

Introduction
The duocarmycins (1 and 2)1 belong to a small family of natural products (Figure 1) that also include yatakemycin (3)2 and CC-1065 (4).3 Their exceptionally potent cytotoxic activity is derived from their ability to bind and alkylate DNA in AT-rich regions of the minor groove.4-8 This family of natural products incorporates a remarkable set of molecular features integrated into compact structures capable of simultaneously expressing multiple functions.9
Figure 1.

Natural products and the DNA alkylation reaction.
Much like their predecessors distamycin and netropsin, the overall curvature and shape of the molecules leads to their preferential binding in the narrower, deeper, AT-rich minor groove where stabilizing van der Waals contacts are maximized (shape-selective recognition).8-11 The alkylation subunit vinylogous amide conveys a remarkable stability to a cross-conjugated and otherwise reactive cyclopropane. We have suggested that the disruption of this vinylogous amide conjugation by way of a DNA minor groove binding-induced conformational change brings the cyclopropane into conjugation with the cyclohexadienone activating it for nucleophilic attack, and provides the catalysis for the DNA alkylation reaction (shape-dependent catalysis, Figure 2).8,12,13 Significantly, these compounds are relatively unreactive until they reach their biological target, where they are selectively activated for DNA alkylation (target-based activation).14 Unique to this class, this activation occurs without a chemical change or reaction of the molecule, rather it occurs simply through a binding-induced conformational change in the compound. Studies that define the key relationships between structure and reactivity and quantitate the magnitude of these effects are detailed herein.
Figure 2.
Shape-dependent catalysis.
Just as important as their unique method of activation is a parabolic relationship between intrinsic reactivity and biological potency that has emerged from studying the natural products and numerous synthetic analogues (Figure 3).15 Presumably this reflects the requirement for sufficient stability for the compounds to reach their biological target balanced against the need for sufficient reactivity to efficiently alkylate DNA once they do. Establishment of this parabolic relationship defined what this optimal balance of reactivity and stability is for this class of natural products and key studies designed to define this parabolic relationship and exploit its use are also reviewed herein. Combined, these studies highlight remarkable fundamental relationships between structure, reactivity, and biological potency embodied in this class of natural products.
Figure 3.
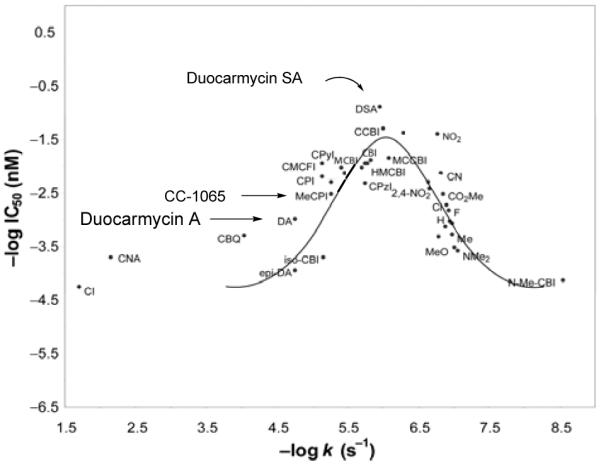
Parabolic relationship between reactivity and potency.
Importance and Role of the Vinylogous Amide: Structure versus Reactivity and Reaction Regioselectivity
The synthesis, reactivity comparison, and correlation with X-ray structures of a key series of alkylation subunit analogues established the magnitude and role of the vinylogous amide stabilization, revealed the stereoelectronic alignment of the reacting cyclopropane that dictates the regioselectivity of ensuing nucleophilic additions, and defined their dominant role in controlling the chemical reactivity and regioselectivity for this class of natural products. Moreover, their side-by-side comparison provides a depiction of the structural changes and reactivity effects that accompany a DNA binding-induced conformational change in 1-4 and indicates that they are of a magnitude to account for catalysis of the DNA alkylation reaction.
Thus, X-ray crystallographic studies of a structurally related and progressive series of 1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI)a analogues afforded information regarding the extent of vinylogous amide conjugation (c bond length), the disposition of the attached acyl groups (χ1 dihedral angle), the orbital alignment of the cyclopropane bonds, and their intimate relationship with both the stability of the molecule and the regioselective addition of nucleophiles to the cyclopropane. This series examined the effects of increasing the C-ring size from a 5-membered ring (CBI) as found in the natural products to a 6- and 7-membered ring system (2,3,10,10a-tetrahydro-1H-cyclopropa[d]benz[f]quinol-5-one (CBQ)16 and 1,2,3,4,11,11a-hexahydrocyclopropa[c]naphtho[2,1-b]azepin-6-one (CNA),17 respectively), Figure 4.
Figure 4.
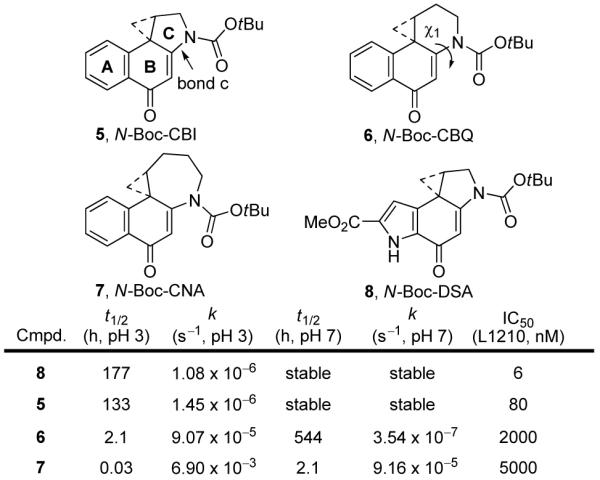
C-Ring analogues of CBI.
The reactivity increases as measured by solvolysis across the series are remarkable (>104) despite the subtle structural changes; CBQ16 and especially CNA17 exhibit substantial reactivity at pH 7 whereas both CBI and 8 are stable at this pH, Figure 4. Beautifully reflecting this remarkable increase in reactivity, the c bond lengths smoothly increase across this series (1.390, 1.415, and 1.428 Å) reflecting the progressive loss in vinylogous amide conjugation (Figure 5). Tracking with this increase in reactivity and loss of vinylogous amide stabilization, the χ1 dihedral angles progressively increase (21°, 34°, and 86°) across the series as well, mimicking the structural and reactivity changes imposed by a DNA binding-induced conformational change that increases the χ1 dihedral angle. Just as significant, the composite bond lengths of the cyclopropane increase as one progresses across the series, reflecting their weaker bond strengths, their increasing reactivity, and their increasing conjugation with the activating cyclohexadienone system. Additionally and unique to this series, the cyclopropane alignments with the cyclohexadienone π-system undergo remarkable changes as well, dictating the regioselectivity of nucleophilic attack.
Figure 5.
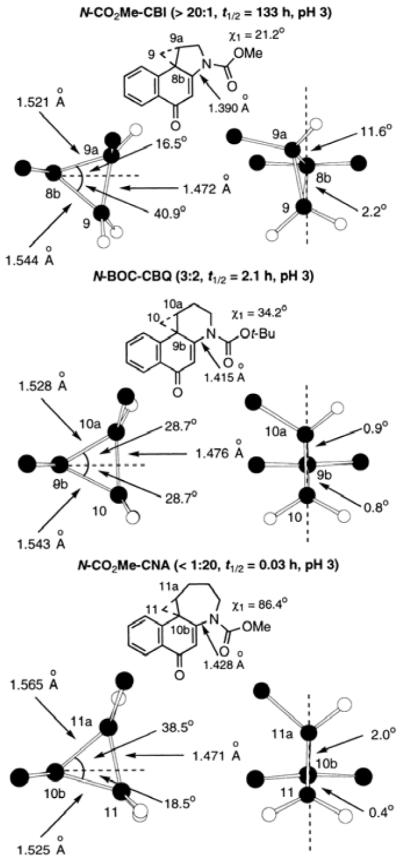
X-ray crystal structure data.
