radic Biol Med. 2006;40:295–302. doi: 10.1016/j.freeradbiomed.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 6.Dezfulian C, Raat N, Shiva S, Gladwin MT. Role of the anion nitrite in ischemia-reperfusion cytoprotection and therapeutics. Cardiovascular Research. 2007;75:327–338. doi: 10.1016/j.cardiores.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB, Hogg N. The reaction between nitrite and deoxyhemoglobin. Reassessment of reaction kinetics and stoichiometry. J Biol Chem. 2005;280:31126–31131. doi: 10.1074/jbc.M501496200. [DOI] [PubMed] [Google Scholar]
- 8.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung KH, Chu K, Ko SY, Lee ST, Sinn DI, Park DK, Kim JM, Song EC, Kim M, Roh JK. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke. 2006;37:2744–2750. doi: 10.1161/01.STR.0000245116.40163.1c. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez FM, Shiva S, Vincent PS, Ringwood LA, Hsu LY, Hon YY, Aletras AH, Cannon RO, 3rd, Gladwin MT, Arai AE. Nitrite anion provides potent cytoprotective and antiapoptotic effects as adjunctive therapy to reperfusion for acute myocardial infarction. Circulation. 2008;117:2986–2994. doi: 10.1161/CIRCULATIONAHA.107.748814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pluta RM, Dejam A, Grimes G, Gladwin MT, Oldfield EH. Nitrite infusions to prevent delayed cerebral vasospasm in a primate model of subarachnoid hemorrhage. Jama. 2005;293:1477–1484. doi: 10.1001/jama.293.12.1477. [DOI] [PubMed] [Google Scholar]
- 12.Shiva S, Wang X, Ringwood LA, Xu X, Yuditskaya S, Annavajjhala V, Miyajima H, Hogg N, Harris ZL, Gladwin MT. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2:486–493. doi: 10.1038/nchembio813. [DOI] [PubMed] [Google Scholar]
- 13.Bryan NS, Calvert JW, Elrod JW, Gundewar S, Ji SY, Lefer DJ. Dietary nitrite supplementation protects against myocardial ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2007;104:19144–19149. doi: 10.1073/pnas.0706579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dejam A, Hunter CJ, Tremonti C, Pluta RM, Hon YY, Grimes G, Partovi K, Pelletier MM, Oldfield EH, Cannon RO, 3rd, Schechter AN, Gladwin MT. Nitrite Infusion in Humans and Nonhuman Primates. Endocrine Effects, Pharmacokinetics, and Tolerance Formation. Circulation. 2007;116:1821–1831. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- 15.Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Bottiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, Kern KB, Laurent I, Longstreth WT, Jr, Merchant RM, Morley P, Morrison LJ, Nadkarni V, Peberdy MA, Rivers EP, Rodriguez-Nunez A, Sellke FW, Spaulding C, Sunde K, Vanden Hoek T. Post-Cardiac Arrest Syndrome. Epidemiology, Pathophysiology, Treatment, and Prognostication A Consensus Statement From the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation. 2008 doi: 10.1016/j.resuscitation.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 16.Nichol G, Thomas E, Callaway CW, Hedges J, Powell JL, Aufderheide TP, Rea T, Lowe R, Brown T, Dreyer J, Davis D, Idris A, Stiell I. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300:1423–1431. doi: 10.1001/jama.300.12.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kofler J, Hattori K, Sawada M, DeVries AC, Martin LJ, Hurn PD, Traystman RJ. Histopathological and behavioral characterization of a novel model of cardiac arrest and cardiopulmonary resuscitation in mice. J Neurosci Methods. 2004;136:33–44. doi: 10.1016/j.jneumeth.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 18.Abella BS, Zhao D, Alvarado J, Hamann K, Vanden Hoek TL, Becker LB. Intra-arrest cooling improves outcomes in a murine cardiac arrest model. Circulation. 2004;109:2786–2791. doi: 10.1161/01.CIR.0000131940.19833.85. [DOI] [PubMed] [Google Scholar]
- 19.Schmitt-Ulms G, Hansen K, Liu J, Cowdrey C, Yang J, DeArmond SJ, Cohen FE, Prusiner SB, Baldwin MA. Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat Biotechnol. 2004;22:724–731. doi: 10.1038/nbt969. [DOI] [PubMed] [Google Scholar]
- 20.MacArthur PH, Shiva S, Gladwin MT. Measurement of circulating nitrite and S-nitrosothiols by reductive chemiluminescence. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;851:93–105. doi: 10.1016/j.jchromb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Pelletier MM, Kleinbongard P, Ringwood L, Hito R, Hunter CJ, Schechter AN, Gladwin MT, Dejam A. The measurement of blood and plasma nitrite by chemiluminescence: pitfalls and solutions. Free Radic Biol Med. 2006;41:541–548. doi: 10.1016/j.freeradbiomed.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci U S A. 2001;98:7212–7217. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ, Gladwin MT. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiravanti E, Samouilov A, Zweier JL. Nitrosyl-heme complexes are formed in the ischemic heart: evidence of nitrite-derived nitric oxide formation, storage, and signaling in post-ischemic tissues. J Biol Chem. 2004;279:11065–11073. doi: 10.1074/jbc.M311908200. [DOI] [PubMed] [Google Scholar]
