

Abstract
Intravital multiphoton microscopy provides a unique opportunity to discover and characterize biological phenomena in the natural context of living organisms. Here we provide an overview of multiphoton microscopy with particular attention to its application for studying host-pathogen interactions.
Humans, like all jawed vertebrates, are confronted with hordes of pathogens seeking a host in which to procreate. Thanks to evolution, we are endowed with a highly sophisticated multilayered response that utilizes numerous strategies for preventing, limiting, and eradicating infections. The initial response is mounted by the innate immune system, which plays a crucial holding action for up to a week to limit pathogen replication to manageable levels. This involves soluble (e.g., interferons) and cellular (e.g., natural killer [NK] cells) elements of the innate immune system. The adaptive immune system initiates lymphocyte responses to generate effector T cells and antibodies within a few hours of infection. When successful, the immune system eliminates the threat and the host survives to pass on its own genes to future generations.
Although there has been tremendous progress in understanding immunity to pathogens, gained in large part through ex vivo approaches, many questions remain. When and where is infection established at the cellular level? How does infection disseminate through the organ/organism? How do infected cells signal to the immune system, and how quickly after infection do cells of the innate immune system respond? How do innate responses to infection shape adaptive responses? How and where do immune effector cells encounter pathogens and/or pathogen-infected cells? What happens next? How do the answers differ between pathogens? A long list to be sure, but longer still as each answer raises its own set of questions.
The most direct approach to understanding the complex cellular events occurring at the organismal level after infection is to simply look at an infection as it progresses. Previously, direct visualization of infection has been essentially limited to static immunofluorescence confocal microscopy imaging of sectioned tissues. Lately, the advent of new technologies such as two-photon (2P) microscopy and whole-body imaging have provided new perspectives on both pathogen behavior and host responses within the live host.
In this Review, we provide a broad overview of 2P microscopy as it relates to imaging infectious organisms, focusing on the benefits, caveats, and pitfalls of this methodology.
Microscopy: One Photon or Two?
From analysis of virion release from cells on a coverslip to imaging bacterial invasion of an entire organism, light microscopy has rapidly advanced our understanding of host-pathogen interactions. Until recently, fluorescence microscopy relied on single-photon excitation–i.e., a photon of a given wavelength excites a fluorophore, resulting in emission of a longer wavelength photon that is then registered by a detector, be it the human retina, film, or photomultiplier array (Figure 1A). In wide field epifluorescence microscopy, the entire microscope field is bathed in fluorescent light, and fluorescent molecules in the optical path are equally excited and detected regardless of their relationship to the focal plane. The distinguishing feature of confocal microscopy is the addition of a confocal pinhole that greatly reduces out-of-focus fluorescence. The result is greatly enhanced image quality, and the ability to computationally generate 3D images by collecting images as the focal plane is precisely moved in the z direction by raising or lowering the microscope objective.
Figure 1. One (A) and Two (B) -Photon Excitation of a Fluorophore.
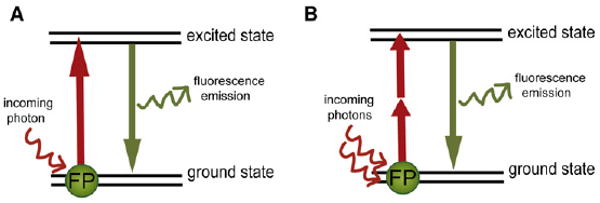
(A) During single-photon excitation, a fluorophore or fluorescent protein (FP) is excited by an incoming photon to a higher vibrational energy level (the excited state, depicted by the top black lines). In less than a picosecond, the fluorophore undergoes vibrational relaxation to the lowest-energy excited state and finally emits fluorescence as it is returning to its unexcited ground state.
(B) When photon densities are high, two photons can combine energy to cause FP transition to the excited state. Vibrational relaxation and fluorescence emission proceeds as if a single photon had excited the FP. Due to the use of mode-locked lasers, the two photons need to excite the FP typically have identical energies equal to half the energy need for 1P excitation, although any two wavelengths summing to the appropriate energy will result in excitation.
Laser scanning confocal microscopy (LSCM) has provided a solid foundation for our understanding of cellular events occurring after infection. Sections can be cut from infected tissues and analyzed for the presence of pathogens as well as immune cell subsets (even endogenous antigen-specific lymphocytes [Khanna et al., 2007]). LSCM has numerous advantages over other ex vivo techniques: (1) instruments, while expensive, are widely available, typically at core facilities that provide expertise and services at affordable rates; (2) it provides an actual image of cell interactions occurring postinfection; (3) a veritable rainbow of colors can be used for imaging, allowing multiple antibodies and stains to be used, including those requiring cell permeabilization, and (4) imaging can be performed at the convenience of the investigator since sections can typically be stored indefinitely. The adaptability to a wide variety of experimental situations has made confocal microscopy the method of choice for many different studies of host-pathogen interplay.
Along with its numerous advantages, however, come some drawbacks: (1) LSCM provides only a static image, making it difficult (at best) and frequently impossible to identify transient events and impossible to observe the movement of immune cells; (2) tissue sectioning can have profound effects on tissue morphology and antigenicity; (3) generating overlapping sections to cover large areas of tissues is technically challenging and labor intensive, as is generating serial sections to overcome the limited working distance of the focal plane (about 100 μM can be imaged in the z plane).
Enter two-photon fluorescence laser scanning microscopy (2PLSM) (Denk et al., 1990), which uses a high-power laser to deliver short (∼100 femtosecond), rapid (∼80 MHz), and intense (∼109 Watts/mm2) pulses of lower-energy (near infrared) photons to a tissue of interest. When two photons hit a fluorophore within an attosecond (10−18) of each other, they combine their energies to excite the fluorophore to a state achieved by a single photon with approximately twice the energy (Figure 1B) (Zipfel et al., 2003). High power is needed, since 2P excitation requires a million-fold higher photon flux than standard 1P excitation to activate the same number of fluors (3P excitation is possible since only a 10-fold higher photon flux is required).
