

Abstract
Heme oxygenase-1 (HO-1), a stress-inducible enzyme anchored in the endoplasmic reticulum (ER) by a single transmembrane segment (TMS) located at the C terminus, interacts with NADPH cytochrome P450 reductase and biliverdin reductase to catalyze heme degradation to biliverdin and its metabolite, bilirubin. Previous studies suggested that HO-1 functions as a monomer. Using chemical cross-linking, co-immunoprecipitation, and fluorescence resonance energy transfer (FRET) experiments, here we showed that HO-1 forms dimers/oligomers in the ER. However, oligomerization was not observed with a truncated HO-1 lacking the C-terminal TMS (amino acids 266–285), which exhibited cytosolic and nuclear localization, indicating that the TMS is essential for the self-assembly of HO-1 in the ER. To identify the interface involved in the TMS-TMS interaction, residue Trp-270, predicted by molecular modeling as a potential interfacial residue of TMS α-helices, was mutated, and the effects on protein subcellular localization and activity assessed. The results showed that the W270A mutant was present exclusively in the ER and formed oligomers with similar activity to those of the wild type HO-1. Interestingly, the W270N mutant was localized not only in the ER, but also in the cytosol and nucleus, suggesting it is susceptible to proteolytic cleavage. Moreover, the microsomal HO activity of the W270N mutant was significantly lower than that of the wild type. The W270N mutation appears to interfere with the oligomeric state, as revealed by a lower FRET efficiency. Collectively, these data suggest that oligomerization, driven by TMS-TMS interactions, is crucial for the stabilization and function of HO-1 in the ER.
Heme oxygenase (HO)3 catalyzes the NADPH cytochrome P450 reductase-dependent oxidative degradation of cellular heme to biliverdin, carbon monoxide (CO), and free iron (1, 2). Biliverdin is subsequently converted to bilirubin by biliverdin reductase in the cytosol. Two HO isoforms have been identified in mammalian systems. HO-1 is a 288 amino acid protein and is expressed at high amounts in a variety of pathological conditions associated with cellular stress. There is compelling evidence that HO-1 induction represents an important cytoprotective defense mechanism against oxidative insults by virtue of the anti-oxidant properties of the bilirubin and the anti-inflammatory effect of the CO produced (2). HO-1 is anchored in the endoplasmic reticulum (ER) through a single transmembrane segment (TMS) located at the C terminus, while the rest of the molecule is cytoplasmic (3). HO-1 is sensitive to proteolytic cleavage (4), and it was recently shown that HO-1 can be proteolytically cleaved from the ER and translocated to the nucleus under certain stress conditions (5). Although the catalytic site in the cytoplasmic domain remains intact, the activity of soluble HO-1 is drastically reduced (5), indicating that ER localization is important for its full enzymatic function.
Self-assembly to form dimers and higher oligomers is a common phenomenon in many membrane proteins (6, 7). Numerous studies have revealed that interactions between TMSs play an important role in the structure and function of many membrane proteins. Examples include receptors, enzymes, neurotransmitter transporters, and ion channels, in which oligomerization is crucial for their proper cellular localization and function (8). HO-1 does not contain any cysteine residues and has therefore been assumed to function as a monomer (1). To determine whether HO-1 forms oligomers in native membranes, in the present study, we performed chemical cross-linking, co-immunoprecipitation, and FRET analysis using fluorescent protein tags fused to the N terminus of HO-1. The results showed that HO-1 formed dimers/oligomers in the ER and that the TMS provided the interface for the protein-protein interactions. Interference with the TMS-TMS interaction resulted in destabilization of HO-1 and a reduction in enzymatic function.