- 25.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 26.Gordon C, Yang Y. Thermoregulatory Response to Chemical Toxicants and Other Insults. Annals of the New York Academy of Sciences. 1997;813:835–848. doi: 10.1111/j.1749-6632.1997.tb51789.x. [DOI] [PubMed] [Google Scholar]
- 27.Teschendorf P, Padosch SA, Spöhr F, Albertsmeier M, Schneider A, Vogel P, Choi Y-H, Böttiger BW, Popp E. Time course of caspase activation in selectively vulnerable brain areas following global cerebral ischemia due to cardiac arrest in rats. Neuroscience Letters. 2008;448:194–199. doi: 10.1016/j.neulet.2008.10.030. [DOI] [PubMed] [Google Scholar]
- 28.Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: Effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. Journal of Molecular and Cellular Cardiology. 2007;42:812–825. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. Journal of Molecular and Cellular Cardiology. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Boucher JL, Moali C, Tenu JP. Nitric oxide biosynthesis, nitric oxide synthase inhibitors and arginase competition for L-arginine utilization. Cell Mol Life Sci. 1999;55:1015–1028. doi: 10.1007/s000180050352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, 3rd, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 32.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Bioscience reports. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 34.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of Electron Transport before Cardiac Ischemia with the Reversible Inhibitor Amobarbital Protects Rat Heart Mitochondria. J Pharmacol Exp Ther. 2006;316:200–207. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 35.Anderson TC, Li CQ, Shao ZH, Hoang T, Chan KC, Hamann KJ, Becker LB, Vanden Hoek TL. Transient and partial mitochondrial inhibition for the treatment of postresuscitation injury: Getting it just right. Crit Care Med. 2006;34:S474–S482. doi: 10.1097/01.CCM.0000246014.19486.A1. [DOI] [PubMed] [Google Scholar]
- 36.Chen Q, Camara AKS, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol. 2007;292:C137–147. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- 37.Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biol Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- 38.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tripatara P, Patel NSA, Webb A, Rathod K, Lecomte FMJ, Mazzon E, Cuzzocrea S, Yaqoob MM, Ahluwalia A, Thiemermann C. Nitrite-Derived Nitric Oxide Protects the Rat Kidney against Ischemia/Reperfusion Injury In Vivo: Role for Xanthine Oxidoreductase. J Am Soc Nephrol. 2007;18:570–580. doi: 10.1681/ASN.2006050450. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez MM, Berg RA, Nadkarni VM, Vianna CB, Kern KB, Timerman S, Ramires JA. Left Ventricular Systolic Function and Outcome After In-Hospital Cardiac Arrest. Circulation. 2008;117:1864–1872. doi: 10.1161/CIRCULATIONAHA.107.740167. [DOI] [PubMed] [Google Scholar]
- 41.Chang WT, Ma MH, Chien KL, Huang CH, Tsai MS, Shih FY, Yuan A, Tsai KC, Lin FY, Lee YT, Chen WJ. Postresuscitation myocardial dysfunction: correlated factors and prognostic implications. Intensive Care Med. 2007;33:88–95. doi: 10.1007/s00134-006-0442-9. [DOI] [PubMed] [Google Scholar]
- 42.Ruiz-Bailen M, Aguayo de Hoyos E, Ruiz-Navarro S, Diaz-Castellanos MA, Rucabado-Aguilar L, Gomez-Jimenez FJ, Martinez-Escobar S, Moreno RM, Fierro-Roson J. Reversible myocardial dysfunction after cardiopulmonary resuscitation. Resuscitation. 2005;66:175–181. doi: 10.1016/j.resuscitation.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiological reviews. 1999;79:609–634. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 44.Sun JZ, Kaur H, Halliwell B, Li XY, Bolli R. Use of aromatic hydroxylation of phenylalanine to measure production of hydroxyl radicals after myocardial ischemia in vivo. Direct evidence for a pathogenetic role of the hydroxyl radical in myocardial stunning. Circ Res. 1993;73:534–549. doi: 10.1161/01.res.73.3.534. [DOI] [PubMed] [Google Scholar]