The X-ray crystal structure of N-CO2Me-CBI (>20:1 favoring ring-opening attack at C9 vs ring-expansion attack at C9a) reveals a favorable overlap between the orbital of the cyclopropane 8b-9 bond and the developing π-system of the phenol solvolysis product, whereas the orbital of the 8b-9a bond is much less effectively aligned to become a part of the developing aromatic π-system. The lengths of these two bonds reflect this variation in alignment, with that of the cleaved and stereoelectronically aligned 8b-9 bond (1.544 Å) being longer and weaker than the 8b-9a bond (1.521 Å). N-Boc-CBQ shows little preference for regioselective addition (3:2 ring-opening attack at C10 vs ring-expansion attack at C10a) and contains a cyclopropane that is nearly ideally conjugated and aligned with the π-system. Both cyclopropane bonds are perfectly bisected by the cyclohexadienone π-system, suggesting that the slight preference for addition to the least-substituted carbon is due to preferential cleavage of the longer and inherently weaker 9b-10 bond (1.543 Å), in addition to a potential steric preference for SN2 addition to the least-substituted C10 carbon. Analysis of N-CO2Me-CNA reveals a situation similar to, but reversed from that of N-CO2Me-CBI. The bond to the more-substituted carbon (10b-11a) is now better aligned with the developing π-system of the product phenol, and it is now the longest (1.565 Å) and weakest of the cyclopropane bonds. These observations correlate beautifully with the observed reaction regioselectivity of the CNA alkylation subunit (<1:20 ring-opening attack at C11 vs ring-expansion attack at C11a), which is completely reversed from that observed with CBI. Notably, in all instances and contrary to an early mischaracterization,18 the ring expansion cleavage of the cyclopropane proceeds with clean inversion of stereochemistry at the reacting center indicative of a SN2, not SN1, nucleophilic addition.19 Thus, in addition to providing insights into their reactivity, the studies highlight the importance of the fused 5-membered C-ring, which serves to enforce a stereoelectronically-controlled nucleophilic addition to the least substituted cyclopropane carbon.
Additional modifications to the structures have provided further insight into the importance of the vinylogous amide and the linking amide (Figure 6).20 Removing the carbonyl of the linking amide in CBI-TMI21 (9) afforded a compound 10 that failed to alkylate DNA even under forcing conditions (37 °C, 14 days, 10-1 M agent vs 25 °C, 24 h, 10-6 M for 9), that was biologically inactive displaying cytotoxic activity that was >105-fold less potent than 9, and that exhibited little solvolysis requiring exposure to pH 1 to detect a measurable reactivity. In addition to establishing the fundamental importance of the linking amide, the comparisons highlight the truly remarkable stability provided to this unusual ring system by full vinylogous amide conjugation that in turn is reduced (tuned) by N-acylation as found in the natural product structures. Its disruption by a DNA binding-induced conformational change is no longer possible with 10, accounting for its complete lack of DNA alkylation. The disparate behavior of 9 versus 10 not only revealed that the linking amide plays an important role activating the alkylation subunit for nucleophilic attack, but that it is critical to catalysis of the DNA alkylation reaction.
Figure 6.
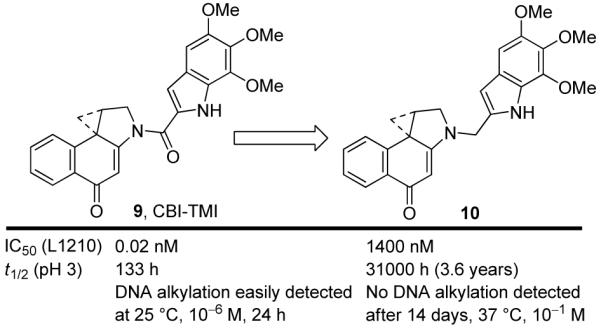
Importance of the linking amide.
Equally revealing was examination of 1,2,9,9a-tetrahydro-1H-cyclopropa[c]benz[e]inden-4-one (CBIn, 11, Figure 7),22 which contains a carbocyclic skeleton lacking the nitrogen crucial for stability of the alkylation subunit. Consistent with intuitive expectations, CBIn proved sufficiently stable for characterization, but remarkably reactive toward nucleophilic addition. CBIn is 3200× less stable than its CBI counterpart at pH 3 and greater than 103-104× more reactive than CBI at pH 7, where CBI is stable. Solvolysis studies conducted over a pH range of 2-12 revealed no acid concentration dependence between pH 4-12 indicating that the reaction is uncatalyzed (versus acid-catalyzed) in this pH range, and suggesting that there is no basis for invoking an acid-catalyzed activation18,23 of these compounds when bound to DNA (pH 7.4, 0.0001-0.00004% protonation of phosphate backbone).
Figure 7.
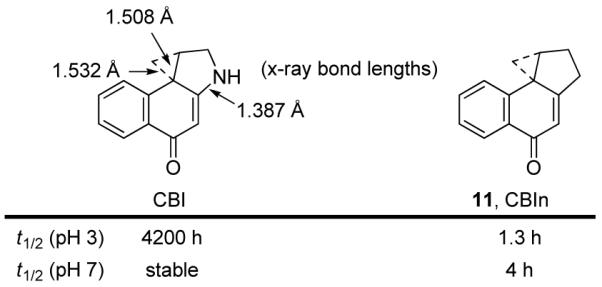
CBI versus CBIn.
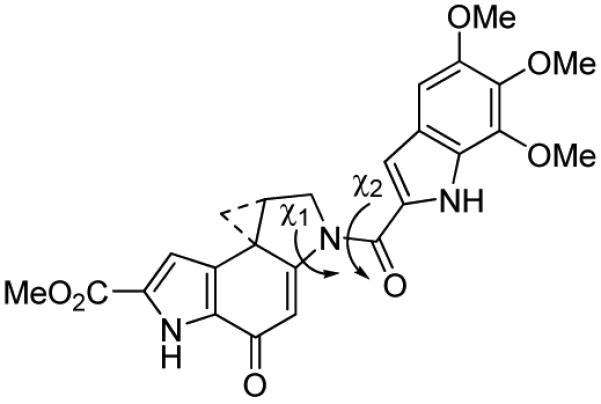
Additional insights into the behavior of this class of natural products was recently reported with the examination of 12, which lacks the characteristic vinylogous amide stabilization and is incapable of activation by C4-carbonyl protonation (Figure 8). Preparation of 12 revealed that spirocyclization can occur through deprotonation of the indole NH, rather than the conventional phenol, but provides an intrinsically much more reactive compound.24 It also exhibits a rate of solvolysis that is independent of pH. Although more reactive than its natural product counterpart, 12 exhibits an identical DNA alkylation selectivity as duocarmycin SA and a reduced, but significant level of biological activity (L1210 IC50 = 5 nM).
Figure 8.
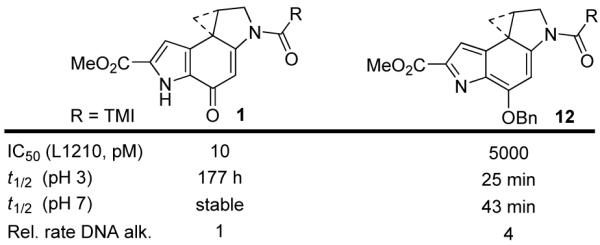
Alternate spirocyclization.
These observations reaffirm several key features shown to contribute to the DNA alkylation selectivity of duocarmycin SA (DSA). First, the DNA alkylation selectivity is derived from their intrinsic non-covalent binding selectivity within the minor groove of DNA,10,13,25 and not from the source of alkylation catalysis, which differs between 1 and 12. Like 12, derivatives that lack the C4-carbonyl,10,25 analogues in which the C4-carbonyl has been relocated,26 and those that utilize an alternative mechanism of catalysis27 exhibit an indistinguishable DNA alkylation selectivity, albeit proceeding at altered rates. Second, the comparable behavior of 1 and 12 indicates that the alkylation selectivity cannot originate from a sequence-dependent DNA backbone phosphate protonation18,23,28 of the C4-carbonyl of 1, as this feature is not present with 12.
Although this alternative spirocyclization plays no present day role in the behavior of duocarmycin SA, it is possible that it is related to an evolutionary origin of 1 and its related natural products, Figure 9. Since it is reasonable to assume that the p-spirocyclization (through displacement of an activated primary alcohol) occurs as the last step of the biosynthesis of duocarmycin SA, it is tempting to suggest that such compounds first emerged without the ability to undergo p-spirocyclization, closing to the cyclopropane with participation of the indole NH as in 13 instead (Figure 9). Evolutionary pressures may have improved on this by subsequent aryl hydroxylation to afford the present day phenol precursors where the resulting increase in potency (10 pM vs 5 nM) would have provided the producing bacteria with a beneficial growth advantage.
Figure 9.
