


Abstract
Initiation of retroviral genomic RNA dimerisation is mediated by the mutual interaction of the dimerisation initiation site (DIS) stem–loops near to the 5′ end of the RNA. This process is thought to involve formation of a transient ‘kissing’ complex over the self-complementary loop bases, which then refolds into a more stable extended interaction. We have developed a novel experimental system that allows us to clearly detect the extended duplex in vitro. Ribozyme sequences were incorporated into or adjacent to the type 1 human immunodeficiency virus DIS stem, leading to the formation of a functional ribozyme only in the extended duplex conformer. Here we show that extended duplex formation results in ribozyme cleavage, thus demonstrating the double-stranded nature of the extended complex and confirming that refolding occurs via melting of the DIS stems. Loop complementarity is essential for extended duplex formation but no sequence requirements for the loops were observed. Efficiency of extended duplex formation is dependent on the strength of the loop–loop interaction, temperature, the magnesium concentration and is strongly accelerated by the viral nucleocapsid protein NCp7. Our ribozyme-coupled approach should be applicable to the analyses of other refolding processes involving RNA loop–loop interactions.
INTRODUCTION
Mature retroviral particles contain two positive-strand genomic RNAs, which are non-covalently linked within a region close to their 5′ ends termed the dimer linkage structure (DLS). Retroviral RNA dimerisation is considered to play an important role in such essential processes like encapsidation, maturation or reverse transcription (1,2). Within the DLS of type 1 human immunodeficiency virus (HIV-1), a highly conserved stem–loop structure was shown to be specifically required for the initiation of genomic RNA dimerisation. Deletion of this sequence element called the dimerisation initiation site (DIS) prevents dimerisation and leads to reduced infectivity as well as drastically reduced replication rates (3–7). The DIS element is a 35 nt stem–loop structure with an internal bulge and a hexameric palindromic sequence within the loop, which is flanked by highly conserved purine nucleotides (Fig. 1a). The most common self-complementary sequences in the DIS loop are GCGCGC (subtype B or LAI variant as well as subtype D) and GUGCAC (subtype A or MAL variant as well as subtype G and C of group O). Two purine nucleotides are usually located 5′ adjacent to the palindromic sequence and one purine nucleotide at the 3′ side. The DIS stem–loops were shown to interact with each other through Watson–Crick base pairing of their palindromic loop sequences forming a structure called a kissing complex. In the current model of DIS-mediated dimerisation, this kissing complex is converted into a more stable extended structure. While the loop–loop base pairing is maintained, the intramolecular base pairs of the DIS stems are opened and become involved in intermolecular interactions, forming a double-stranded RNA (8,9).
Figure 1.
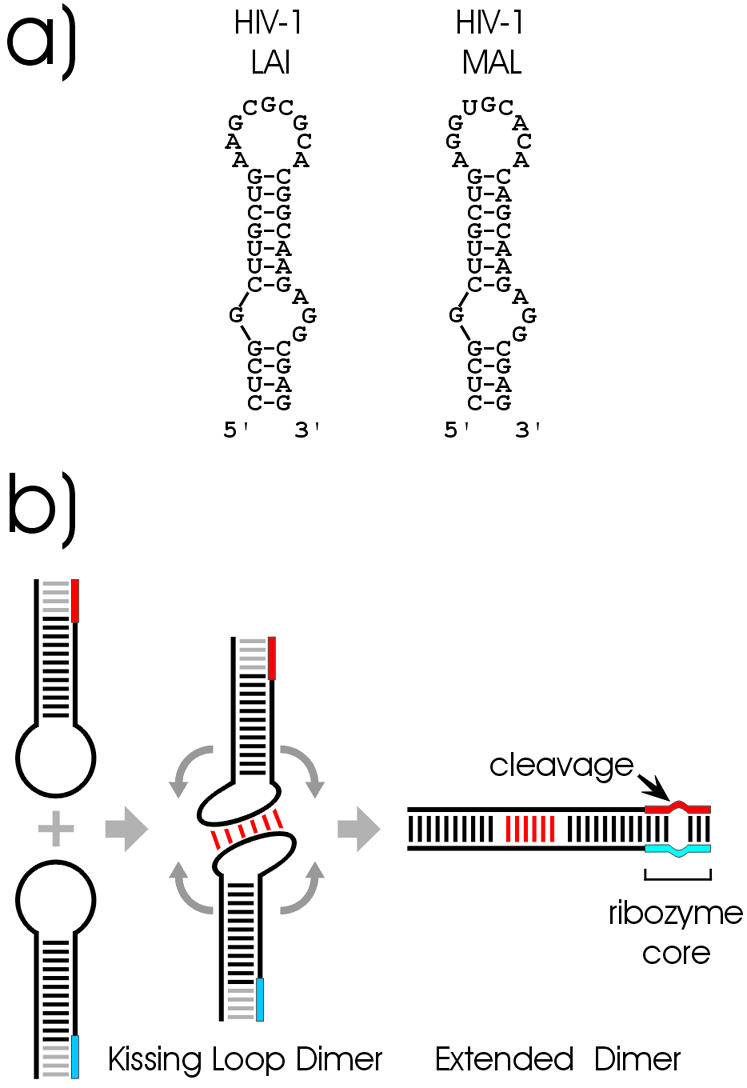
(a) Secondary structure of wild-type HIV-1 LAI and HIV-1 MAL DIS hairpin loops. (b) Principle of the ribozyme coupled dimerisation assay. Heterodimers are formed by kissing interaction between the loop bases of two RNA stem–loops, each containing one part of a ribozyme (red and blue boxes). Only structural rearrangement into a double-stranded extended dimer, but not the kissing complex, leads to the formation of a functional ribozyme.