2P excitation mode confers significant advantages over standard LSCM. Longer wavelengths scatter less and are not absorbed as readily by endogenous chromophores, resulting in deeper tissue penetration (up to 1 mm in some studies) and less phototoxicity than standard 1P excitation. Since simultaneous absorption of two photons is highly dependent on photon flux, and essentially only occurs at the focal plane of the objective, confocality is achieved without using a pinhole, increasing detection sensitivity and limiting photoxicity to the focal plane itself (Figure 2).
Figure 2. Two-Photon Fluorophore Excitation Is Limited to the Focal Plane.
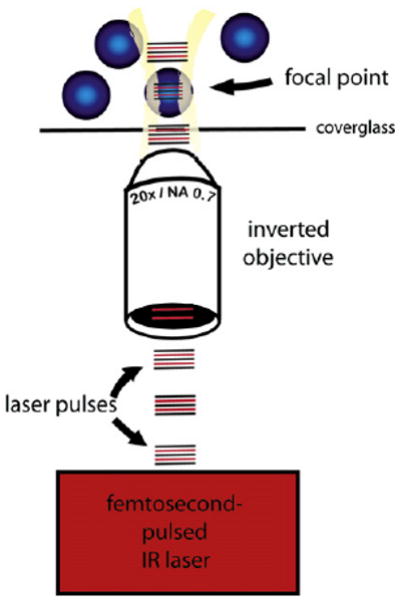
During multiphoton excitation, short, high-intensity pulses are delivered to the tissue or cells being examined. Because two photons must be absorbed within an attosecond to accomplish fluorophore excitation, this is only accomplished within the focal plane. Cells outside of the focal plane are not excited, and a traditional confocal pinhole is eliminated during this mode of microscopy.
These factors make 2P microscopy the choice for imaging living tissues. When combined with a new arsenal of fluorescent pathogens, this technique enables imaging of infections in real time in living animals. Leeuwenhoek, who first reported the microscopic world of bacteria and protists in 1676, would surely be impressed.
Minimizing Buyer's Remorse: What to Purchase?
There are myriad choices for 2P microscopes, ranging from turnkey off-the-shelf models to home-built custom instruments, with major microscope and laser manufacturers eager to contribute to (and profit from) 2P evolution. Understanding the underlying principles of MP and their application to living specimens will help you to select the microscope system and lasers best suited to your studies.
Let There Be Light
As discussed above, 2PLSM relies on enormous photon fluxes, typically generated by Ti:sapphire lasers emitting ultrashort pulses of light (this is termed “mode locking”). Photon flux, which governs effective excitation (and imaging) depth, is expressed in terms of laser power. Laser power should be considered over the entire working range (in terms of wavelength) of the laser, as power typically peaks at 800 nm and declines precipitously with increasing and decreasing wavelengths. Although usable power in vital imaging is ultimately limited by phototoxicity, high power is needed for imaging at longer wavelengths (typically used to excite red fluorescent proteins [FPs]) and in applications such as photobleaching or uncaging, which demand intense excitation.
Most Ti:sapphire lasers are tunable between ∼700 to 1000 nm but have the highest power output at 800 nm. While many fluorescent dyes can be excited at ∼800 nm, some red FPs require excitation at longer wavelengths of >1000 nm. On the horizon are ytterbium-doped lasers that should provide greater power at long wavelengths.
Generating a sufficiently strong signal for deep-tissue imaging is a significant hurdle in 2PLSM. 2P lasers can deliver pulses ranging from picoseconds to femtoseconds, with shorter pulses carrying higher peak intensities. Most 2P microscopists therefore use femtosecond-pulsed lasers (which is currently patented by Zeiss). As photons move through a tissue, they are dispersed inversely proportional to pulse length (a phenomenon called group velocity dispersion [GVD] or chirp), mitigating the advantages of femtosecond versus picosecond excitation. This can be corrected by automatic compensation (termed “pre-chirp” or “negative chirp”), a feature now routinely offered by 2P laser manufacturers.
Optimal laser power varies with depth of imaging, as does detector gain. Appropriate settings for deep tissues (e.g., high gain and laser power) result in detector saturation at shallower depths. To image throughout thick sections, some systems include software that modulates laser power and detector sensitivity with the depth of imaging, automatically correcting for the depth of the focal plane. This useful feature should be considered when selecting a system.
Finally, although Ti:Sapphire lasers are tunable, they do not rapidly switch wavelengths during acquisition, limiting each imaging series to a single wavelength. To increase the number of colors that can be acquired simultaneously, the latest 2P microscopes can be equipped with two lasers, optimized for output at low versus high wavelengths. No doubt a good thing, but there are the inevitable costs both monetary (lasers are expensive, as are service contracts) and technical: (1) systems are more difficult to install and operate, (2) two lasers operating simultaneously cause twice the phototoxicity, (3) two lasers operating sequentially slow the acquisition rate two-fold, and (4) data file sizes increase with additional colors.
Upright or Inverted?