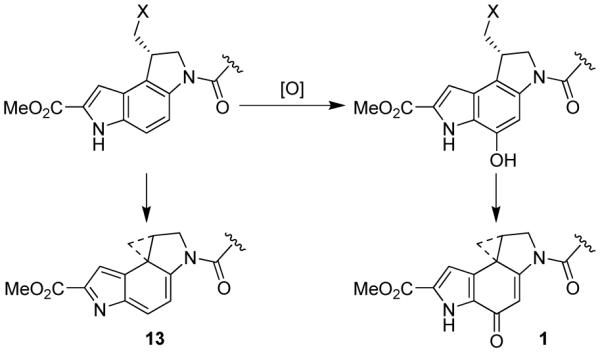
Potential ancestral role of the alternative spirocyclization.
A provocative indication that there is perhaps merit to this proposal emerges from the examination of the correlation between biological potency (cytotoxic activity, -log IC50) and chemical stability (-log ksolv) where 13, like 12, would be projected to lie at an early stage of Nature’s steps in the optimization of this class of DNA alkylating agents, Figure 10.
Figure 10.
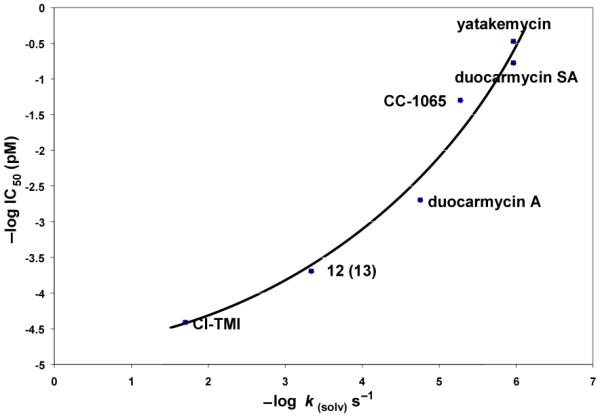
Plot of -log IC50 (L1210, cytotoxic activity) versus -log ksolv (pH 3) that includes the natural products and a representative related compound illustrating the direct correlation between stability and biological potency and highlighting Nature’s potential steps in the optimization of this class of DNA alkylating agents.
Fundamental Parabolic Relationship Between Reactivity and Biological Activity
A wide variety of synthetic duocarmycin SA analogues have been prepared and studied.29 The 1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) alkylation subunit has been the most extensively examined, due to the relative ease of preparation and its impressive properties compared to the natural products.30 Initial studies described the effects of C7-substitution, which is para to the C4-carbonyl. The electronic effects of groups at this position were expected to probe their impact on reactivity and to address the question of whether protonation of the carbonyl was important for activation of these agents. Comparison of the parent unsubstituted CBI subunit with two derivatives, 7-cyano-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (CCBI, 14)31 and 7-methoxy-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (MCBI, 15,32 Figure 11) established that the electronic nature of C7-subsituents did not have a significant effect on the biological potency or intrinsic reactivity.
Figure 11.
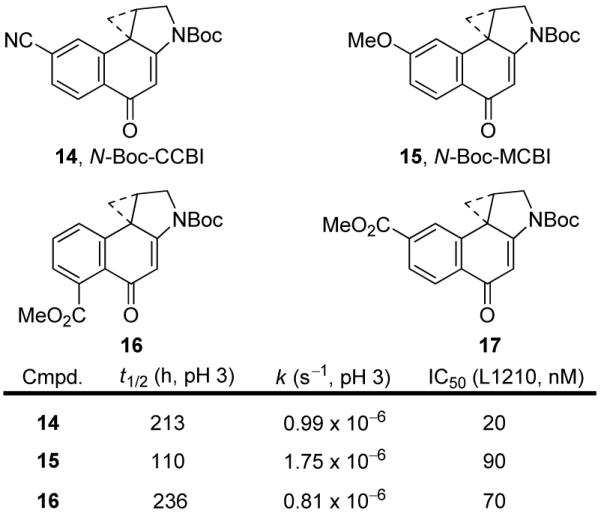
A-Ring derivatives of CBI.
Despite the opposing electronic nature of the substitutents found in CCBI and MCBI, only a 2-fold difference in solvolysis reactivity between the N-Boc derivatives was observed. The cytotoxic activity mirrors this trend, with N-Boc-CCBI being only 4-fold more potent than N-Boc-MCBI, and the full length agents (TMI, indole2) were essentially indistinguishable from each other. In fact, a Hammett analysis of the acid-catalyzed solvolysis of MCBI, CBI, and CCBI revealed a very small ρ value (-0.30 at pH 3), indicative of little substituent effect on the reactivity.
More interestingly, both substituents had a beneficial impact on the DNA alkylation properties of the fully elaborated analogues. The DNA alkylation properties (rate, efficiency, and selectivity) of such MCBI and CCBI derivatives were essentially indistinguishable, but both alkylated DNA at rates and efficiencies that exceed the CBI derivatives. This suggested that it is not the electronic nature of the substituent, but rather the simple presence of a C7-substituent that serves to improve the DNA alkylation rate and efficiency. We have suggested that this is the result of the substituent extending the rigid length of the agent and increasing the extent of the DNA binding-induced conformational change when bound in the minor groove.33 Consistent with this interpretation, incorporation of an electron-withdrawing C5-substituent (CO2Me, 16) subtly increased the intrinsic stability, but slowed the rate and efficiency of DNA alkylation, whereas the corresponding C7-derivative (17) displayed an analogous subtle decrease in reactivity, but displayed the increased rate and efficiency of DNA alkylation observed with both 14 and 15.34
In contrast to A-ring substituents, N2 substitutents have a huge impact on the properties of the molecules by virtue of the direct impact on the vinylogous amide. In addition to those highlighted in Figures 6 and 7, early studies enlisting a series of modified acyl derivatives of CBI indicated that their impact on reactivity was extraordinarily large and linear in a Hammett analysis (ρ = -3.0, r = 0.983) with the stronger electron-withdrawing substituents imparting the greater stability, and that this increased stability directly correlated with a near linear and equally large increase in cytotoxic potency (r = 0.979), Figure 12.35
Figure 12.
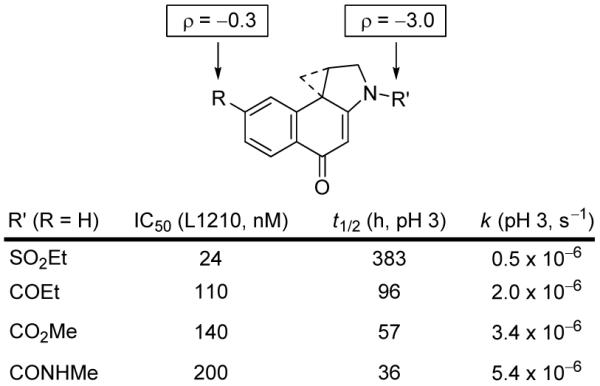
N2-Acyl derivatives of CBI.
Until recently, the relationship between chemical stability and biological potency was restricted to the examination of such derivatives that possessed sufficient reactivity to alkylate DNA and was interpreted to reflect the ability of the chemically more stable derivatives to more effectively reach their biological target (DNA).36 However, as the number of such compounds examined increased, the plot of such a correlation more closely resembled a parabolic relationship, but one in which the derivatives lying on the “too stable” side of the relationship were scarce. Consequently, in our efforts to define the full nature of this relationship, we began examining CBI derivatives that might accurately reflect the behavior of less reactive derivatives. A unique series of N-aryl CBI derivatives, in which the electronic properties of the aryl p-substituent could be systematically varied to alter the reactivity, were found to be remarkably stable relative to the typical N-acyl derivatives.15 This reactivity followed a well-defined correlation with σp (ρ= 0.17) in which electron-withdrawing substituents enhance and electron-donating substituents decrease the reactivity (solvolysis at pH 2, Figure 13).
Figure 13.
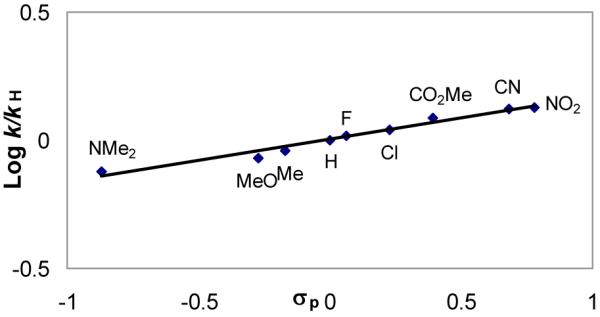
Hammett plot of para substituted N-aryl-CBI derivatives.