EXPERIMENTAL PROCEDURES
Plasmid Construction
cDNAs for human full-length HO-1 (HO-1-FL) and HO-1 with a 42-amino acid truncation at the C terminus (HO-1-CΔ42) were amplified by the polymerase chain reaction (PCR) and subcloned into the PLNCX2 vector. Flag-tagged HO-1 was constructed by subcloning full-length HO-1 cDNA into the pFLAG-CMV-2 expression vector. Full-length HO-1 and C-terminal 24 amino acid truncated HO-1 (HO-1-CΔ24) with CFP or YFP fused to the N terminus were constructed by subcloning the respective cDNAs into the pECFP-C1 or pEYFP-C1 vector (BD Bioscience Clontech). The CFP or YFP fusion HO-1-W270A and HO-1-W270N mutant constructs were prepared by site-directed mutagenesis using PCR (9).
Cell Transfection
HEK293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 4.5 g/l glucose, 10% fetal bovine serum, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. The cells were cultured at 37 °C in an environment of 5% CO2, 95% air in a humidified atmosphere. Cell transfection was performed using Effectene® transfection reagent (Qiagen) following the manufacturer's instructions. For confocal microscopy and FRET assay, cells were plated on poly l-lysine-coated glass cover slips (42 mm) (Carl Zeiss, Inc.) and transiently transfected with an equimolar ratio of CFP and YFP fusion HO-1 plasmids and were used 24 h after transfection. To examine localization of the protein in the ER, cells were cotransfected with an equimolar ratio of YFP fusion HO-1 plasmid and pDsRed-ER(BD Biosciences Clontech), a plasmid coding for a red fluorescent protein expressed specifically in the ER.
Subcellular Fractionation
Cells (3 × 106) were washed twice with cold phosphate-buffered saline, scraped into 500 μl of cold hypotonic buffer (50 mm Tris-HCl, pH 7.4, 0.32 m sucrose, and a 1:100 dilution of protease inhibitor mixture set III (Calbiochem), and homogenized by 30 passages through a 27-gauge needle. The lysates were centrifuged at 800 × g for 10 min at 4 °C, and the pellets washed once with hypotonic buffer and nuclear proteins extracted for 20 min on ice using hypotonic buffer containing 420 mm NaCl. The extracts were then centrifuged at 13,200 × g for 10 min at 4 °C, and the supernatants collected as the nuclear extract. The supernatants from the first centrifugation were subjected to further centrifugation at 227,000 × g for 1 h at 4 °C to separate the cytosol and microsomes.
Chemical Cross-linking
The cells were rinsed twice with phosphate-buffered saline, incubated for 30 min at room temperature with 1 mm dithiobis[succinimidylpropionate] (DSP) (Pierce), and the cross-linker quenched with 20 mm Tris-HCl, pH 7.0. The cells were then lysed with SDS sample buffer and subjected to electrophoresis on 10% SDS-polyacrylamide gels in the presence or absence of 10 mm dithiothreitol, then the proteins were transblotted onto nitrocellulose membranes, and immunoblotted with anti-HO-1 antibody as described previously (10).
Immunoprecipitation
HEK293 cells transfected with the Flag-HO-1 plasmid and/or YFP-HO-1 plasmid for 24 h were lysed with lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.5% octyl-β-d-glucopyranoside, and a 1:100 dilution of protease inhibitor mixture set III), and the lysate centrifuged at 13,200 × g for 15 min at 4 °C. The supernatant was removed, and the Flag HO-1 was purified using ANTI-FLAG® M2 Affinity Gel (Sigma). Briefly, prior to the incubation with cell lysate, the affinity gel was washed twice with 20× volume of TBS buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl), followed by a wash with 0.1 m glycine, pH 3.5. The gel was then washed three times again with TBS buffer containing 0.5% octyl-β-d-glucopyranoside. The cell lysate was incubated with the washed gel overnight at 4 °C with continuous shaking. The gel was then washed three times with lysis buffer and bound proteins eluted by boiling in 2× SDS sample buffer for 3 min.
Confocal Microscopy
Confocal images were obtained using a PerkinElmer Ultra-view confocal microscope (PerkinElmer Life Sciences).