Next decision: Will the laser light be delivered through an upright or inverted microscope (also a vexing decision for regular confocal microscopes). Each design has its advantages, as each is optimally suited for particular organs/tissues–the key word being optimal, since each can be adapted to image just about anything. For instance, mouse inguinal lymph nodes (LNs) are optimally imaged by an inverted microscope, while an upright microscope is better suited for popliteal LNs. Two key parameters are determined by microscope orientation: the lenses that can be used and the physical structures that can be imaged (e.g., mouse knees only bend one way). Dipping lenses (i.e., lenses designed to image while immersed in aqueous solutions) are difficult to employ with inverted microscopes, which is a significant disadvantage as dipping lenses typically feature the longest working distances (i.e., the longest distance from the objective that can be focused). While inverted scopes require less elaborate means of tissue immobilization (Sumen et al., 2004) as the mouse is simply resting on glass placed on the objective, some organs are more easily imaged when the objective can just be placed on top of the organ. An objective inverter is commercially available (LSM TECH, Etters, PA) to convert an inverted microscope into an upright objective orientation, but at cost of light excitation and detection intensity.
Store Bought or Homemade?
Of course, building a 2P microscope system requires far more knowledge and technical expertise than purchasing a turnkey system. The do-it-yourself advantages include (1) cost (homemade systems are usually far cheaper than turnkey systems); (2) optimized light path for vital imaging (turnkey systems are typically built to also perform standard LSCM, which requires compromises in the light path); and (3) hard-earned knowledge (by the time you are done, you will undoubtedly learn a great deal about customizing your microscope to your particular application).
Avoiding Overbytes
Data management is not to be taken lightly when embarking upon your 2PLSM adventure. A byproduct of the most exciting feature of 2PLSM–the generation of both 3D and 4D (the fourth dimension being time) data sets–is enormous file sizes. A typical data file for a 3D reconstruction of a 30 min imaging sequence is 500 MB. Given that 10–20 sequences will typically be generated in a single day, 100 gB of data can easily accumulate within weeks by a keen researcher. It is wise to devise a plan for data storage (and backup!) at the outset of MP experiments.
All those bytes must be processed and analyzed. The most widely used software platforms for 2PLSM analysis are Imaris (Bitplane AG, Zurich, Switzerland) and Volocity (Improvision, Coventry, England). Each program has many useful features including (1) manipulation of large data sets, (2) filtering of data to remove background noise (often a reality due to low signal levels at increasing depths), (3) 3D rendering and movie generation, and (4) object tracking over time. Both Imaris and Volocity support user programming allowing the researcher to tailor the program to their needs, e.g., analysis of the time of contact between two cell types. These features allow statistical analysis of the behavior of cells under different experimental conditions (such as in the presence of pathogen-derived antigen or not). While these software platforms are superb, they are expensive and require high-performance computers with high-end graphics cards to work efficiently. Additionally, to generate appropriately annotated movies for presentation and publication (publication tip: journal reviewers love legends within movies!), video editing software is generally necessary (of course, the movie files themselves should not be manipulated!).
Location, Location, Location
OK, you're up to speed with 2PLSM hardware and data analysis software. Now for the rasion d'être, imaging host-pathogen interactions. This will require all of your expertise in your chosen experimental mouse infection system. The first decision is what organ to image. Many mouse tissues and sites have been imaged to date (Sumen et al., 2004). Pathogen-dependent organ tropism in combination with input dose and route of infection will determine which tissues are infected and the relevant immune organs that can be imaged (Figure 3). While you should (obviously) aim for the most-physiological route of infection and input infectious dose, this is not always feasible, particularly at the start. For example, small numbers of microorganisms located in a large organ (such as a liver) will frustrate imaging efforts since few, if any organisms will be located with the typical 300 μM working depth of the microscope.
Figure 3. Routes of Infection Determine Imaging Location.

Vaccinia virus expressing nuclear-localized green fluorescent protein was delivered to mice through intradermal, subcutaneous, or intraperitoneal injection, and the relevant areas were imaged. Vaccinia virus-infected cells are shown in green. The signal from second harmonic generation is shown in blue. Subcutanous injection in the flank results in virally infected cells in the inguinal lymph node or fat pad, while intradermal injection produces skin infection. Intraperitoneal injection results in infection of peritoneal muscle and ovaries.
Choosing a physiological infectious dose is frequently hampered by ignorance of the details of natural pathogen transmission–e.g., the number of infectious virions transmitted by a mosquito bite (not necessarily a small number! [Styer et al., 2007]). In common with many experimental systems, initial 2PLSM analysis of host-pathogen interactions typically uses relatively high numbers of pathogens delivered by hypodermic syringe either intravenously, in the skin, or directly to a relevant organ. With experience, the system can be made increasingly more physiological (and relevant) by lowering the dose and modifying the route of infection. As an example, Jin et al. (2007) used traditional fluorescence microscopy to image the delivery of malaria parasites to a mouse ear by the bite of an infected mosquito.
Pathogen Painting
One of the most critical and challenging aspects of 2PLSM imaging is optimizing fluorescent molecules for imaging. Bacteria and parasites can usually be genetically engineered to constitutively express sufficient copies of a fluorescent protein (FP) to enable direct visualization of the pathogen in tissues. This allows direct observation of the input pathogen itself and also its progeny.
Viruses are typically more challenging to label. A few large viruses can potentially be engineered to express a fluorescent structural protein that is incorporated in virions in sufficient copy number to enable direct visualization of virions. Most viruses, however, probably cannot accommodate such a drastic structural change. While viruses can be directly labeled using fluorescent membrane (for enveloped viruses) or protein dyes, labeling can drastically reduce infectivity and, in any event, will enable visualization only of input virus and will provide limited (if any) information regarding the expression of viral gene products in host cells. No matter the virion labeling strategy, nonfilamentous viruses are smaller than the resolution of 2PLS microscopes and will be visualized as geometric points.
Virus-based 2PLSM studies will typically utilize viruses that encode FPs synthesized by infected cells but excluded from virions. In general, as strong a promoter as possible should be chosen to express the protein, with the caveat that foreign gene expression should not interfere with the infectious cycle. Greater fluorescent reporter expression will maximize detection of cells that are “weakly” infected, facilitate detection of infected cell extensions (e.g., detection of cytoplasmic or membrane bound FPs in dendritic cell processes), and improve image quality and depth of detection.