Short synthetic RNAs containing the DIS hairpin are able to dimerise spontaneously in vitro, whereas RNAs resembling the retroviral 5′ region but lacking this hairpin fail to do so. In vitro experiments revealed that RNAs corresponding to the whole DIS stem–loop structure are able to form complexes of different stabilities depending on the incubation conditions. At 37°C, a less stable complex, formed via base pairing of the complementary loop sequences, is found and corresponds to the kissing complex. Incubation of these RNAs at 55°C generates a more stable complex (10).
X-ray and NMR structural data is available for subtypes A and B HIV-1 DIS both in the kissing complex as well as in the extended conformation (11–13). The crystal structures obtained from 23mer RNAs that contain the loop as well as the apical stem of the kissing complex of both viral subtypes (MAL and LAI) are virtually identical. In both cases, the two hairpins are in perfect coaxial alignment, thereby strongly resembling the structure of the extended duplex. In contrast, the solution structure obtained by NMR reveals a different picture, with the loop closing bases unpaired and the kissing loop helix not in A-form geometry, but distorted and kinked. Compared with the X-ray structure, the NMR structure shows an entirely different orientation of the 5′ unpaired loop purines, suggesting that these bases are involved in non-canonical base pairing with partial stacking (11).
Recently, fluorescence techniques were used to demonstrate the refolding of small DIS-like RNAs in the presence of the HIV nucleocapsid protein NCp7 (14). In the work presented here, we developed an experimental system in which ribozyme activity is dependent on the formation of the extended duplex, allowing its detection via the formation of a cleavage product (Fig. 1b). Two different ribozyme systems were used in our study, the hammerhead ribozyme (15) and the synthetic leadzyme, which had been isolated via in vitro selection (16). Using these two systems, we investigated structural requirements, the influence of temperature and magnesium concentration as well as the impact of the nucleocapsid protein NCp7 (17,18) on extended duplex formation in vitro.
MATERIALS AND METHODS
Strains and plasmids
Escherichia coli strain DH5α™ and pUC19 plasmid were used for all the cloning work. Synthetic DNA oligonucleotides were purchased from VBC Genomics. Double-stranded DNA fragments were obtained by PCR and used for cloning. The oligonucleotides used for cloning of H1 and H2 were: H1a, 5′-CCCAAGCTTCCAAGTAATACGACTCACTATAGCGTCGGCTTGCTGAAGCGCGCACGGC-3′; H1b, 5′-CGGGGTACCGATATCGCGAGACCACGGTGGTTTCGGCGTCGC CTCTTGCCGTGCGCGCTTCAGCAAGCCG-3′; H2a, 5′-CCCAAGCTTCCAAGTAATACGACTCACTATAGCGACTGATGAGGCGTCGGCTTGCTGAAGCGCGC-3′; H2b, 5′-CCAATGCATTGGTTCTGCAGCGTCGCCTCTTGCCGTGCGCGCTTCAGCAAGCCGACGCCTCATCAG-3′.
The oligonucleotides were annealed and filled in with T4 DNA polymerase. The resulting double-stranded DNA contained restriction sites (HindIII and KpnI for H1, HindIII and PstI for H2) that were used to clone the fragment into the pUC19 plasmid. The resulting clones were then sequenced. Transcription was performed after a PCR amplification step using the following primers: H1fw, 5′-CCAAGTAATACGACTCACTATAGGCGTCGGCTTGCTGAACGCGCACGG-3′; H1rev, 5′-TTTTTTCGCGAGACCACGGTGGT TTCGGCGTCGCCTCTTGCCGTGCGCGCTTCAGCAAGCC-3′; H2fw, 5′-CCAAGTAATACGACTCACTATAGGCGACTGATGAGGCGTCGGC TTG-3′; H2rev, 5′-GCGTCG CCTCTTGCCGTGCGCGCTTCAGCAAGCCGACGCCTCATCAGTCGC-3′.
The following oligonucleotides were used for construction of L1 and L2: L1a, 5′-CCCAAGCTTCCAAGTAATACGACTCACTATAGCGACCGAGCCAGCTGAAGCGCGCACGGCTGGCGTCGCGAATTCC-3′; L1b, 5′-GGAATTC GCGACGCCAGCCGTGCGCGCTTCAGCTGGCTCGGTCGCTATAGTGAGTCGTATTACTTGGAAGCTTGGG-3′; L2a, 5′-CCCAAGCTTCCAAGTAATACGACTCACTATAGCGACGCCAGCUGAAGCGCGCACGGCUGGGAGUCGCGAATTCC-3′; L2b, 5′-GGAATTCGCGACTCCCA GCCGTGCGCGCTTCAGCTGGCGTCGCTATAGTGAGTCGTATTACTTGGAAGCTTGGG-3′.
The oligonucleotides were annealed and the resulting double-stranded DNA was cleaved (HindIII and EcoRI) and cloned into the pUC19 plasmid. The following primers were used to amplify the template for transcription: T7fw, 5′-CCAAGTAATACGACTCACTATAG-3′; L1rev, 5′-GCGACGCCAGCCGTGCG-3′; L2rev, 5′-GCGACTCCCAGCCGTGCGCGC-3′.
GC1 mutant constructs were obtained from cloned wild-type sequences by removing four of the six palindromic loop bases using BssH II cleavage subsequent treatment with nuclease S1 as described by Berkhout and van Wamel (5).
In vitro transcription and labelling
Template DNA for in vitro transcription was amplified by PCR from plasmid vectors with the required insert. The gel-purified DNA template was transcribed using the T7 Megashortscript Kit from Ambion. The transcribed RNA was purified by denaturing PAGE and eluted from the gel with 0.3 M NaOAc. After precipitation, RNA was redissolved in water and concentrations were determined by UV spectrometry. Subsequently, H1 or L1 RNAs were dephosphorylated using alkaline phosphatase and 5′-labelled using T4 polynucleotide kinase and [γ-32P]ATP following the manufacturer’s instructions.