X-ray structures of the full series of derivatives revealed that increasingly electron-withdrawing p-substituents effectively increase the diagnostic c bond length, decrease the d bond length, decrease the f bond length, and increase the χ1 dihedral angle, all of which indicate an effect of diminishing the conjugation of the vinylogous amide (Figure 14), leading to increases in reactivity. Conversely, the presence of electron-donating p-substituents had the opposite effect, effectively increasing the conjugation of the vinylogous amide and further stabilizing the alkylation subunit to nucleophilic attack. Most significantly, the correlation of reactivity with cytotoxic activity is now reversed with the more reactive derivatives in this unusually stable series exhibiting the more potent activity. Beautifully, this series served to complete the parabolic relationship between intrinsic reactivity (stability) and cytotoxic activity providing the data shown in the shaded box in Figure 14.
Figure 14.

Structural and reactivity effects of para substituted N-aryl-CBI derivatives.
In addition to the impact of attached substituents, there are structural features embedded in the skeletons of the natural products that also impact the reactivity of these compounds. It has been suggested that an internal hydrogen bond between the pyrrole NH and the C4-carbonyl of duocarmycin SA serves to activate the agent for nucleophilic attack.37 To probe this feature, iso-duocarmycin SA (iso-DSA, 27) was prepared and evaluated. In addition to addressing this question of H-bond activation, examination of this analogue would also serve to probe the effects of incorporating a H-bond donor directed towards the floor of the minor groove, and would determine whether a second vinylogous amide system would serve to further stabilize or destabilize the alkylation subunit.38
Solvolysis studies revealed N-Boc-iso-DSA (26) to be slightly more reactive by a factor of two than its parent N-Boc-DSA (8). As a result, the iso-duocarmycin SA series was predictably less potent than the natural product, Figure 15. N-Boc-iso-DSA (26) was found to be 5-fold less potent than its natural product counterpart, nicely reflecting the 2-fold difference observed in the solvolytic reactivity and the same trend was observed for iso-duocarmycin SA (27). When 26 was placed on the plot of reactivity versus potency and given the anomalously potent activity of duocarmycin SA, its relative cytotoxic activity even more closely approached that expected of a derivative exhibiting its relative reactivity (Figure 16).
Figure 15.
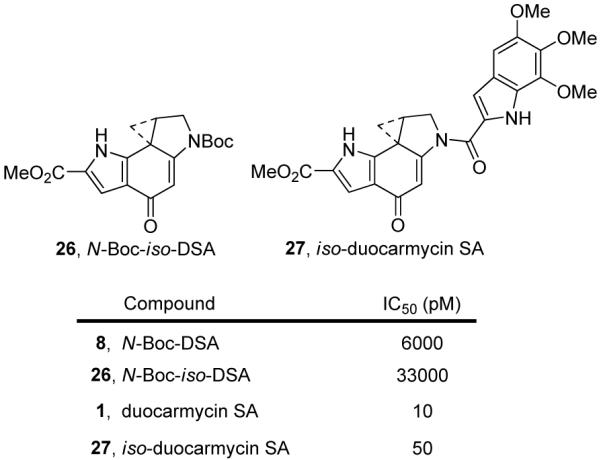
N-Boc-iso-DSA and iso-duocarmycin SA.
Figure 16.
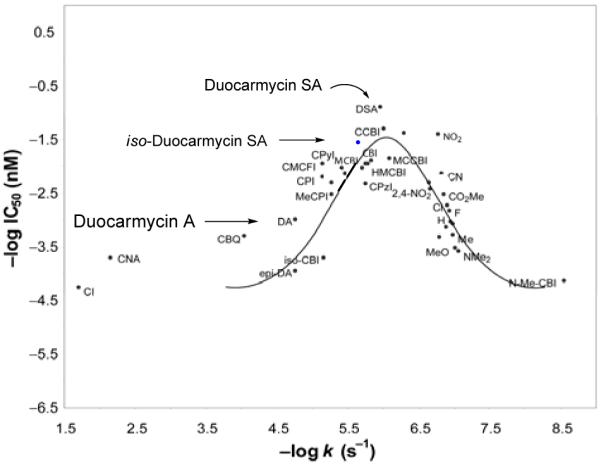
Placement of N-Boc-iso-DSA on the parabolic curve.
Thus, the enhanced reactivity of 26 versus 8 indicates that the putative internal H-bond of 8, which has been removed in 26, does not contribute to the activation of the compound towards nucleophilic addition. In addition, the incorporation of a second vinylogous amide in the molecule does not further stabilize the reactive cyclopropane, which would lead to greater potency and reduced solvolysis reactivity. In fact, the opposite appears to be true, suggesting that the additional cross-conjugated and competing vinylogous amide destabilizes the alkylation subunit.
Using the Parabolic Relationship: Rational Design of an Improved CC-1065
The parabolic relationship between reactivity and potency that emerged from these studies provides the opportunity to rationally design analogues that would sit at or near the pinnacle of the parabola, with a reactivity/potency relationship similar to that of duocarmycin SA. In our efforts, a series of benzothiophene-based analogues were designed and evaluated.39 Intuitively, the replacement of the pyrrole NH of CC-1065 with the larger sulfur atom was expected to release some of the ring strain inherent with the pyrrole, creating a ring system closer in structure to that of CBI, and increasing the stability of the molecule. Additionally, the increased electronegativity of sulfur versus the pyrrole nitrogen was expected to further contribute to an increasingly stable alkylation subunit whose activity was expected to surpass that of its parent compound CC-1065. These intuitive arguments were supported by computational studies (AM1, MNDO) that predicted that the substitution of sulfur for NH in the CPI structure would increase the stability of the alkylation subunit.
The synthesis and examination of 7-methyl-1,2,8,8a-tetrahydrocyclopropa[c]thieno[3,2-e]indol-4-one (MeCTI, 30) and 6-methyl-1,2,8,8a-tetrahydrocyclopropa[c]thieno[2,3-e]indol-4-one (iso-MeCTI, 31) confirmed these expectations.37 Solvolysis of N-Boc-MeCTI (30) and N-Boc-iso-MeCTI (31) revealed two compounds whose stabilities (pH 3, t1/2 = 206 and 209 h, respectively) were substantially increased over that of their parent compound N-Boc-MeCPI (29, t1/2 = 37 h). They also proved slightly more stable than N-Boc-DSA (8, t1/2 = 177 h) and N-Boc-CBI (5, t1/2 = 133 h), making them among the most stable alkylation subunits explored to date. Significantly, these compounds exhibited substantially increased biological potency versus CC-1065 and its derivatives. The natural enantiomers of N-Boc-MeCTI (30, IC50 = 30 nM) and N-Boc-iso-MeCTI (31, IC50 = 25 nM) were 10-fold more potent than N-Boc-MeCPI (29, IC50 = 330 nM) and 2-3-fold more potent than N-Boc-CBI (5, IC50 = 80 nM), beautifully falling in line with expectations based on their relative solvolytic reactivities. This is illustrated on the parabolic curve, where MeCTI and iso-MeCTI sit at or near the pinnacle of the relationship, just below the anomalously potent DSA alkylation subunit (Figure 18).
Figure 18.
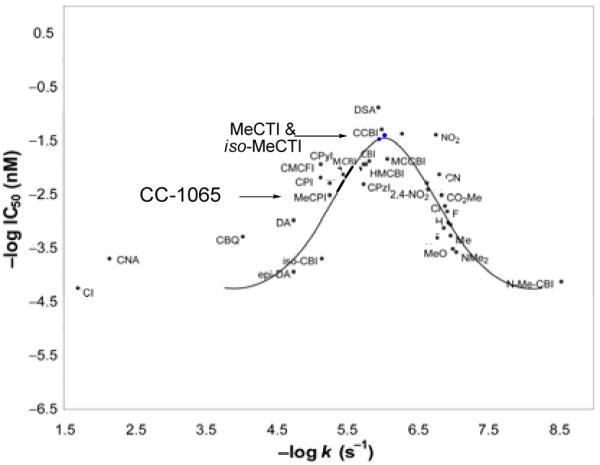
MeCTI and iso-MeCTI on the plot of reactivity versus potency.
MeCTI-PDE2 (28), the sulfur-based analogue of CC-1065 (4), displayed an identical DNA alkylation selectivity relative to the parent compound, displaying the characteristic enantiomeric differences. Significant in these comparisons is the observation that a single atom change in the skeleton of the alkylation subunit structure of CC-1065 (NH→S) predictably increased the stability of the natural product that in turn reliably led to a substantial increase in its biological potency.