Considerable thought should be given to the form of antigen expressed. If pathogen interaction with T cells is a goal of the study, the FP can be fused with sequences that encode one or more determinants recognized by MHC class I- or class II-restricted TCR-transgenic mice. While most fluorophores used in 2PSLM result in cytoplasmic staining of the cell of interest, targeting of the FP to various cellular organelles has advantages/disadvantages. Nuclear targeting concentrates the FP in a relatively small volume, increasing the sensitivity of detection and providing clear demonstration that cells are truly infected and have not endocytosed FP from other infected cells. Membrane targeting enables clear delineation of infected cell borders and facilitates observations of cell-surface interactions between infected cells and responding immune cells, but can be weak in comparison to nuclear/cytoplasmic staining. Mitochondrial targeting is of great use if detecting virus-induced apoptosis is a central goal of the study.
Similarly, in viral systems with temporally controlled gene expression, it may be possible to kinetically control virus-driven FP expression (by placement after a regulated promoter). Generally, expression throughout the infectious cycle will probably be easiest for the identification of infected cells. As confidence in the system grows, it is possible to refine the experiments by temporally limiting FP expression to early or late parts of the infectious cycle.
Some viruses cannot be engineered to express FPs while maintaining full infectivity, because their genomes cannot accept additional genetic information without replacing essential genes (e.g., influenza A virus). For pathogens that fall into this dreaded category, an alternative (as-yet-unreported) strategy is to rely on virus-induced transactivation to induce FP expression in host cells. For instance, a very early event in most viral infections is the production of type I interferons. Transgenic mice that express a FP under control of an interferon-responsive promoter should enable detection of local infection, with the obvious caveat that neighboring cells exposed to interferons will also express the FP. Promoter leakiness could also wreak havoc on such a system. The discovery of more specific promoters should facilitate the identification of infected cells using this strategy.
Once you have arrived upon a suitable method of labeling your pathogen, you must decide which FP to use. The standard FP in 2PLSM (and all) imaging is eGFP. eGFP is a safe choice for initial studies, as it is brighter and more photostable than many alternative FPs and has been used by many researchers who can offer advice. A close relative of eGFP, Venus-eYFP, is a bright alternative in the green/yellow range (Giepmans et al., 2006). Cyan FP (eCFP) has been used for imaging, but has a lower quantum yield (i.e., is less bright) than green/yellow FPs. Red FPs (such as dsRed, tdTomato, and mCherry) have also been utilized, but are more difficult to excite using 2P, have lower quantum yields, and may photobleach rapidly. Although a far red FP now exists (mPlum), it has been difficult to excite using standard 2P lasers. The principal reason for turning to eGFP alternatives for pathogen tagging is the growing number of eGFP/eYFP-expressing transgenic mice that can be used to image host responses to infection in combination with blue or red pathogens.
FP-expressing pathogens used in intravital imaging studies reported to date are listed in Table 1.
Table 1. Pathogens Studied in flagrante delicto.
| Pathogen | Fluorophore | Comments | Reference |
|---|---|---|---|
| Viruses | |||
| Vaccinia | eGFP | Static image of infection using 2PLSM | (Norbury et al., 2002) |
| Vaccinia | eGFP | 2PLSM movies of lymph node infection | (Hickman et al., 2008) |
| Vesicular stomatits | eGFP | 2PLSM movies of lymph node infection | (Hickman et al., 2008) |
| UV-VSV | AlexaFluor-568/488 | 2PLSM movies of inactivated virions in the lymph node | (Junt et al., 2007) |
| Prokaryotes | |||
| Salmonella typhimurium | DsRed | 2PLSM movies of mucosal DC response | (Chieppa et al., 2006) |
| Mycobacterium bovis | eGFP or DsRed | 2PLSM movies of liver infection | (Egen et al., 2008) |
| Escherichia coli | GFP | 2PLSM movies of kidney tubule infection | (Mansson et al., 2007) |
| Borrelia burgdorferi | GFP | 2PLSM movies of spirochetes in skin microvasculature | (Moriarty et al., 2008) |
| Eukaryotes | |||
| Plasmodium berghei | RedStar | Intravital movies of parasites in the liver | (Frevert et al., 2005) |
| Plasmodium yoelii | GFP (GFPmut3) | Intravital movies of parasite late liver stages | (Tarun et al., 2006) |
| Plasmodium yoelii | GFP | Intravital movies of parasite release from the liver | (Baer et al., 2007) |
| Leishmania major | RFP | 2PLSM movies of parasites in the skin | (Peters et al., 2008) |
| Toxoplasma gondii | tdTomato | 2PLSM movies of parasites in the lymph node | (Chtanova et al., 2008) |
Visualizing Host Cells
Now that you can “see” your pathogen, what about the host response? The easiest and often the most physiological way to image the host cellular immune response is to use transgenic mice expressing FPs labeling specific cell populations. The most commonly used mice for immunological 2PLSM studies (and some that will undoubtedly soon be used) are listed in Table 2. Most of the mice express eGFP, necessitating the use of an alternate FP for pathogen expression (note, however, that while suboptimal, it is possible to use a pathogen also expressing eGFP). In some cases, weak transgene expression will significantly limit imaging.