In vitro dimerisation assay
Both RNAs (H1 and H2 or L1 and L2) were heated separately for 1 min at 90°C and immediately chilled on ice water in order to prevent homodimerisation. After 5 min on ice, the RNAs were added to the reaction mixture. The reaction was performed in 50 mM Tris pH 7.5 and 10 mM MgCl2 unless indicated otherwise. A 100-fold excess of unlabelled L2 RNA (usually 10 nM L1 and 1000 nM L2) for the leadzyme system and a 20-fold excess of unlabelled H2 RNA (10 nM H1 and 200 nM H2) for the hammerhead system were regularly used for heterodimer formation. The reaction was then allowed to progress at the indicated temperature and samples were taken at different time points. In the case of the leadzyme system, lead acetate (200 µM PbOAc) was added to these samples after the refolding reaction and cleavage was allowed to occur for 30 min at room temperature. The reaction was stopped by adding RNA loading buffer (7 M urea) and the reaction products were separated by denaturing PAGE (20% polyacrylamide). The results were visualised and quantified using a phosphoimager. In each case at least two independent experiments were performed. The cleavage rates (Kobs) were determined assuming a simple exponential decay of the full-length RNA and the data were fitted using the least-squares method with the Kaleidagraph software.
Semi-native polyacrylamide gel
The 15% (w/v) polyacrylamide gel was poured and assembled using a BioRad Mighty Small gel apparatus. Samples were taken up in 15% glycerol and the gel was run at 100 V using 1× TBE (89 mM Tris, 89 mM borate, 2 mM EDTA) running buffer. Unlabelled nucleic acids were stained by soaking the gel in 1× TBE containing ethidium bromide (10 µg/ml) and subsequent washing with water.
Partial RNAse T1 digest
End-labelled RNA (0.2 pmol) was incubated together with 3 µg of tRNA and 15 U of RNAseT1 in loading buffer (total volume 10 µl) for 10 min at 50°C and directly applied onto a denaturing 20% polyacrylamide gel.
Dimerisation in the presence of NCp7 protein
Lyophilised, chemically synthesised NCp7 [obtained from L. Scherer and J. Rossi, Beckman Research Center, City of Hope (19)] was dissolved and stored in degassed storage buffer (20 mM Tris pH 7.5, 50 mM NaCl, 10 mM DTT, 0.25 mM PMSF, 10% glycerol and 20 µM ZnCl2). Dimerisation assays with NCp7 were performed the same way as described above. The protein was added on ice immediately before the reaction was started (10 µM NCp7, a 10-fold excess of protein over RNA). The reaction was performed at 37°C. Five microlitre samples were taken at different time points, proteinase K was added and incubated for 1 h at 37°C. PbOAc was then added to a final concentration of 200 µM and cleavage was allowed to occur for 30 min at room temperature. The reaction was stopped with RNA loading buffer and samples kept on ice until they were loaded on a denaturing 20% polyacrylamide gel.
RESULTS
Construction of HIV-1-like DIS stem–loops containing ribozyme domains
RNA molecules containing DIS stem–loop elements (Fig. 2), which fold into active ribozymes were constructed with the loop sequences of HIV-1 (5′-aaGCGCGCa-3′ of subtype B, LAI variant) using both the hammerhead- (Fig. 2a) or the Pb2+-dependent (leadzyme) (Fig. 2b) ribozymes (15,16). The entire wild-type DIS stem–loop was fused to either 5′ or 3′ parts of the hammerhead ribozyme (Fig. 2a) and in the second approach, leadzyme sequences were directly incorporated into DIS-like stem–loops (Fig. 2b). While the hammerhead system uses the complete DIS wild-type sequence including the G/AGG internal bulge and has partially single-stranded ribozyme sequences attached, the DIS stem–loop in the leadzyme system differs from the wild-type stem–loop in sequence but is structurally similar as the RNAs are fully closed stem–loops. Additionally, the Pb2+ dependence of the leadzyme allows us to uncouple refolding from the cleavage step.
Figure 2.

Hammerhead ribozyme and leadzyme systems. (a) H1 and H2 RNAs containing parts of the hammerhead ribozyme (left) and the hammerhead forming extended dimer (right). (b) L1 and L2 RNAs containing parts of the leadzyme (left) and the leadzyme forming extended dimer (right). Leadzyme and hammerhead core ribozyme sequences are shown in blue and red, respectively. The arrows indicate the cleavage sites of the ribozymes.
Extended duplex formation results in specific ribozyme cleavage
To determine whether heterodimerisation and subsequent refolding leads to specific ribozyme cleavage, we performed dimerisation assays at 37 and 55°C as shown in Figure 3. In vitro transcribed H1 and L1 RNAs, which should be cleaved upon formation of functional ribozymes, were radioactively labelled at their 5′ ends. We regularly used a 20- (hammerhead system) to 100-fold (leadzyme system) excess of unlabelled H2 or L2 RNAs to compete out H1 or L1 homodimer formation. We observed the appearance of ribozyme-specific cleavage products for the hammerhead and leadzyme systems (Fig. 3). The observed products correspond to the predicted 54 nt shortened H1 RNA or 6 nt L1 fragment as confirmed by RNAse T1 analysis. The amount of cleavage product and therefore the efficiency of extended duplex formation are strongly increased when the reaction is carried out at 55°C compared with 37°C as shown for the hammerhead system in Figure 3a (right). This is in agreement with previous data that show induction of extended duplex formation of DIS stem–loop RNAs at 55°C (10). Furthermore, specific ribozyme cleavage is dependent on the presence of both L1 and L2 (Fig. 3b) or H1 and H2 (data not shown) RNAs. Additionally, cleavage products are formed only when Pb2+ is added to the leadzyme system (Fig. 3b, compare lanes 13 and 14) thereby demonstrating that the observed cleavage products result from the formation of a functional ribozyme as a consequence of extended duplex formation.