Fluorescence Resonance Energy Transfer (FRET) Measurement
Epi-fluorescence images were acquired using a Zeiss Axiovert 200 m microscope (Carl Zeiss, Inc.) equipped with a mercury lamp and the following filter cubes (nm): (1) CFP cube: EX 436/20, EM 480/40, DCLP 455; (2) YFP cube: EX 500/20, EM 535/30, DCLP 515; (3) FRET cube: EX 436/20, EM 535/30, DCLP 455 (Omega Optical). Images were recorded with a CoolView-IDI intensifier CCD (Photonic Science). The FR was determined using the 3-cube FRET approach (11). The FR is equal to the fractional increase in YFP emission due to FRET and is calculated in Equation 1.
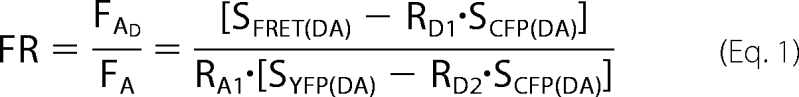 |
SCUBE(SPECIMEN) denotes the image measurement, where CUBE indicates the filter cube (CEP, YFP, or FRET), and SPECIMEN indicates whether the cell expresses the donor (D; CFP), acceptor (A; YFP), or both (DA). RD1 = SFRET(D)/SCFP(D) = 0.50 ± 0.002 (n = 21), RD2 = SYFP(D)/SCFP(D) = 0.0051 ± 0.0004 (n = 21), and RA1 = SFRET(A)/SYFP(A) = 0.26 ± 0.01 (n = 23) are constants predetermined from measurements on cells expressing only YFP or CFP. The effective FRET efficiency (FE) was determined in Equation 2.
The bracketed term is the ratio of the YFP and CFP molar extinction coefficients scaled for the FRET cube excitation filter (11) and was previously determined to be 0.094 (11).
Circular Dichroism (CD) Spectroscopy
The C-terminal 25-residue peptide of human HO-1 (QAPLLRW270VLTLSFLVATVAVGLYAM) and two peptides containing W270A and W270N mutations, respectively, were synthesized by JPT Peptide Technologies, Inc (Berlin, Germany) and purchased through its distributor, Prisma Biotech Co. (Taipei, Taiwan). Peptide purity was ∼91% based on HPLC analysis, and the sequence was confirmed by mass spectrometry. CD experiments were performed on an AVIV 202 SF CD spectrometer (Lakewood, NJ) calibrated with d-10-camphorsulfonic acid at 25 °C. The synthesized wild-type peptide (20 μm) dissolved in 20 mm phosphate buffer (pH 7.0) containing 10 mm sodium dodecyl sulfate (SDS) or dodecylphosphocholine (DPC) in a 1-mm pathlength cuvette was used for CD experiments. In experiments comparing the wild-type and mutated peptides, SDS was increased to 200 mm to enhance the solubility of the mutated peptides. The steady-state CD spectra were recorded from 180 to 260 nm at a scanning rate of 38 nm/min with a wavelength step of 0.5 nm and time constant of 100 ms. All CD data were obtained from an average of 3 scans with a 1-nm bandwidth. After background subtraction and smoothing, all the CD data were converted from the CD signal (millidegree) into the mean residue ellipticity (deg cm2 dmol−1). The quantitative estimation of secondary structure content was performed using three programs, CONTIN/LL, SELCON3, and CDSSTR, accordingly (12).
HO Activity Measurement
HEK293 cells were transfected with indicated HO-1 plasmid constructs for 24 h. Cells were then rinsed twice with ice-cold PBS and harvested by centrifugation at 1,000 × g for 5 min at 4 °C. The whole cell lysate was prepared and HO activity was determined as described previously (13). To determine the microsomal HO activity, the subcellular fractionation was performed as described above to isolate microsomal fraction. 100 μg of microsomal proteins were then used for HO activity assay. The HO activities in different microsomal preparations were normalized to the corresponding HO-1 protein levels determined by Western blotting.
Statistical Analysis
Data are expressed as the mean ± S.E. Group data were analyzed by one-way analysis of variance. A value of p < 0.05 was considered statistically significant.