Table 2. FP-Expressing Transgenic Mice.
| CD11c-DTR-eGFP | DC | Weak cell surface eGFP | (Jung et al., 2002) |
| CD11c-eYFP | DC | Venus eYFP | (Lindquist et al., 2004) |
| CCL17-GFP | Lymph node DC | GFP | (Alferink et al., 2003) |
| Yet40 | DC, macrophages | eYFP | (Reinhardt et al., 2006) |
| MHC II-eGFP | DC, B cells (intermediate), macrophages (dim) | eGFP | (Boes et al., 2002) |
| CD11b-DTR-eGFP | Macrophages | Weak cell surface eGFP | (Cailhier et al., 2005) |
| MaFIA | Macrophages, GR1+ lung cells, DC (weaker) | eGFP | (Burnett et al., 2004) |
| CX3CR1-GFP | Monocytes, some NK cells, microglia, DC (intermediate) | Variable eGFP expression | (Jung et al., 2000) |
| 7.2fms-eGFP | Mononuclear phagocytes | eGFP | (Sasmono et al., 2003) |
| Lysozyme-eGFP | Neutrophils, monocytes, macrophages | eGFP | (Faust et al., 2000) |
| T-GFP | Naive T cells | eGFP | (Manjunath et al., 1999) |
| T-Red | T cells, plasmacytoid DC | dsRedII | (Mempel et al., 2006) |
| DPE-GFP | T cells | eGFP | (Mempel et al., 2006) |
| 4get | IL-4 producing cells, CD4+ T cells | eGFP | (Mohrs et al., 2001) |
| Cxcr6 | Activated/memory T cells | eGFP | (Unutmaz et al., 2000) |
| FoxP3 | Regulatory T cells | eGFP | (Haribhai et al., 2007) |
| CD2-eGFP | CD8+ T cells, CD4+ T cells and NK cells weak | Variable eGFP expression | (Mempel et al., 2006) |
| meGFP/mb-1(inv) | B cells | eGFP | (Pelanda et al., 2002) |
| Yeti | NK and NKT cells; some activated T cells | eYFP | (Stetson et al., 2003) |
| Ncr1-GFP | NK cells (the Ncr1+ subset) | GFP | (Gazit et al., 2006) |
| Ubi-GFP | Ubiquitous, can be used to identify stromal cells | GFP | (Schaefer et al., 2001) |
| Beta-actin eGFP | Ubiquitous, can be used to identify stromal cells | eGFP | (Okabe et al., 1997) |
| FoxP3 | Regulatory T cells | eGFP | (Haribhai et al., 2007) |
A second arrow in the 2P immunologist's quiver is adoptive transfer of labeled cell populations. Immune cells are removed from one mouse, labeled with a cell-permeant fluorescent dye (discussed below), and then transferred into a recipient mouse. Both B and T cells can be visualized in this way, and it can be quite useful to transfer antigen-specific cells from T cell-receptor or B cell-receptor transgenic mice. Likewise, dendritic cells (either cultivated in vitro from bone marrow precursors or purified from spleens) can be matured, labeled, and transferred into a mouse where they will migrate to the regional draining lymph node and can be imaged.
Adoptive transfer offers a number of advantages. First, any cell population that can be purified can be studied. Second, cells can be manipulated ex vivo prior to transfer (treated with various agonists, pulsed with antigenic peptides, etc.), facilitating detailed functional analysis. Third, for cell types whose natural low abundance in a given tissue limits imaging, it can be possible to increase cell numbers to facilitate imaging.
Inevitably, adoptive transfer has its limitations. Certain cell types may be difficult or impossible to purify from donor mice due to low numbers or difficulty to isolate in a functional state (Bonasio and von Andrian, 2006). Manipulation-induced alteration of transferred cell function is common (and can sometimes be visualized as macrophages–experts at engulfing apoptotic cells–become fluorescent as they phagocytose dye-laden apoptotic fragments of your precious labeled cell populations). When establishing cell isolation procedures, it is prudent to characterize isolated cells by flow cytometry and, when possible, functional assays, to assess whether they retain their normal phenotype and function. Maintaining a useful functional state can be difficult, as some cells (e.g., naive dendritic cells or effector memory T cells) fail to localize to the desired imaging area, or even their source organ, after transfer (Bonasio and von Andrian, 2006). For adoptive transfer of antigen-specific B and T cells from receptor-transgenic mice, a mainstay of vital imaging studies, the increased precursor frequency of antigen-specific cells relative to naive mice can affect the kinetics and character of responses (Badovinac et al., 2007). Finally, the necessity to transfer cells in an arbitrary order, such as antigen-loaded DC prior to naive T cells, may influence the area in which cells may interact.
When the cell-purification and dye-labeling stars align, beautiful images can result, due to intense labeling that optimizes 2PLSM imaging. Cells are labeled by simply incubating with dyes for 10–45 min, taking care to first determine the maximal labeling level that can be achieved without compromising cell function. This must be determined for each dye-cell type combination, and typically must be determined for each new batch of dye due to lot to lot variation (a particular problem with CFSE). The most common dyes are CMF2HC (blue Cell Tracker [Invitrogen, Carlsbad, CA]), CFSE (standard green dye that has been used extensively for cell proliferation experiments [available from multiple sources]), CMTMR (orange/red dye [multiple sources]), and CMTPX (red Cell Tracker [Invitrogen]). These dyes are all membrane permeant and react with either amino- or sulfhydryl-containing intracellular macromolecules that retain the dyes for long periods. Fluorescent dyes that indicate pH (such as Snarf [Invitrogen]) or calcium flux (such as Fura-2 [Invitrogen]) have also been used for 2PLSM studies, and yield additional information about cellular functionality in vivo.
Imaging multiple labels can be challenging, especially when using a single laser. In this case (the usual scenario), an excitation wavelength should be selected that simultaneously excites both fluors, shading the wavelength to the weaker fluor to equalize detection. For example, we typically image eGFP and CMTPX labels by exciting at 850 nm, halfway between the 800 nm needed for CMTPX and 900 nm needed for eGFP.