Figure 3.

Dimerisation assay with hammerhead and leadzyme constructs. (a) Secondary structure of H1 RNA as proposed by mfold (33) and confirmed by RNase T1 digestion (left). Extended duplex formation between H1 and H2 RNAs at 37 and 55°C. Cleavage products of radiolabelled H1 RNA are separated by denaturing PAGE (right). (b) Secondary structure of L1 RNA (left). Denaturing PAGE analysis of leadzyme-dependent cleavage reaction at 55°C of L1 and L2 RNAs in the presence (lanes 8–13) or absence (lane 14) of 200 µM Pb2+. Control reaction with L1 RNA alone in the presence (lanes 1–6) or absence (lane 7) of Pb2+. T1, OH– and UT lanes represent RNA partially digested with RNAse T1, partial alkaline hydrolysis or untreated RNA, respectively. Red (DIS loop) and orange (ribozyme sequences) dots indicate T1 cleavage sites on the gels and the corresponding G residues within the secondary structures; blue arrows mark sites of specific ribozyme cleavage. Asterisks indicate radioactively labelled RNAs.
To test the starting state of the hairpin RNAs, a semi-native PAGE analysis was performed showing that by heating the RNAs to 90°C followed by immediate cooling on ice water, the RNAs are monomers (Fig. 4a). Lanes 1 and 2 show homodimer formation after slow cooling and incubation at 37°C, while lanes 3 and 4 show only monomers after fast cooling by putting samples on ice water immediately after heating.
Figure 4.

Influence of loop complementarity on extended duplex formation. (a) Semi-native PAGE of H1 (lanes 1–4) and H1GC1 (lanes 5–8) RNAs. All samples were heated for 1 min to 90°C and subsequently cooled slowly to 37°C (lanes 1 and 5), cooled slowly to 37°C and incubated at 37°C for 2 h (lanes 2 and 6), put on ice water for 2 min (lanes 3 and 7) or put on ice water and incubated at 37°C for 2 h (lanes 4 and 8). The closed arrow indicates homodimeric and the open arrow the monomeric form of H1. (b) Secondary structure representations of H2GC1 and H1GC1 RNAs and quantification of cleavage efficiency following dimerisation assays at 37°C with different hammerhead construct combinations (right). (c) L1/L2 and LA/L2 dimerisation assays at 55°C (left). Secondary structure representations of L1A, L2GC1 and L1GC1 RNAs (middle). Quantification of cleavage efficiency following dimerisation assays with wild-type loop sequences (L1/L2) and sequences with reduced (L1GC1/L2GC1) or no complementarity (L1A/L2) (right). The remaining labelling is as described in Figure 3.
Kissing loop interaction is essential for subsequent extended duplex formation
If extended duplex formation is dependent on a preceding kissing complex, mutations that reduce or abolish loop complementarity should affect reaction efficiencies. We used the previously described GC1 mutant which lacks four of the six palindromic loop nucleotides (H1GC1, H2GC1 and L1GC1, L2GC1 constructs, Fig. 4) (5). Compared with wild-type loop sequences, GC1 constructs showed strongly reduced reaction efficiencies but were still capable of forming extended duplexes. This is observed at 37°C in the presence of 50 mM MgCl2 as shown with the hammerhead system (Fig. 4b) as well as at 55°C in the presence of 10 mM MgCl2 as shown with leadzyme RNAs (Fig. 4c). We also tested combinations of H1 and H2GC1 or H2 and H1GC1 RNAs, which have only two complementary nucleotides in their loops, and the extended dimers of these two combinations contain two unpaired GC pairs. These combinations show a strongly reduced capacity to form duplexes compared with the wild type. These results indicate that a reduced complementarity is sufficient to induce extended duplex formation, although the stability of the kissing complex influences the efficiency.
To determine whether the complete abolishment of loop–loop complementarity would also abrogate extended duplex formation, the wild-type DIS loop was substituted by six adenosines. This construct, termed L1A, is unable to form loop base pairs with L2 RNA. By testing dimerisation at 55°C, the L1A/L2 RNAs do not show extended duplex formation at all (Fig. 4c). Therefore, kissing complex formation is an essential prerequisite for extended duplex formation.
Influence of different loop sequences on extended duplex formation
Of 64 possible palindromic loop sequences, only two have been found to occur naturally, namely 5′-GCGCGC-3′ and 5′-GUGCAC-3′ (20). To compare extended duplex formation of the two major HIV-1 subtypes, the AAGCGCGCA sequence of subtype B was substituted with the loop sequence of the other major HIV-1 subtype AGGUGCACA (subtype A). The constructs derived from L1 and L2 were termed L1MAL and L2MAL, respectively. The efficiency of extended duplex formation is comparable with that observed for L1 and L2 RNAs, although the activity of the MAL constructs was slightly, but reproducibly less, efficient than the L1 and L2 RNAs with LAI sequences (compare Fig. 5a with Fig. 4c).
Figure 5.

Influence of different loop and apical stem sequences on extended duplex formation. (a) Secondary structure representation of L1MAL/L2MAL and L1nsc/L2nsc RNAs (left) and corresponding cleavage rates in dimerisation assays at 55°C (right). (b) Secondary structure of constructs with shorter apical stems (L1s/L2s) derived from L1/L2 (left) and cleavage efficiency following dimerisation assays at 37 or 55°C (right).