Conclusions
The small family of natural products that include the duocarmycins, yatakemycin, and CC-1065 incorporate a remarkable combination of molecular features integrated into their compact structures controlling their DNA binding and ensuing alkylation reaction and their resulting exceptionally potent biological activity. The systematic total synthesis of an extensive series of analogues containing deep-seated structural changes have been utilized to define subtle relationships between structure, reactivity, reaction regioselectivity and biological potency using fundamental chemical principles. Superimposed on a skeleton that selectively binds DNA minor groove AT-rich sequences (shape-selective recognition), the compounds incorporate a spirocyclopropylcyclohexadienone whose reactivity has been masked by a cross-conjugated and remarkably stabilizing vinylogous amide, taming what would ordinarily be an exceptionally reactive electrophile. We have suggested that disruption of this vinylogous amide conjugation through a DNA binding-induced conformational change activates the cyclopropane for a stereoelectronically-controlled nucleophilic attack serving as the catalysis for the DNA alkylation reaction (shape-dependent catalysis). These studies highlight the remarkable design Nature utilized to mask a reactive electrophile that is subsequently capable of selective activation by a unique mechanism upon reaching its biological target (target-based activation).
In addition to elucidating the mechanism of activation of these natural products, a detailed understanding of the relationship between intrinsic reactivity and biological potency resulted from studying analogues with deep-seated structural modifications. These studies defined a fundamental parabolic relationship between chemical reactivity and biological potency (cytotoxic activity) and identified the optimal balance between reactivity—stability, reflecting a stability required to effectively reach their biological target in a biological milieu while maintaining a sufficient reactivity to effectively alkylate DNA once they do. Remarkably, but perhaps not surprisingly, duocarmycin SA and yatakemycin incorporate an alkylation subunit that lies at the pinnacle of this relationship reflecting Nature’s exquisite optimization of their DNA alkylation properties. Unique insights into Nature’s potential steps in this evolutionary optimization were revealed with the discovery of an alternative, but less productive indole N5H spirocyclization of duocarmycin SA lacking a C4-phenol. Finally, as a result of the increased understanding of these compounds (reactivity vs potency), we described the rational design of a synthetic alkylation subunit (CTI) incorporating a single skeletal atom change that maintains the DNA alkylation properties of its parent natural product (CC-1065), but exhibited a greater chemical stability and resulting increased biological potency. This remarkably stable alkylation subunit analogue lies at the pinnacle of the parabolic relationship and constitutes one of the most stable derivatives to be characterized to date.
Figure 17.
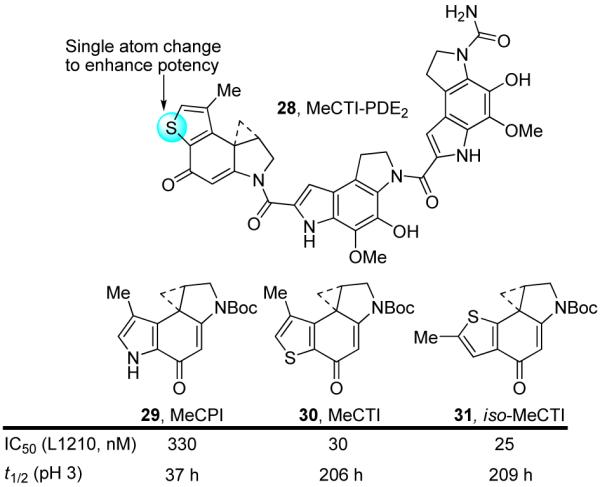
Rationally designed analogues of CC-1065.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA041986) and the Skaggs Institute for Chemical Biology. KSM is a Skaggs Fellow.
Biography
Dale L. Boger received his B.S. degree in Chemistry from the University of Kansas in 1975, and Ph. D. degree from Harvard University in 1980. Following graduate school, he returned to the University of Kansas as a member of the faculty in the Department of Medicinal Chemistry, moved to Purdue University in 1986, and joined the newly founded Department of Chemistry at The Scripps Research Institute in 1991 as the Richard and Alice Cramer Professor of Chemistry. His research interests span the fields of organic and bioorganic chemistry, medicinal chemistry, the study of DNA-agent and protein-ligand interactions, and antitumor agents.
Karen S. MacMillan received her B.S. degree in Chemistry from the University of California, Davis in 2002 and her Ph.D. degree in Chemistry from The Scripps Research Institute in 2009 under the guidance of Professor Dale L. Boger. Currently, she is working as an Assistant Instructor in the Chemistry Core Facility within the Department of Biochemistry at the University of Texas Southwestern Medical Center.
Footnotes
- DSA
- duocarmycin SA
- DA
- duocarmycin A
- epi-DA
- epi-duocarmycin A
- CBI
- 1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one
- CPI
- 1,2,8,8a-tetrahydrocyclopropa[c]pyrrolo[3,2-e]indol-4-one
- MeCPI
- 7-methyl-1,2,8,8a-tetrahydrocyclopropa[c]pyrrolo[3,2-e]indol-4-one
- CI
- 1,2,7,7a-tetrahydrocyclopropa[1,2-c]indol-4-one
- CNA
- 1,2,3,4,11,11a-hexahydrocyclopropa[c]naphtho[2,1-b]azepin-6-one
- CBQ
- 2,3,10,10a-tetrahydro-1H-cyclopropa[d]benzo[f]quinol-5-one
- iso-CBI
- 1,2,9,9a-tetrahydrocyclopropa[c]benzo[f]indol-8-one
- CMCFI
- 6-carbomethoxy-1,2,8,8a-tetrahydrocyclopropa[c]furano[3,2-e]indol-4-one
- CPyI
- methyl 1,2,9,9a-tetrahydrocyclopropa[c]pyrido[3,2-e]indol-4-one-7-carboxylate
- MCBI
- 7-methoxy-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one
- CCBI
- 7-cyano-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one
- MCCBI
- 5-methoxycarbonyl-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one
- HMCBI
- 5-hydroxymethyl-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one
- CPzI
- 7-methyl-1,2,8,8a-tetrahydrocyclopropa[c]pyrazolo[4,3-e]indol-4-one
- CBIn
- 1,2,9,9a-tetrahydro-1H-cyclopropa[c]benz[e]inden-4-one
- iso-DSA
- 6-methoxycarbonyl-1,2,8,8a-tetrahydrocyclopropa[c]pyrrolo[2,3-g]indol-4-one
- MeCTI
- 7-methyl-1,2,8,8a-tetrahydrocyclopropa[c]thieno[3,2-e]indol-4-one
- iso-MeCTI
- 6-methyl-1,2,8,8a-tetrahydrocyclopropa[c]thieno[2,3-e]indol-4-one
- TMI
- 5,6,7-trimethoxyindole-2-carboxylate
References
- (1).(a) Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, Takahashi I, Nakano H. Duocarmycin SA, A New Antitumor Antibiotic from Streptomyces sp. J. Antibiot. 1990;43:1037–1038. doi: 10.7164/antibiotics.43.1037. [DOI] [PubMed] [Google Scholar]; (b) Takahashi I, Takahashi K, Ichimura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H. Duocarmycin A, A New Antitumor Antibiotic from Streptomyces. J. Antibiot. 1988;41:1915–1917. doi: 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]
- (2).Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai T. Yatakemycin, A Novel Antifungal Antibiotic Produced by Streptomyces sp. TP-A0356. J. Antibiot. 2003;56:107–113. doi: 10.7164/antibiotics.56.107.Structure revision:Tichenor MS, Kastrinsky DB, Boger DL. Total Synthesis, Structure Revision, and Absolute Configuration of (+)-Yatakemycin. J. Am. Chem. Soc. 2004;126:8396–8398. doi: 10.1021/ja0472735.
- (3).Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J. CC-1065 (NSC 298223), A Potent New Antitumor Agent. Improved Production and Isolation, Characterization and Antitumor Activity. J. Antibiot. 1981;34:1119–1125. doi: 10.7164/antibiotics.34.1119. [DOI] [PubMed] [Google Scholar]
- (4).Duocarmycin SA:Boger DL, Johnson DS, Yun W. (+)- and ent-(-)-Duocarmycin SA and (+)- and ent-(-)-N-Boc-DSA DNA Alkylation Properties. Alkylation Site Models That Accommodate the Offset AT-Rich Adenine N3 Alkylation Selectivity of the Enantiomeric Agents. J. Am. Chem. Soc. 1994;116:1635–1656. doi: 10.1016/s0968-0896(00)82007-6.