Surgery
Imaging of internal organs requires surgical exposure of the tissue. To attain maximal imaging depth, the microscope objective must either directly contact the organ (for a dipping lens), or contact 0.17 mm thickness glass (#1 cover glass) pressed against the organ. One of the most challenging aspects of intravital microscopy is optimizing surgery to expose and stabilize the tissue of interest with the goal of minimizing tissue disruption and maintaining normal flow of blood and lymph. This is particularly relevant to imaging infectious processes, since surgical preparation can modify pathogen dissemination and virulence.
Prior to surgery, mice are anesthetized using either inhaled or injected anesthetics to achieve a suitable plane of anesthesia, which entails complete cessation of voluntary muscle movements while maintaining breathing and minimally affecting cellular function. 2PLSM studies typically utilize isoflurane continuously administered intranasally via a vaporizer. Alternatively, avertin can be intermittently injected intraperitoneally to maintain anesthesia, using a syringe attached via a tube to a 27 gauge needle placed in the abdomen to avoid moving the mouse during imaging. Of course, any surgical procedures performed should first be cleared with your institutional animal care and use committee.
Anesthetized mice cannot maintain their body temperature, which plummets, affecting immune cells and lymph flow (Miller et al., 2002). The optimal solution to this problem is to enclose the microscope in an environmental chamber that regulates temperature. Heating blankets with feedback thermometers are much cheaper, but can only be placed on one side of a mouse that is being imaged, thereby overheating one side and under heating the other.
Once the mouse is anesthetized, it is prepped for surgery. The first step is shaving the surgical site, typically using an electric shaver, followed by fine trimming with a razor blade if necessary. The utmost caution must be applied to remove shaved, highly autofluorescent hairs from the imaging site. The surgical site is cleansed with alcohol or another nonfluorescent sterilizing solution, and surgery begins. As mentioned above, a myriad of tissues and organs can be examined via 2PLSM, and each has its own surgical strategy and requirements. There are, however, some common elements. Surgeries are typically performed with the aid of a dissecting microscope, which can be equipped to detect fluorescence. Surgery can be facilitated using microsurgical instruments designed for use on mice (available from multiple companies).
Surgery must be optimized for each organ examined, with the goal being to expose the area for imaging with minimal alteration of blood and lymph supply. Exposed organs should be immersed in sterile saline to prevent tissue desiccation. There are several methods to monitor collateral damage. Blood flow can be monitored by i.v. injection of fluorescent dextran, beads, or serum albumin. Blood vessels in the organ being imaged should appear within seconds after intravenous injection (Movie S1). Disruption of blood flow also results in diminished movement of mobile cells in the affected tissue, e.g., lymphocytes in lymph nodes. Preservation of lymph flow is monitored by intradermal or subcutaneous injection of fluorescent particles, which enter the lymph node subcapsular sinus within seconds and are rapidly phagocytosed by macrophages.
Mice with eGFP knocked in to the lysozyme M locus have green neutrophils and monocytes and are extremely useful for monitoring tissue damage, which triggers neutrophil diapedesis (Faust et al., 2000). Here, we demonstrate the phototoxic effects of the MP laser on the skin using low versus high laser intensities (Movie S2). Tie-2 GFP mice (Motoike et al., 2000) with eGFP-expressing endothelial cells can be used to determine the extent of endothelial disruption during surgery. Performing surgical prep and 2PLSM in these reporter mice, even if not ultimately utilized for the actual experiments, are useful tools for fine-tuning experimental parameters.
Image quality is utterly dependent on minimizing the motion of the imaged tissue. In contrast to static microscopy, frame averaging (the process of acquiring multiple images of the same field and averaging them to remove noise) cannot be performed in 2PLSM due to the constant movement of cells in the living animal. Although fully anesthetized mice exhibit no voluntary motion, their heart and lungs continue to function, dislocating contiguous structures. Distant tissues can exhibit slight involuntary movement difficult to visualize by the naked eye that still degrade imaging and introduce serious errors into cell movement calculations. Movements can be minimized by custom building a stabilizer for each tissue, which can require a great deal of tinkering (and patience!). Additionally, internal organs (such as the spleen and ovaries) generally suffer less from movement artifacts because they are generally stabilized in some way when they are exteriorized for imaging. On the horizon are new, high-speed imaging modalities that may circumvent issues with motion, but timing will still be a key concern.
Imaging and Interpretation
At this point you may be relieved to know that imaging is actually the easiest part of 2P vital microscopy. The precise imaging parameters will depend on the volume examined and the goals of the experiment. The variables include laser power, objective magnification, size and depth of field, collection interval, and number of colors. Keep in mind that virtually every decision entails optimizing opposing factors. For example, collecting more z-sections will increase imaging time, phototoxicity, and file size. To capture rapid events, it is best to perform more frequent imaging of a smaller volume. To capture slow events, such as pathogen migration throughout an organ, longer intervals over a prolonged period are appropriate. In general, start as simple as possible and gradually increase imaging complexity. Over the course of repeat experiments you should aim to refine the protocol and imaging settings to optimize your observations.
Once you have navigated all of the pitfalls and difficulties and generated beautiful 4D movies, you still must decide what it all means. Always keep in mind that what you can see in any image is far less than what you can't see. Every attempt should be made to minimize subjective conclusions by using software-based quantitation and calculations to generate data suitable for statistical analysis. For instance, concluding that a given set of cells contact each other (or migrate, etc.) more than another set, which may appear obvious when viewing the movies, should be based on statistical comparison between the contact times generated by image analysis software. Most 2PLS microscopists use Imaris (Bitplane), Volocity (Improvision), or Metamorph (Molecular Devices) to accomplish preliminary analyses on raw microscopic data, but a secondary statistical program (such as GraphPad's Prism) is often additionally needed to compare results.