In order to investigate whether extended duplex formation is possible with non-naturally occurring sequences another set of constructs was designed. L1nsc and L2nsc RNAs contain the non-self complementary loop sequences 5′-AAGAGAGGA-3′ and 5′-AACUCUCCA-3′, respectively (Fig. 5a). These non-self-complementary sequences are unable to form homodimers, which might interfere with the dimerisation reaction. Additionally, these RNAs do not have a central GC pair within the complementary hexanucleotide sequence of the loop, which had previously been suggested to be essential for dimerisation (8,9,21). As shown in Figure 5a, extended duplex formation of the nsc constructs is slightly more efficient than that of the MAL constructs. Therefore, dimerisation can be promoted by kissing complexes formed by sequences other than the wild-type palindromes. Neither self-complementarity nor the central GC nucleotides are required for the refolding reaction.
Finally, L1MAL and L2nsc RNAs were tested for their ability to form extended duplexes. These two constructs are only complementary for two loop bases with four mismatched bases. As for the GC1 RNAs, this sequence combination still shows extended duplex formation to some extent although the reaction efficiency is strongly reduced (Fig. 5a). This demonstrates that the strength of the loop–loop interaction is a key parameter for dimerisation.
Influence of temperature, magnesium concentration and stem length on extended duplex formation
As mentioned previously, incubation at 55°C enhances extended duplex formation in various small DIS-like RNAs. Some of these RNAs with short stems form the extended dimer even at lower temperatures (22). This is not unexpected as extended duplex formation requires opening of all DIS stem base pairs (12 in the case of L1 RNA) so they can then undergo intermolecular base pairing. This refolding process represents a kinetic barrier that has to be overcome to establish the thermodynamically more stable extended duplex conformation. We wanted to test the effect of higher temperature and of a shortened stem on extended duplex formation as well as the influence of the Mg2+ concentration on duplex formation.
RNAs derived from L1 and L2 constructs (Fig. 5b) were designed in which 2 bp from the upper DIS stem were deleted. These shorter constructs, termed L1s and L2s, were tested at 37 and 55°C for their ability to form the extended duplex. At 37°C, both L1/L2 and L1s/L2s show similar behaviour. At 55°C we observed a slight but reproducible increase in cleavage efficiency that lies within the error range of the experiments. Therefore, weakening of the upper stem by only 2 bp is not sufficient to significantly increase the efficiency of extended duplex formation (Fig. 5b).
Figure 6a shows that extended duplex formation of L1 and L2 RNAs is enhanced with increasing temperature up to 55°C. A strong temperature dependence was also observed for the hammerhead system, which shows complete extended duplex formation at 55°C after 90 min (Fig. 3a and data not shown). Since at any temperature the known rate constants for ribozyme cleavage far exceed the rates of cleavage observed in our systems, the limiting step appears to be refolding rather than catalysis. Therefore, temperature-dependent differences in the observed rate constants likely reflect the rate of refolding.
Figure 6.
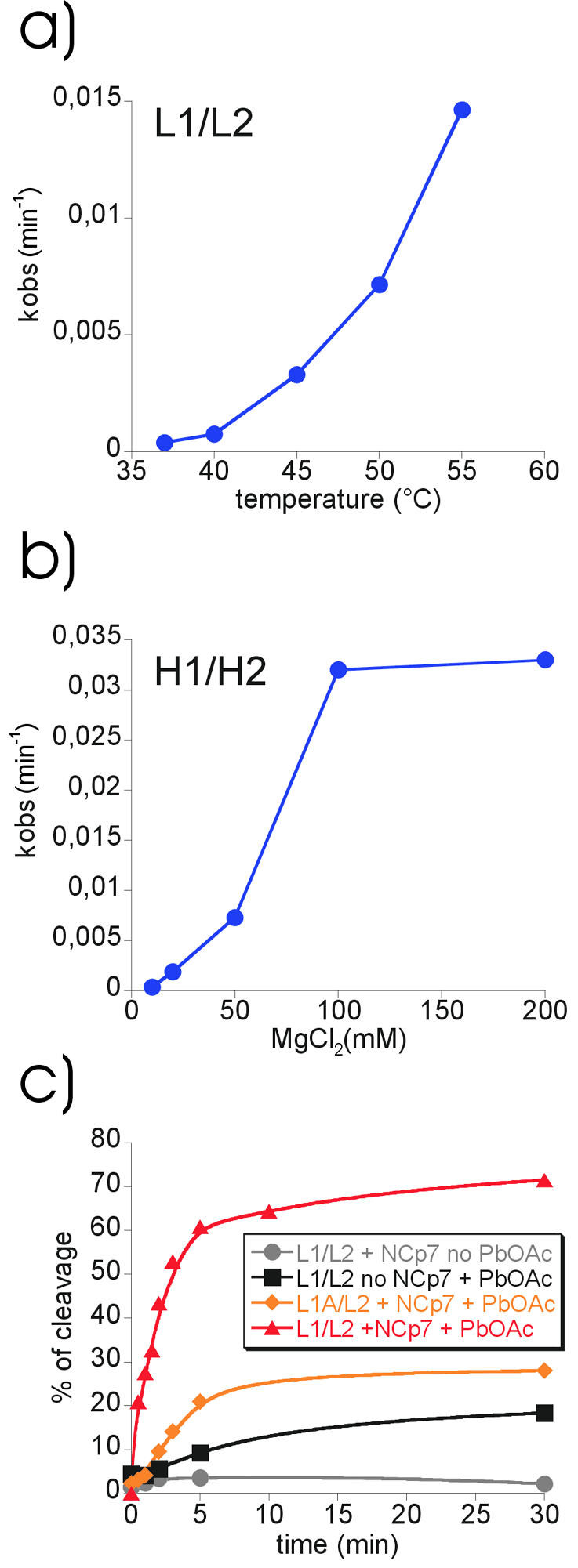
Influence of external parameters on extended dimer formation. (a) Cleavage efficiencies of leadzyme L1/L2 constructs following dimerisation at different temperatures. Kobs (min–1) values were plotted against temperature. (b) Influence of magnesium concentration on the dimerisation of H1/H2 hammerhead constructs. Kobs (min–1) values were plotted against magnesium concentration. (c) Influence of added HIV-1 NCp7 protein on extended dimer formation at 37°C. L1/L2 or L1A/L2 dimerisation in the absence or presence of the nucleo capsid protein NCp7 or Pb2+. Quantification of cleavage efficiencies was plotted against time.