- (5).Yatakemycin:Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarishi Y, Boger DL. DNA Alkylation Properties of Yatakemycin. J. Am. Chem. Soc. 2003;125:10971–10976. doi: 10.1021/ja035984h.Trzupek JD, Gottesfeld JM, Boger DL. Alkylation of Duplex DNA in Nucleosome Core Particles by Duocarmycin SA and Yatakemycin. Nature Chem. Biol. 2006;2:79–82. doi: 10.1038/nchembio761.Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. Asymmetric Total Synthesis of (+)- and ent-(-)-Yatakemycin and Duocarmycin SA: Evaluation of Yatakemycin Key Partial Structures and Its Unnatural Enantiomer. J. Am. Chem. Soc. 2006;128:15683–115696. doi: 10.1021/ja064228j.Tichenor MS, MacMillan KS, Trzupek JD, Rayl TJ, Hwang I, Boger DL. Systematic Exploration of the Structural Features of Yatakemycin Impacting DNA Alkylation and Biological Activity. J. Am. Chem. Soc. 2007;129:10858–10869. doi: 10.1021/ja072777z.
- (6).CC-1065:Hurley LH, Lee C-S, McGovren JP, Warpehoski MA, Mitchell MA, Kelly RC, Aristoff PA. Molecular Basis for Sequence-Specific DNA Alkylation by CC-1065. Biochemistry. 1988;27:3886–3892. doi: 10.1021/bi00410a054.Hurley LH, Warpehoski MA, Lee C-S, McGovren JP, Scahill TA, Kelly RC, Mitchell MA, Wicnienski NA, Gebhard I, Johnson PD, Bradford VS. Sequence Specificity of DNA Alkylation by the Unnatural Enantiomer of CC-1065 and Its Synthetic Analogues. J. Am. Chem. Soc. 1990;112:4633–4649.Boger DL, Johnson DS, Yun W, Tarby CM. Molecular Basis for Sequence Selective DNA Alkylation by (+)- and ent-(-)-CC-1065 and Related Agents: Alkylation Site Models that Accommodate the Offset AT-rich Adenine N3 Alkylation Selectivity. Bioorg. Med. Chem. 1994;2:115–135. doi: 10.1016/s0968-0896(00)82007-6.Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. Synthesis and Evaluation of Aborted and Extended CC-1065 Functional Analogues: (+)- and (-)-CPI-PDE-I1, (+)- and (-)-CPI-CDPI1, and (+)- and (-)-CPI-CDPI3. Preparation of Key Partial Structures and Definition of an Additional Functional Role of the CC-1065 Central and Right-Hand Subunits. J. Am. Chem. Soc. 1990;112:4623–4632.
- (7).Duocarmycin A:Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. Duocarmycin-Pyrindamycin DNA Alkylation Properties and Identification, Synthesis, and Evaluation of Agents Incorporating the Pharmacophore of the Duocarmycin-Pyrindamycin Alkylation Subunit. Identification of the CC-1065-Duocarmycin Common Pharmacophore. J. Am. Chem. Soc. 1990;112:8961–8971.Boger DL, Ishizaki T, Zarrinmayeh H. Isolation and Characterization of the Duocarmycin-Adenine DNA Adduct. J. Am. Chem. Soc. 1991;113:6645–6649.Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. DNA Alkylation Properties of the Duocarmycins: (+)-Duocarmycin A, epi-(+)-Duocarmycin A, ent-(-)-Duocarmycin A and epi,ent-(-)-Duocarmycin A. Bioorg. Med. Chem. Lett. 1992;2:759–765.Boger DL, Yun W. Reversibility of the Duocarmycin A and SA DNA Alkylation Reaction. J. Am. Chem. Soc. 1993;115:9872–9873.Boger DL, McKie JA, Nishi T, Ogiku T. Total Synthesis of (+)-Duocarmycin A, epi-(+)-Duocarmycin A and Their Unnatural Enantiomers: Assessment of Chemical and Biological Properties. J. Am. Chem. Soc. 1997;119:311–325.Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. Synthesis and Preliminary Evaluation of Agents Incorporating the Pharmacophore of the Duocarmycin/Pyrindamycin Alkylation Subunit: Identification of the CC-1065/Duocarmycin Common Pharmacophore. J. Org. Chem. 1990;55:4499–4502.
- (8).Reviews:Boger DL, Johnson DS. CC-1065 and the Duocarmycins: Understanding Their Biological Function Through Mechanistic Studies. Angew. Chem. Int. Ed. Engl. 1996;35:1438–1474.Boger DL. The Duocarmycins: Synthetic and Mechanistic Studies. Acc. Chem. Res. 1995;28:20–29.Boger DL, Johnson DS. CC-1065 and the Duocarmycins: Unraveling the Keys to a New Class of Naturally Derived DNA Alkylating Agents. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3642–3649. doi: 10.1073/pnas.92.9.3642.Boger DL, Garbaccio RM. Shape-Dependent Catalysis: Insights into the Source of Catalysis for the CC-1065 and Duocarmycin DNA Alkylation Reaction. Acc. Chem. Res. 1999;32:1043–1052.Tichenor MS, Boger DL. Yatakemycin: Total Synthesis, DNA Alkylation, and Biological Properties. Natural Prod. Rep. 2008;25:220–226. doi: 10.1039/b705665f.
- (9).Tse WC, Boger DL. Sequence-selective DNA Recognition: Natural Products and Nature’s Lessons. Chem. Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- (10).(a) Boger DL, Coleman RS, Invergo BJ, Zarrinmayeh H, Kitos PA, Thompson SC, Leong T, McLaughlin LW. A Demonstration of the Intrinsic Importance of Stabilizing Hydrophobic Binding and Non-covalent van der Waals Contacts Dominant in the Non-covalent CC-1065/B-DNA Binding. Chem.-Biol. Interact. 1990;73:29–52. doi: 10.1016/0009-2797(90)90107-x. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. Demonstration of a Pronounced Effect of Noncovalent Binding Selectivity on the (+)-CC-1065 DNA Alkylation and Identification of the Pharmacophore of the Alkylation Subunit. Proc. Natl. Acad. Sci. U.S.A. 1991;88:1431–1435. doi: 10.1073/pnas.88.4.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boger DL, Munk SA, Zarrinmayeh H. (+)-CC-1065 DNA Alkylation: Key Studies Demonstrating a Noncovalent Binding Selectivity Contribution to the Alkylation Selectivity. J. Am. Chem. Soc. 1991;113:3980–3983. [Google Scholar]; (d) Boger DL, Johnson DS. Second Definitive Test of Proposed Models for the Origin of the CC-1065 and Duocarmycin DNA Alkylation Selectivity. J. Am. Chem. Soc. 1995;117:1443–1444. [Google Scholar]; (e) Boger DL, Zhou J, Cai H. Demonstration and Definition of the Noncovalent Binding Selectivity of Agents Related to CC-1065 by an Affinity Cleavage Agent: Noncovalent Binding Coincidental with Alkylation. Bioorg. Med. Chem. 1996;4:859–868. doi: 10.1016/0968-0896(96)00073-9. [DOI] [PubMed] [Google Scholar]
- (11).(a) Boger DL, Fink BE, Brunette SR, Tse WC, Hedrick MP. A Simple, High-Resolution Method for Establishing DNA Binding Affinity and Sequence Selectivity. J. Am. Chem. Soc. 2001;123:5878–5891. doi: 10.1021/ja010041a. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Tse WC. Thiazole Orange as the Fluorescent Intercalator in a High Resolution FID Assay for Determining DNA Binding Affinity and Sequence Selectivity of Small Molecules. Bioorg. Med. Chem. 2001;9:2511–2518. doi: 10.1016/s0968-0896(01)00243-7. [DOI] [PubMed] [Google Scholar]; (c) Tse WC, Boger DL. A Fluorescent Intercalator Displacement Assay for Establishing DNA Binding Selectivity and Affinity. Acc. Chem. Res. 2004;37:61–69. doi: 10.1021/ar030113y. [DOI] [PubMed] [Google Scholar]
- (12).Boger DL, Garbaccio RM. Catalysis of the CC-1065 and Duocarmycin DNA Alkylation Reaction: DNA Binding Induced Conformational Change in the Agent Results in Activation. Bioorg. Med. Chem. 1997;5:263–276. doi: 10.1016/s0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]