Tricks of the Trade
Autofluorescence
All tissues exhibit some level of autofluorescence when excited by light of the appropriate wavelength. While this can cause severe headaches (and depression) when trying to resolve low-level fluorescence signal from background “noise,” it can be exploited to image skin substructure, ovaries, and fat (among others). Eosinophil granules are autoflourescenet and can be excited using 2P (Richards-Kortum and Sevick-Muraca, 1996). Elastic fibers throughout the body exhibit high autofluorescence, as do macrophages within the lymph nodes. Based on autofluorescence, elastin-fibril containing areas of the skin and macrophage-rich regions of the lymph node, respectively, can be readily distinguished. As 2P experience grows, the catalog of useful autofluorescent structures will no doubt expand accordingly.
Second Harmonic Generation
Autofluorescent signals become even more powerful when combined with a secondary mode of natural fluorescence, second harmonic generation (SHG), an imaging modality that can be used to visualize ordered and repetitive proteins such as collagen, tubulin, elastin, and myosin (Zoumi et al., 2002). Regardless of the target, its SHG emission wavelength is uniformly half of the excitation wavelength–e.g., collagen can be excited at 800 nm and detected at 400 nm. SHG can be used to image skin, the collagenous capsule of the lymph node and ovaries, and muscle tissue. In the lymph node, collagen-containing fibers visualized by 2P SHG have been shown to form highways for motile cells (Bajenoff et al., 2006).
Instant ID
Delivery of fluorescent compounds can delineate specific cell populations, anatomical structures, or regions of organs populated by specific cells. Fluorescent dextran delivered subcutaneously drains to the regional lymph node and labels endocytic cells within the node. Dextran given intravenously demarcates blood vessels. Nile Red is a lipophilic dye that can be administered or painted on skin to label adipocytes. Certain lectins, such as wheat germ agglutinin, introduced subcutaneously drain via the lymphatics to the sinuses of the lymph node, outlining those structures.
Fluorescent antibodies can also be extremely useful. Intravenous administration of fluorescent Meca-79, an antibody against peripheral node addressin (pNAD), labels high endothelial venules by binding to pNAD in the vein lumen. Subcutaneous injection of LYVE-1 can likewise label lymph vessels by binding to lymph-specific endothelium. Injection of some antibodies can also be used to label lymphocytes, although this is usually limited to what can be accessed via the blood.
Synchronization
Many cellular behaviors change depending on the time postinfection. Starting with a synchronized population of cells can greatly aid statistical analyses of temporal behavioral changes. One of the easiest strategies is to adoptively transfer cells at an appropriate time postinfection, but this can be complicated during infection as the transferred cells would naturally progress through different functional states as well. For synchronized analyses of T cell behavior in lymph nodes, the CD62L-specific mAb Mel-14 can be given intravenously to block T cell entry into lymph nodes (Mempel et al., 2004). An alternative approach involves the administration of high doses of MHC class II-binding peptide into a mouse that has only class II positive dendritic cells (Celli et al., 2007). To simultaneously expose cells to a compound while imaging, a “caged” form can be given that is activated by 2P illumination to release the biologically active form (Furuta et al., 1999).
No doubt many additional strategies for enhancing vital imaging experiments will be added as 2PLSM imaging enters the mainstream of experimental biology. As a budding 2PLS microscopist, a sense of adventure will serve you well. Some reagents and strategies that shouldn't work will, and some that should won't. Out-of-the-box thinking should be encouraged; if you have a clever idea, give it a try.
Future Prospects
Intravital 2PLSM has come a long way since its initial description in 1990 and will be increasingly important in understanding host-pathogen interactions. New strategies and technologies will be developed to label pathogens that currently cannot be labeled. Novel FPs and probes and in combination with transgenic technology will allow simultaneous identification of multiple cell populations, furthering our understanding of the choreography of the cellular immune response to infection. Increasing depth of laser penetration, enhanced detector sensitivity, in combination with fiber optic-based endomicroscope objectives will allow us to examine regions of tissues that are currently off limits, including tissues in large animals such as primates.
While the future is bright, the present is no less bright: there are plenty of questions ripe for the pickin' with today's standard systems. Fire up the laser, and start harvesting.
Supplementary Material
Footnotes
Supplemental Data: Supplemental Data include two movies and can be found online at http://www.cell.com/cellhostandmicrobe/supplemental/S1931-3128(08)00407-1.