The dependence of extended duplex formation on Mg2+ concentration was tested using the hammerhead system because high Mg2+ concentrations interfere with lead cleavage (data not shown). The optimal Mg2+ concentration for hammerhead ribozyme activity is usually 10 mM MgCl2 (23). We found that extended duplex formation is strongly enhanced by the presence of high Mg2+ concentrations up to 100 mM MgCl2 (Fig. 6b). Mg2+ might stabilise the kissing complex, which would increase the likelihood of subsequent extended duplex formation.
NCp7 strongly accelerates extended duplex formation
It has been suggested that NCp7 plays a critical role in HIV genomic RNA maturation. Indeed, it has been shown that NCp7 is able to convert low stability retroviral RNA dimers into more stable dimers in vitro (24–27). NCp7 also catalyses conversion of small DIS-like RNAs (<25 nt) into the extended duplex form (14). However, the specificity of this interaction is uncertain since NCp7 also acts as a general nucleic acid chaperone. It has been shown to posses nucleic acid annealing and strand exchange activity in vitro (28,29), RNA chaperone activity in vivo (30) and it also accelerates cleavage of the hammerhead ribozyme (19).
We set out to test whether the NCp7 protein would also enhance extended duplex formation in our system. To discriminate between general RNA–RNA annealing activity and a more specific action we used L1 and L2 RNAs but also the combination L1A and L2, which completely lacks loop complementarity. As shown in Figure 6c, NCp7 strongly increases extended duplex formation between L1 and L2 RNAs. Sixty percent of the RNAs are refolded after only 5 min at 37°C compared with 10% in the absence of NCp7 (Fig. 6c, compare triangles and squares). The reaction levels off to reach ∼70% completion after 30 min. L1A/L2 RNAs, which lack loop complementarity and do not show any extended duplex formation at 55°C in the absence of NCp7, do react in the presence of NCp7. We conclude that NCp7 strongly enhances specific extended duplex formation in our system, but to some extent also of RNAs without loop complementarity via its general nucleic acid structure destabilising activity.
DISCUSSION
Here we present a novel experimental approach to monitor refolding of two RNA hairpins into an extended duplex. The system consists of two HIV-1-derived DIS hairpins, each fused to one half of a ribozyme. Refolding of the heterodimeric DIS kissing complex into a partially double-stranded extended RNA duplex leads to formation of an active ribozyme and a specific cleavage event. The advantage of this system over previous analysis methods using native gel electrophoresis is the clear discrimination between kissing complex and extended duplex due to the formation of a cleavage product. The reversibility of the folding process reduces the applicability of native gel electrophoresis. The ribozyme system results in an irreversible and easily detectable product allowing a wide range of external parameters and structural requirements to be tested. Our approach clearly demonstrates that ribozymes are useful tools for the monitoring of RNA refolding processes.
Formation of the active ribozyme after refolding of the hairpins into the extended duplex clearly demonstrated the double-stranded nature of the extended dimers. Our results further confirm the current model of HIV-1 DIS dimerisation involving refolding of the kissing complex with melting of the stems into a partially double-stranded dimer and rule out a hypothetical mechanism for extended duplex formation involving symmetric cleavage of both chains and subsequent religation (13). From crystal structure data, it was found that the 2′ OH group of the second purine residue 5′ to the complementary sequence is in favourable position for an in line attack on the neighbouring 3′ phosphate. A mechanism involving a nucleophilic attack leading to strand scission, followed by a dangling movement of this base and cross religation of both RNAs was proposed. Such a mechanism would join both DIS stem–loop RNAs within their loop regions keeping the initial stems intact. If this were true, both parts of the ribozyme would remain separated from each other and would be unable to form a functional ribozyme. Our observation that extended duplex formation leads indeed to ribozyme cleavage therefore strongly supports the refolding model with the melting of the stems.
We further show that formation of a kissing complex through interaction of complementary loop nucleotides is an essential prerequisite for extended duplex formation. In contrast to previous reports investigating viral RNA dimerisation in vitro, we found that neither self-complementarity nor the central GC pairs within the palindrome are essential for refolding. It has been suggested that the central GC pairs are essential for efficient dimerisation in vitro possibly through the involvement in non-canonical base pair interactions by forming a potential nucleation point of the dimer (8,9,21). Our results are in accordance with in vitro selection data that show that this dinucleotide favours dimerisation but is not essential (31,32). Self-complementarity of the loops is evidently required for viral RNA dimerisation of homologous strands, but apparently not for the refolding process per se. These observations are particularly interesting given the fact that only two different loop sequences have been found in HIV-1 isolates, raising the question on the nature of the selective pressure for these two sequences since they can be substituted by other sequences for extended duplex formation in vitro. It has been shown that the entire HIV 5′ leader RNA can adopt two different conformations, suggesting that sequence conservation might reflect the need to preserve these two structures (33).