RESULTS
Biochemical Assessment of HO-1 Oligomerization
To test the possibility that HO-1 forms oligomers in the ER, we expressed full-length human HO-1 (HO-1-FL) and a soluble form lacking the C-terminal 42 residues (HO-1-CΔ42) in HEK293 cells. The cells were then incubated for 30 min at room temperature with a thiol-cleavable, amine-reactive cross-linker, DSP. As shown in Fig. 1A, HO-1 dimers and higher oligomers were formed in cells expressing HO-1-FL, but not those expressing HO-1-CΔ42, and the HO-1 dimers and oligomers were reduced to monomers by dithiothreitol. Oligomerization was observed not only in cells with high, but also moderate levels of HO-1 expression, indicating that it was not caused by the aggregation of overexpressed protein. HO-1 protein-protein interactions were also demonstrated in cells co-expressing yellow fluorescent protein-tagged full-length HO-1 (YFP-HO-1-FL) and Flag-tagged full-length HO-1 (Flag-HO-1-FL). As shown in Fig. 1B, when Flag-HO-1-FL was immunoprecipitated from these cells using anti-Flag antibody-conjugated agarose, YFP-HO-1-FL was detected in the immunoprecipitate by Western blot analysis. However, YFP-HO-1-FL was not co-immunoprecipitated from a mixture of cell lysates containing either Flag-HO-1-FL or YFP-HO-1-FL, supporting the idea that the interaction of Flag-HO-1-FL and YFP-HO-1-FL observed in the co-expressing cells occurred prior to detergent solubilization.
FIGURE 1.

HO-1 protein-protein interactions revealed by chemical cross-linking and co-immunoprecipitation. A, HEK293 cells were transfected with the indicated amounts of plasmids coding for HO-1-CΔ42 or HO-1-FL for 24 h, followed by cross-linking with DSP, then cell lysates were subjected to SDS-PAGE in the presence or absence of 10 mm dithiothreitol and HO-1 protein examined by Western blot analysis. B, HEK293 cells were transfected with plasmids carrying Flag-HO-1 or/and YFP-HO-1 cDNA for 24 h, then were solubilized with lysis buffer as described under “Experimental Procedures.” Equal amounts of cell lysates were then subjected to immunoprecipitation using an anti-Flag affinity gel. Both crude lysates and immunoprecipitates from cells transfected with YFP-HO-1 cDNA (lanes 1 and 4), Flag-HO-1 cDNA (lanes 2 and 5), or YFP-HO-1 and Flag-HO-1 cDNAs together (lanes 3 and 7) were subjected to Western blotting using anti-HO-1 antibodies to detect the presence of YFP-HO-1 and Flag-HO-1. Lane 6 represents the immunoprecipitate from a mixture of lysates from cells transfected with Flag-HO-1-cDNA or YFP-HO-1-cDNA alone (mixed in a 1:1 ratio).
HO-1 Oligomerization Revealed by FRET Analysis
To examine the intermolecular interactions of HO-1 in living cells, we next performed FRET analysis using the three cube method (11). The cyan fluorescent protein (CFP)- and YFP-tagged versions of full-length HO-1 (CFP-HO-1-FL and YFP-HO-1-FL) and a second truncated form lacking the C-terminal 24 residues (CFP-HO-1-CΔ24 and YFP-HO-1-CΔ24) were used in the experiments. HEK293 cells expressing YFP and CFP were used as the background control. The subcellular localization of the two YFP-fused HO-1 proteins expressed in HEK293 cells was first examined by confocal microscopy. Fig. 2A shows that YFP-HO-1-FL was found exclusively in the ER, whereas YFP-HO-1-CΔ24 was found in the cytosol and nucleus, but not the ER. The HO activity determined in cells overexpressing YFP-HO-1-CΔ24 was significantly lower as compared with that in cells overexpressing YFP-HO-1-FL (Fig. 2B). Fig. 2C shows the epi-fluorescence images obtained using the CFP, YFP, or FRET cube in cells expressing the indicated proteins and the corresponding FRET ratio (FR), the fractional increase in YFP emission due to