References
- Alferink J, Lieberam I, Reindl W, Behrens A, Weiss S, Huser N, Gerauer K, Ross R, Reske-Kunz AB, Ahmad-Nejad P, et al. Compartmentalized production of CCL17 in vivo: strong inducibility in peripheral dendritic cells contrasts selective absence from the spleen. J Exp Med. 2003;197:585–599. doi: 10.1084/jem.20021859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badovinac VP, Haring JS, Harty JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer K, Klotz C, Kappe SH, Schnieder T, Frevert U. Release of hepatic Plasmodium yoelii merozoites into the pulmonary microvasculature. PLoS Pathog. 2007;3:e171. doi: 10.1371/journal.ppat.0030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boes M, Cerny J, Massol R, Op den Brouw M, Kirchhausen T, Chen J, Ploegh HL. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 2002;418:983–988. doi: 10.1038/nature01004. [DOI] [PubMed] [Google Scholar]
- Bonasio R, von Andrian UH. Generation, migration and function of circulating dendritic cells. Curr Opin Immunol. 2006;18:503–511. doi: 10.1016/j.coi.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, Cohen DA. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol. 2004;75:612–623. doi: 10.1189/jlb.0903442. [DOI] [PubMed] [Google Scholar]
- Cailhier JF, Partolina M, Vuthoori S, Wu S, Ko K, Watson S, Savill J, Hughes J, Lang RA. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J Immunol. 2005;174:2336–2342. doi: 10.4049/jimmunol.174.4.2336. [DOI] [PubMed] [Google Scholar]
- Celli S, Lemaitre F, Bousso P. Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity. 2007;27:625–634. doi: 10.1016/j.immuni.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Chieppa M, Rescigno M, Huang AY, Germain RN. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203:2841–2852. doi: 10.1084/jem.20061884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chtanova T, Schaeffer M, Han SJ, van Dooren GG, Nollmann M, Herzmark P, Chan SW, Satija H, Camfield K, Aaron H, et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity. 2008;29:487–496. doi: 10.1016/j.immuni.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008;28:271–284. doi: 10.1016/j.immuni.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–726. [PubMed] [Google Scholar]
- Frevert U, Engelmann S, Zougbede S, Stange J, Ng B, Matuschewski K, Liebes L, Yee H. Intravital observation of Plasmodium berghei sporozoite infection of the liver. PLoS Biol. 2005;3:e192. doi: 10.1371/journal.pbio.0030192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta T, Wang SS, Dantzker JL, Dore TM, Bybee WJ, Callaway EM, Denk W, Tsien RY. Brominated 7-hydroxycoumarin-4-ylmethyls: photolabile protecting groups with biologically useful cross-sections for two photon photolysis. Proc Natl Acad Sci USA. 1999;96:1193–1200. doi: 10.1073/pnas.96.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. 2006;7:517–523. doi: 10.1038/ni1322. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol. 2007;178:2961–2972. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, Barber GN, Bennink JR, Yewdell JW. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol. 2008;9:155–165. doi: 10.1038/ni1557. [DOI] [PubMed] [Google Scholar]
- Jin Y, Kebaier C, Vanderberg J. Direct microscopic quantification of dynamics of Plasmodium berghei sporozoite transmission from mosquitoes to mice. Infect Immun. 2007;75:5532–5539. doi: 10.1128/IAI.00600-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los SK, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450:110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- Khanna KM, McNamara JT, Lefrancois L. In situ imaging of the endogenous CD8 T cell response to infection. Science. 2007;318:116–120. doi: 10.1126/science.1146291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, Nussenzweig MC. Visualizing dendritic cell networks in vivo. Nat Immunol. 2004;5:1243–1250. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- Manjunath N, Shankar P, Stockton B, Dubey PD, Lieberman J, von Andrian UH. A transgenic mouse model to analyze CD8(+) effector T cell differentiation in vivo. Proc Natl Acad Sci USA. 1999;96:13932–13937. doi: 10.1073/pnas.96.24.13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansson LE, Melican K, Boekel J, Sandoval RM, Hautefort I, Tanner GA, Molitoris BA, Richter-Dahlfors A. Real-time studies of the progression of bacterial infections and immediate tissue responses in live animals. Cell Microbiol. 2007;9:413–424. doi: 10.1111/j.1462-5822.2006.00799.x. [DOI] [PubMed] [Google Scholar]
- Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases 2. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, von Andrian UH. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. 2006;25:129–141. doi: 10.1016/j.immuni.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- Mohrs M, Shinkai K, Mohrs K, Locksley RM. Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity. 2001;15:303–311. doi: 10.1016/s1074-7613(01)00186-8. [DOI] [PubMed] [Google Scholar]
- Moriarty TJ, Norman MU, Colarusso P, Bankhead T, Kubes P, Chaconas G. Real-time high resolution 3D imaging of the lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog. 2008;4:e1000090. doi: 10.1371/journal.ppat.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoike T, Loughna S, Perens E, Roman BL, Liao W, Chau TC, Richardson CD, Kawate T, Kuno J, Weinstein BM, et al. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3:265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Pelanda R, Hobeika E, Kurokawa T, Zhang Y, Kuppig S, Reth M. Cre recombinase-controlled expression of the mb-1 allele. Genesis. 2002;32:154–157. doi: 10.1002/gene.10070. [DOI] [PubMed] [Google Scholar]
- Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321:970–974. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt RL, Hong S, Kang SJ, Wang ZE, Locksley RM. Visualization of IL-12/23p40 in vivo reveals immunostimulatory dendritic cell migrants that promote Th1 differentiation. J Immunol. 2006;177:1618–1627. doi: 10.4049/jimmunol.177.3.1618. [DOI] [PubMed] [Google Scholar]
- Richards-Kortum R, Sevick-Muraca E. Quantitative optical spectroscopy for tissue diagnosis. Annu Rev Phys Chem. 1996;47:555–606. doi: 10.1146/annurev.physchem.47.1.555. [DOI] [PubMed] [Google Scholar]
- Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101:1155–1163. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, Kronenberg M, Locksley RM. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med. 2003;198:1069–1076. doi: 10.1084/jem.20030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styer LM, Kent KA, Albright RG, Bennett CJ, Kramer LD, Bernard KA. Mosquitoes inoculate high doses of West Nile virus as they probe and feed on live hosts. PLoS Pathog. 2007;3:1262–1270. doi: 10.1371/journal.ppat.0030132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumen C, Mempel TR, Mazo IB, von Andrian UH. Intravital microscopy: visualizing immunity in context. Immunity. 2004;21:315. doi: 10.1016/j.immuni.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Tarun AS, Baer K, Dumpit RF, Gray S, Lejarcegui N, Frevert U, Kappe SH. Quantitative isolation and in vivo imaging of malaria parasite liver stages. Int J Parasitol. 2006;36:1283–1293. doi: 10.1016/j.ijpara.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, Littman DR. The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol. 2000;165:3284–3292. doi: 10.4049/jimmunol.165.6.3284. [DOI] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol. 2003;21:1369–1377. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- Zoumi A, Yeh A, Tromberg BJ. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proc Natl Acad Sci USA. 2002;99:11014–11019. doi: 10.1073/pnas.172368799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.