Extended duplex formation was still possible when only partially complementary sequences or sequences with strongly reduced loop complementarity were used. This suggests that loop–loop interaction through partial base pairing itself is sufficient for extended duplex formation at 55°C, although full complementarity, as is the case for wild-type loop sequences, strongly enhances the efficiency of the refolding process. Assuming that reduction of loop complementarity does not affect refolding itself but only reduces kissing complex stability, then the previous findings of Berkhout and van Wamel (5) might be explained. These authors reported strongly reduced infectivity when two GC pairs were deleted from the palindrome of HIV-1 LAI subtype; however, purified virions still contained stable dimeric RNA. Reduced stability of the kissing complex might lead to a strong impairment of the packaging efficiency, but any loose dimers that happened to be encapsidated were still able to refold, forming stable dimers.
The study on the influence of external parameters on the refolding process revealed that its efficiency is temperature dependent, suggesting that refolding proceeds by the destabilisation of the stems. Additionally, it could be shown that the presence of magnesium ions enhances extended duplex formation although it is not essential for this process. High magnesium concentrations accelerated the reaction in a way that very efficient extended duplex formation could already be observed at 37°C. This could be explained by a more efficient stabilisation of the kissing complex than that of the stems. Indeed, such a strong stabilisation of the kissing complex compared with the stem is observed in the presence of magnesium ions in UV melting experiments (A.Weixlbaumer and R.Schroeder, unpublished results).
Transition from a kissing complex to an extended duplex in vitro is clearly observed at physiological temperatures in the presence of the viral nucleocapsid protein NCp7. This small basic 55 amino acid long highly conserved RNA binding protein was reported to have general RNA chaperone activity (19,28). In vivo experiments demonstrated that a viral protease-dependent maturation resulting in the cleavage of the gag precursor to generate nucleocapsid protein is accompanied by conversion of the dimeric RNA particles into a more heat stable dimer (24,34). Our data show that NCp7-induced extended duplex formation is extremely efficient compared with the temperature-induced refolding. The amount of extended duplex formed by constructs lacking loop complementarity can be explained by the general RNA–RNA annealing activity of NCp7 (19). This might propose a model for how extended duplex formation is promoted by NCp7. It is conceivable that NCp7 induces kissing complex formation by its RNA–RNA annealing activity and/or by its helix destabilising activity (29). The kissing interaction might provide a stable and specific interaction surface as a starting point for an efficient refolding. In contrast, it might be the temporal availability and spatial location of NCp7 during the viral maturation cycle that allows it to use its general nucleic acid refolding activity in a specific manner. However, it should be mentioned that it is currently unclear how the data on DIS-mediated dimerisation obtained in vitro translate to the in vivo situation, given that there is no direct evidence that the mature form of the dimer inside the viral particle is an extended duplex.
RNA loop–loop interactions are involved in a variety of biological processes and some of these interactions are accompanied by structural rearrangements leading to more stable structures (reviewed in 20). It is often difficult to distinguish between the two alternative conformations. Since the sequence requirements of the leadzyme are relatively small, derivates from this system could be easily used to investigate whether refolding follows RNA loop–loop interactions in other biological contexts such as antisense regulation, or bicoid mRNA localisation. Using the hammerhead system, investigation of refolding processes following RNA loop–loop interactions could be addressed in vivo as well.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Lisa Scherer and John Rossi for generously providing NCp7 peptide and Lisa Scherer for help with the handling of NCp7. We are grateful to Tim Skern for useful comments and revision. The input of all members of the Schroeder laboratory is gratefully appreciated. This work was funded by the Austrian Science Foundation FWF grant no. P16026 to R.S.
REFERENCES
- 1.Coffin J.M., Hughes,S.H. and Varmus,H. (1997) Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed] [Google Scholar]
- 2.Berkhout B. (1996) Structure and function of the human immunodeficiency virus leader RNA. Prog. Nucleic Acid Res. Mol. Biol., 54, 1–34. [DOI] [PubMed] [Google Scholar]
- 3.Paillart J.C., Marquet,R., Skripkin,E., Ehresmann,B. and Ehresmann,C. (1994) Mutational analysis of the bipartite dimer linkage structure of human immunodeficiency virus type 1 genomic RNA. J. Biol. Chem., 269, 27486–27493. [PubMed] [Google Scholar]
- 4.Paillart J.C., Marquet,R., Skripkin,E., Ehresmann,C. and Ehresmann,B. (1996) Dimerization of retroviral genomic RNAs: structural and functional implications. Biochimie, 78, 639–653. [DOI] [PubMed] [Google Scholar]
- 5.Berkhout B. and van Wamel,J.L. (1996) Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J. Virol., 70, 6723–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laughrea M. and Jette,L. (1996) Kissing-loop model of HIV-1 genome dimerization: HIV-1 RNAs can assume alternative dimeric forms and all sequences upstream or downstream of hairpin 248–271 are dispensable for dimer formation. Biochemistry, 35, 1589–1598. [DOI] [PubMed] [Google Scholar]