- (13).(a) Boger DL, Hertzog DL, Bollinger B, Johnson DS, Cai H, Goldberg J, Turnbull P. Duocarmycin SA Shortened, Simplified, and Extended Agents: A Systematic Examination of the Role of the DNA Binding Subunit. J. Am. Chem. Soc. 1997;119:4977–4986. [Google Scholar]; (b) Boger DL, Bollinger B, Hertzog DL, Johnson DS, Cai H, Mesini P, Garbaccio RM, Jin Q, Kitos PA. Reversed and Sandwiched Analogs of Duocarmycin SA: Establishment of the Origin of the Sequence-Selective Alkylation of DNA and New Insights into the Source of Catalysis. J. Am. Chem. Soc. 1997;119:4987–4998. [Google Scholar]
- (14).Wolkenberg SE, Boger DL. Mechanisms of in Situ Activation for DNA-Targeting Antitumor Agents. Chem. Rev. 2002;102:2477–2496. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- (15).(a) Parrish JP, Hughes TV, Hwang I, Boger DL. Establishing the Parabolic Relationship between Reactivity and Activity for Derivatives and Analogues of the Duocarmycin and CC-1065 Alkylation Subunits. J. Am. Chem. Soc. 2004;126:80–81. doi: 10.1021/ja038162t. [DOI] [PubMed] [Google Scholar]; (b) Parrish JP, Trzupek JD, Hughes TV, Hwang I, Boger DL. Synthesis and Evaluation of N-Aryl and N-Alkenyl CBI Derivatives. Bioorg. Med. Chem. 2004;12:5845–5856. doi: 10.1016/j.bmc.2004.08.032. [DOI] [PubMed] [Google Scholar]
- (16).(a) Boger DL, Mesini P, Tarby CM. Chemical and Structural Comparison of N-Boc-CBQ and N-Boc-CBI: Identification and Structural Origin of an Unappreciated but Productive Stability of the CC-1065 and Duocarmycin SA Alkylation Subunits. J. Am. Chem. Soc. 1994;116:6461–6462. [Google Scholar]; (b) Boger DL, Mesini P. Design, Synthesis, and Evaluation of CC-1065 and Duocarmycin Analogs Incorporating the 2,3,10,10a-Tetrahydro-1H-cyclopropa[d]benzo[f]quinol-5-one (CBQ) Alkylation Subunit: Identification and Structural Origin of Subtle Stereoelectronic Features That Govern Reactivity and Regioselectivity. J. Am. Chem. Soc. 1994;116:11335–11348. [Google Scholar]; (c) Boger DL, Mesini P. DNA Alkylation Properties of CC-1065 and Duocarmycin Analogs Incorporating the 2,3,10,10a-Tetrahydrocyclopropa[d]benzo[f]quinol-5-one Alkylation Subunit: Identification of Subtle Structural Features That Contribute to the Regioselectivity of the Adenine N3 Alkylation Reaction. J. Am. Chem. Soc. 1995;117:11647–11655. [Google Scholar]
- (17).Boger DL, Turnbull P. Synthesis and Evaluation of CC-1065 and Duocarmycin Analogs Incorporating the 1,2,3,4,11,11a-Hexahydrocyclopropa[c]naphtha[2,1-b]azepin-6-one (CNA) Alkylation Subunit: Structural Features that Govern Reactivity and Reaction Regioselectivity. J. Org. Chem. 1997;62:5849–5863. [Google Scholar]
- (18).(a) Warpehoski MA, Harper DE. Acid-Dependent Electrophilicity of Cyclopropylpyrroloindoles. Nature’s Masking Strategy for a Potent DNA Alkylator. J. Am. Chem. Soc. 1994;116:7573–7580. [Google Scholar]; (b) Warpehoski MA, Harper DE. Enzyme-like Rate Acceleration in the DNA Minor Groove. Cyclopropylpyrroloindoles as Mechanism-Based Inactivators of DNA. J. Am. Chem. Soc. 1995;117:2951–2952. [Google Scholar]
- (19).For example:Boger DL, Goldberg J, McKie JA. A Comparative Study of the Solvolysis Reactivity, Regioselectivity, and Stereochemistry of the Duocarmycin A and SA Alkylation Subunits. Bioorg. Med. Chem. Lett. 1996;6:1955–1960.
- (20).(a) Boger DL, Santillan A, Jr., Searcey M, Jin Q. Critical Role of the Linking Amide in CC-1065 and the Duocarmycins: Implications on the Source of DNA Alkylation Catalysis. J. Am. Chem. Soc. 1998;120:11554–11557. [Google Scholar]; (b) Boger DL, Santillan A, Jr., Searcey M, Jin Q. Synthesis and Evaluation of Duocarmycin and CC-1065 Analogues Containing Modifications in the Subunit Linking Amide. J. Org. Chem. 1999;64:5241–5244. doi: 10.1021/jo990452y. [DOI] [PubMed] [Google Scholar]
- (21).Boger DL, Yun W. CBI-TMI: Synthesis and Evaluation of a Key Analog of the Duocarmycins. Validation of a Direct Relationship Between Chemical Solvolytic Stability and Cytotoxic Potency and Confirmation of the Structural Features Responsible for the Distinguishing Behavior of Enantiomeric Pairs of Agents. J. Am. Chem. Soc. 1994;116:7996–8006. [Google Scholar]
- (22).Boger DL, Turnbull P. Synthesis and Evaluation of a Carbocyclic Analogue of the CC-1065 and Duocarmycin Alkylation Subunits: Role of the Vinylogous Amide and Implications on DNA Alkylation Catalysis. J. Org. Chem. 1998;63:8004–8011. [Google Scholar]
- (23).(a) Lin CH, Beale JM, Hurley LH. Structure of the (+)-CC-1065-DNA Adduct: Critical Role of Ordered Water Molecules and Implications for Involvement of Phosphate Catalysis in the Covalent Reaction. Biochemistry. 1991;30:3597. doi: 10.1021/bi00229a002. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Boyce CW, Johnson DS. pH Dependence of the Rate of DNA Alkylation for (+)-Duocarmycin SA and (+)-CCBI-TMI. Bioorg. Med. Chem. Lett. 1997;7:233–238. [Google Scholar]
- (24).MacMillan KS, Boger DL. An Additional Spirocyclization for Duocarmycin SA. J. Am. Chem. Soc. 2008;130:16521–16523. doi: 10.1021/ja806593w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Boger DL, Munk SA, Zarrinmayeh H, Ishizaki T, Haught J, Bina M. An Alternative and Convenient Strategy for Generation of Substantial Quantities of Singly 5′-32P-End-labeled Double-stranded DNA for Binding Studies: Development of a Protocol for Examination of Functional Features of (+)-CC-1065 and the Duocarmycins That Contribute to Their Sequence-Selective DNA Alkylation Properties. Tetrahedron. 1991;47:2661–2682. [PubMed] [Google Scholar]
- (26).(a) Boger DL, Garbaccio RM, Jin Q. Synthesis and Evaluation of CC-1065 and Duocarmycin Analogues Incorporating the iso-CI and iso-CBI Alkylation Subunits: Impact of Relocation of the C-4 Carbonyl. J. Org. Chem. 1997;62:8875–8891. [Google Scholar]; (b) Boger DL, Garbaccio RM. A Novel Class of CC-1065 and Duocarmycin Analogues Subject to Mitomycin-Related Reductive Activation. J. Org. Chem. 1999;64:8350–8362. doi: 10.1021/jo991301y. [DOI] [PubMed] [Google Scholar]
- (27).(a) Boger DL, Boyce CW. Selective Metal Cation Activation of a DNA Alkylating Agent: Synthesis and Evaluation of Methyl 1,2,9,9a-Tetrahydrocyclopropa[c]pyrido[3,2-e]indol-4-one-7-carboxylate (CPyI) J. Org. Chem. 2000;65:4088–4100. doi: 10.1021/jo000177b. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Wolkenberg SE, Boyce CW. A New Method of in Situ Activation for a Novel Class of DNA Alkylating Agents: Tunable Metal Cation Complexation and Activation. J. Am. Chem. Soc. 2000;122:6325–6326. [Google Scholar]; (c) Ellis DA, Wolkenberg SE, Boger DL. Metal Cation Complexation and Activation of Reversed CPyI Analogues of CC-1065 and Duocarmycin SA: Partitioning the Effects of Binding and Catalysis. J. Am. Chem. Soc. 2001;123:9299–9306. doi: 10.1021/ja010769r. [DOI] [PubMed] [Google Scholar]