- 7.Laughrea M., Jette,L., Mak,J., Kleiman,L., Liang,C. and Wainberg,M.A. (1997) Mutations in the kissing-loop hairpin of human immunodeficiency virus type 1 reduce viral infectivity as well as genomic RNA packaging and dimerization. J. Virol., 71, 3397–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clever J.L., Wong,M.L. and Parslow,T.G. (1996) Requirements for kissing-loop-mediated dimerization of human immunodeficiency virus RNA. J. Virol., 70, 5902–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paillart J.C., Westhof,E., Ehresmann,C., Ehresmann,B. and Marquet,R. (1997) Non-canonical interactions in a kissing loop complex: the dimerization initiation site of HIV-1 genomic RNA. J. Mol. Biol., 270, 36–49. [DOI] [PubMed] [Google Scholar]
- 10.Muriaux D., Fosse,P. and Paoletti,J. (1996) A kissing complex together with a stable dimer is involved in the HIV-1Lai RNA dimerization process in vitro. Biochemistry, 35, 5075–5082. [DOI] [PubMed] [Google Scholar]
- 11.Mujeeb A., Clever,J.L., Billeci,T.M., James,T.L. and Parslow,T.G. (1998) Structure of the dimer initiation complex of HIV-1 genomic RNA. Nature Struct. Biol., 5, 432–436. [DOI] [PubMed] [Google Scholar]
- 12.Ennifar E., Yusupov,M., Walter,P., Marquet,R., Ehresmann,B., Ehresmann,C. and Dumas,P. (1999) The crystal structure of the dimerization initiation site of genomic HIV-1 RNA reveals an extended duplex with two adenine bulges. Struct. Fold Des., 7, 1439–1449. [DOI] [PubMed] [Google Scholar]
- 13.Ennifar E., Walter,P., Ehresmann,B., Ehresmann,C. and Dumas,P. (2001) Crystal structures of coaxially stacked kissing complexes of the HIV-1 RNA dimerization initiation site. Nature Struct. Biol., 8, 1064–1068. [DOI] [PubMed] [Google Scholar]
- 14.Rist M.J. and Marino,J.P. (2002) Mechanism of nucleocapsid protein catalyzed structural isomerization of the dimerization initiation site of HIV-1. Biochemistry, 41, 14762–14770. [DOI] [PubMed] [Google Scholar]
- 15.Symons R.H. (1992) Small catalytic RNAs. Annu. Rev. Biochem., 61, 641–671. [DOI] [PubMed] [Google Scholar]
- 16.Pan T. and Uhlenbeck,O.C. (1992) A small metalloribozyme with a two-step mechanism. Nature, 358, 560–563. [DOI] [PubMed] [Google Scholar]
- 17.Darlix J.L., Lapadat-Tapolsky,M., de Rocquigny,H. and Roques,B.P. (1995) First glimpses at structure-function relationships of the nucleocapsid protein of retroviruses. J. Mol. Biol., 254, 523–537. [DOI] [PubMed] [Google Scholar]
- 18.You J.C. and McHenry,C.C. (1994) Human immunodeficient virus nucleocapsid protein accelerates strand tranfer of the terminally redundant sequences involved in reverse transcription. J. Biol. Chem., 269, 31491–31495. [PubMed] [Google Scholar]
- 19.Bertrand E.L. and Rossi,J.J. (1994) Facilitation of hammerhead ribozyme cleavage catalysis by the nucleocapsid protein of HIV-1 and the heterogeneous nucleo ribonucleoprotein A1. EMBO J., 13, 2904–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunel C., Marquet,R., Romby,P. and Ehresmann,C. (2002) RNA loop–loop interactions as dynamic functional motifs. Biochimie, 84, 925–944. [DOI] [PubMed] [Google Scholar]
- 21.Jossinet F., Paillart,J.C., Westhof,E., Hermann,T., Skripkin,E., Lodmell,J.S., Ehresmann,C., Ehresmann,B. and Marquet,R. (1999) Dimerization of HIV-1 genomic RNA of subtypes A and B: RNA loop structure and magnesium binding. RNA, 5, 1222–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi K.I., Baba,S., Chattopadhyay,P., Koyanagi,Y., Yamamoto,N., Takaku,H. and Kawai,G. (2000) Structural requirement for the two-step dimerization of human immunodeficiency virus type 1 genome. RNA, 6, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahm S. and Uhlenbeck,O.C. (1991) Role of divalent metal ions in the hammerhead RNA cleavage reaction. Biochemistry, 30, 9464–9469. [DOI] [PubMed] [Google Scholar]
- 24.Fu W., Gorelick,R.J. and Rein,A. (1994) Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J. Virol., 68, 5013–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Y.X., Copeland,T.D., Henderson,L.E., Gorelick,R.J., Bosche,W.J., Levin,J.G. and Rein,A. (1996) HIV-1 nucleocapsid protein induces ‘maturation’ of dimeric retroviral RNA in vitro. Proc. Natl Acad. Sci. USA, 93, 7577–7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lapadat-Tapolsky M., Pernelle,C., Borie,C. and Darlix,J.L. (1995) Analysis of the nucleic acid annealing activities of nucleocapsid protein from HIV-1. Nucleic Acids Res., 23, 2434–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muriaux D., De Rocquigny,H., Roques,B.P. and Paoletti,J. (1996) NCp7 activates HIV-1Lai RNA dimerization by converting a transient loop–loop complex into a stable dimer. J. Biol. Chem., 271, 33686–33692. [DOI] [PubMed] [Google Scholar]
- 28.Tsuchihashi Z., Khosla,M. and Herschlag,D. (1993) Protein enhancement of hammerhead ribozyme catalysis. Science, 262, 99–102. [DOI] [PubMed] [Google Scholar]
- 29.Tsuchihashi Z. and Brown,P.O. (1994) DNA strand exchange and selective DNA annealing promoted by the human immunodeficiency virus type 1 nucleocapsid protein. J. Virol., 68, 5863–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clodi E., Semrad,K. and Schroeder,R. (1999) Assaying RNA chaperone activity in vivo using a novel RNA folding trap. EMBO J., 18, 3776–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lodmell J.S., Ehresmann,C., Ehresmann,B. and Marquet,R. (2000) Convergence of natural and artificial evolution on an RNA loop–loop interaction: the HIV-1 dimerization initiation site. RNA, 6, 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lodmell J.S., Ehresmann,C., Ehresmann,B. and Marquet,R. (2001) Structure and dimerization of HIV-1 kissing loop aptamers. J. Mol. Biol., 311, 475–490. [DOI] [PubMed] [Google Scholar]
- 33.Berkhout B. and van Wamel,J.L. (2000) The leader of the HIV-1 RNA genome forms a compactly folded tertiary structure. RNA, 6, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu W. and Rein,A. (1993) Maturation of dimeric viral RNA of Moloney murine leukemia virus. J. Virol., 67, 5443–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]