- (28).(a) Ambroise Y, Boger DL. The DNA Phosphate Backbone is Not Involved in Catalysis of the Duocarmycin and CC-1065 DNA Alkylation Reaction. Bioorg. Med. Chem. Lett. 2002;12:303–306. doi: 10.1016/s0960-894x(01)00740-5. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Garbaccio RM. Are the Duocarmycin and CC-1065 DNA Alkylation Reactions Acid-Catalyzed? Solvolysis pH-Rate Profiles Suggest They Are Not. J. Org. Chem. 1999;64:5666–5669. doi: 10.1021/jo990762g. [DOI] [PubMed] [Google Scholar]
- (29).Boger DL, Boyce CW, Garbaccio RM, Goldberg J. CC-1065 and the Duocarmycins: Synthetic Studies. Chem. Rev. 1997;97:787–828. doi: 10.1021/cr960095g. [DOI] [PubMed] [Google Scholar]
- (30).(a) Boger DL, Ishizaki T, Wysocki RJ, Jr., Munk SA, Kitos PA, Suntornwat O. Total Synthesis and Evaluation of (±)-N-(tert-Butyloxycarbonyl)-CBI, (±)-CBI-CDPI1, and (±)-CBI-CDPI2: CC-1065 Functional Agents Incorporating the Equivalent 1,2,9,9a-Tetrahydrocycloprop[1,2-c]benz[1,2-e]indol-4-one (CBI) Left-Hand Subunit. J. Am. Chem. Soc. 1989;111:6461–6463. [Google Scholar]; (b) Boger DL, Ishizaki T, Kitos PA, Suntornwat O. Synthesis of N-(tert-Butyloxycarbony1)-CBI, CBI, CBI-CDPI1, and CBI-CDPI2: Enhanced Functional Analogues of CC-1065 Incorporating the 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) Left-Hand Subunit. J. Org. Chem. 1990;55:5823–5832. [Google Scholar]; (c) Boger DL, Munk SA. DNA Alkylation Properties of Enhanced Functional Analogs of CC-1065 Incorporating the 1,2,9,9a-Tetrahydrocycloprop[1,2-c]benz[1,2-e]indol-4-one (CBI) Alkylation Subunit. J. Am. Chem. Soc. 1992;114:5487–5496. [Google Scholar]; (d) Boger DL, Yun W, Teegarden BR. An Improved Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI): A Simplified Analogue of the CC-1065 Alkylation Subunit. J. Org. Chem. 1992;57:2873–2876. [Google Scholar]; (e) Boger DL, McKie JA. An Efficient Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI): An Enhanced and Simplified Analog of the CC-1065 and Duocarmycin Alkylation Subunits. J. Org. Chem. 1995;60:1271–1275. [Google Scholar]; (f) Boger DL, McKie JA, Boyce CW. Asymmetric Synthesis of the CBI Alkylation Subunit of the CC-1065 and Duocarmycin Analogs. Synlett. 1997:515–517. [Google Scholar]; (g) Boger DL, Boyce CW, Garbaccio RM, Searcey M. Synthesis of CC-1065 and Duocarmycin Analogs via Intramolecular Aryl Radical Cyclization of a Tethered Vinyl Chloride. Tetrahedron Lett. 1998;39:2227–2230. [Google Scholar]; (h) Kastrinsky DB, Boger DL. Effective Asymmetric Synthesis of 1,2,9,9a-Tetrahydrocyclopropa[c]benzo[e]indol-4-one (CBI) J. Org. Chem. 2004;69:2284–2289. doi: 10.1021/jo035465x. [DOI] [PubMed] [Google Scholar]; (i) Boger DL, Yun W, Han N. 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) Analogs of CC-1065 and the Duocarmycins: Synthesis and Evaluation. Bioorg. Med. Chem. 1995;3:1429–1453. doi: 10.1016/0968-0896(95)00130-9. [DOI] [PubMed] [Google Scholar]
- (31).Boger DL, Han N, Tarby CM, Boyce CW, Cai H, Jin Q, Kitos PA. Synthesis, Chemical Properties, and Preliminary Evaluation of Substituted CBI Analogs of CC-1065 and the Duocarmycins Incorporating the 7-Cyano-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one Alkylation Subunit: Hammett Quantitation of the Magnitude of Electronic Effects on Functional Reactivity. J. Org. Chem. 1996;61:4894–4912. doi: 10.1021/jo952033g. [DOI] [PubMed] [Google Scholar]
- (32).(a) Boger DL, McKie JA, Cai H, Cacciari B, Baraldi PG. Synthesis and Properties of Substituted CBI Analogs of CC-1065 and the Duocarmycins Incorporating the 7-Methoxy-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one Alkylation Subunit: Magnitude of Electronic Effects on the Functional Reactivity. J. Org. Chem. 1996;61:1710–1729. doi: 10.1021/jo952033g. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, McKie JA, Han N, Tarby CM, Riggs HW, Kitos PA. A Hammett Correlation for CC-1065 and Duocarmycin Analogs: Magnitude of Substituent Electronic Effects on Functional Reactivity. Bioorg. Med. Chem. Lett. 1996;6:659–664. [Google Scholar]
- (33).(a) Boger DL, Stauffer F, Hedrick MP. Substituent Effects Within the DNA Binding Subunit of CBI Analogues of the Duocarmycins and CC-1065. Bioorg. Med. Chem. Lett. 2001;11:2021–2024. doi: 10.1016/s0960-894x(01)00372-9. [DOI] [PubMed] [Google Scholar]; (b) Parrish JP, Kastrinsky DB, Stauffer F, Hedrick MP, Hwang I, Boger DL. Establishment of Substituent Effects in the DNA Binding Subunit of CBI Analogues of the Duocarmycins and CC-1065. Bioorg. Med. Chem. 2003;13:3815–3838. doi: 10.1016/s0968-0896(03)00194-9. [DOI] [PubMed] [Google Scholar]
- (34).Boger DL, Hughes TV, Hedrick MP. Synthesis, Chemical Properties, and Biological Evaluation of CC-1065 and Duocarmycin Analogues Incorporating the 5-Methoxycarbonyl-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one Alkylation Subunit. J. Org. Chem. 2001;66:2207–2216. doi: 10.1021/jo001772g. [DOI] [PubMed] [Google Scholar]
- (35).Boger DL, Yun W. Role of the CC-1065 and Duocarmycin N2 Substituent: Validation of a Direct Relationship Between Solvolysis Chemical Stability and in Vitro Biological Potency. J. Am. Chem. Soc. 1994;116:5523–5524. [Google Scholar]
- (36).(a) Boger DL, Ishizaki T. Resolution of a CBI Precursor and Incorporation into the Synthesis of (+)-CBI, (+)-CBI-CDPI1, (+)-CBI-CDPI2: Enhanced Functional Analogs of (+)-CC-1065. A Critical Appraisal of a Proposed Relationship Between Electrophile Reactivity, DNA Binding Properties, and Cytotoxic Potency. Tetrahedron Lett. 1990;31:793–796. [Google Scholar]; (b) Boger DL, Ishizaki T, Sakya SM, Munk SA, Kitos PA, Jin Q, Besterman JM. Synthesis and Preliminary Evaluation of (+)-CBI-indole2: An Enhanced Functional Analog of (+)-CC-1065. Bioorg. Med. Chem. Lett. 1991;1:115–120. [Google Scholar]; (c) Boger DL, Munk SA, Ishizaki T. (+)-CC-1065 DNA Alkylation: Observation of an Unexpected Relationship between Cyclopropane Electrophile Reactivity and the Intensity of DNA Alkylation. J. Am. Chem. Soc. 1991;113:2779–2780. [Google Scholar]
- (37).LaBarbera DV, Skibo EB. Solution Kinetics of CC-1065 A-Ring Opening: Substituent Effects and General Acid/Base Catalysis. J. Am. Chem. Soc. 2006;128:3722–3727. doi: 10.1021/ja057289a. [DOI] [PubMed] [Google Scholar]
- (38).MacMillan KS, Nguyen T, Hwang I, Boger DL. Total Synthesis and Evaluation of iso-Duocarmycin SA and iso-Yatakemycin. J. Am. Chem. Soc. 2009;131:1187–1194. doi: 10.1021/ja808108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tichenor MS, MacMillan KS, Stover JS, Wolkenberg SE, Pavani MG, Zanella L, Zaid AN, Spalluto G, Rayl TJ, Hwang I, Baraldi PG, Boger DL. Rational Design, Synthesis, and Evaluation of Key Analogues of CC-1065 and the Duocarmycins. J. Am. Chem. Soc. 2007;129:14092–14099. doi: 10.1021/ja073989z. [DOI] [PMC free article] [PubMed] [Google Scholar]