FRET. In the absence of FRET, the FR is 1, while, when FRET increases, the FR is greater than 1 and reaches a theoretical maximum of 12 if the fluorescence pairs undergo FRET with 100% FRET efficiency (FE) (11). Fig. 2D shows the averaged FR and the corresponding FE. The FR of control cells co-expressing CFP and YFP was 1.14 ± 0.04, with an FE of 1.3 ± 0.1%, indicating there was no interaction between free CFP and YFP, even though both were overexpressed. In cells co-expressing CFP-HO-1-FL and YFP-HO-1-FL (“HO-1-FL”), the averaged FR was 2.01 ± 0.10 and the FE 9.5 ± 0.1%, supporting the idea of intermolecular interactions of HO-1 molecules in the ER membrane. However, in cells co-expressing CFP-HO-1- CΔ24 and YFP-HO-1-CΔ24 (“HO-1- CΔ24”), the FR was 1.20 ± 0.02 and the FE 1.9 ± 0.1%, indicating that the TMS is crucial not only for ER localization, but also for the protein-protein interaction of HO-1. When fluorescent proteins are overexpressed, concentration-dependent FRET may occur (14). Because HO-1 was expressed in large amounts in the ER membrane, we next determined whether the high FR ratio observed in cells co-expressing CFP-HO-1-FL and YFP-HO-1-FL was due to a concentration-dependent artifact. Fig. 2E shows the relationship between the FR and the fluorescence intensity, which is proportional to the protein concentration. We found that the FR was not higher in cells expressing higher concentrations of CFP-HO-1-FL, CFP-HO-1-CΔ24, or CFP. In addition, even though the fluorescence intensity of CFP was as high as that of CFP-HO-1-FL, the FR was much lower in cells co-expressing CFP and YFP than in those co-expressing CFP-HO-1-FL and YFP-HO-1-FL. These results show that the high FR between CFP-HO-1-FL and YFP-HO-1-FL is not a concentration-dependent artifact.
FIGURE 2.

C-terminal TM truncation reduces HO-1 activity and abolishes oligomerization. A, confocal images of HEK293 cells 24 h after co-transfection with pDsRed-ER and plasmid coding for YFP-HO-1-FL or YFP-HO-1-CΔ24. The ER and HO-1 images were obtained using the ER or YFP filter set, respectively. B, HO activities in cells transfected with YFP-HO-1-FL or YFP-HO-1-CΔ24 plasmids were determined. Data are the mean ± S.E. of four independent experiments. *, p < 0.01 versus cells overexpressing YFP-HO-1-FL. C, epi-fluorescent images of HEK293 cells 24 h after co-transfection with plasmids coding for CFP and YFP (CFP+YFP), CFP-HO-1-FL and YFP-HO-1-FL (HO-1-FL), or CFP-HO-1-CΔ24 and YFP-HO-1-CΔ24 (HO-1-CΔ24). Images were obtained using the CFP, YFP, or FRET cube as indicated. D, averaged FR and FE calculated using Equations 1 and 2. The number of cells quantified in each group is shown in parenthesis. ***, p < 0.005 versus control cells transfected with CFP + YFP. E, plot of FR versus CFP cube intensity per area from cells coexpressing the indicated proteins.
Prediction of HO-1 TMS-TMS Interaction Interface
The C-terminal amino acid sequence of HO-1 is highly conserved across different species (Fig. 3A). However, the tertiary structure of the HO-1 TMS has not been determined empirically. We synthesized the C-terminal 25-residue peptide of human HO-1 and performed CD experiments in the presence of SDS or DPC, both of which can form micelles and are commonly used to mimic membrane environments. We found that the CD spectra of the peptide in the presence of SDS or DPC were similar at different pH values (pH 3.0–9.0) (data not shown), indicating that the secondary structure of the peptide was pH-independent. Also, the CD spectra of the peptide in SDS or DPC showed two pronounced CD troughs at 208 and 222 nm and a peak at 191.5 nm (Fig. 3B), showing that the peptide was mainly in the form of α-helix in a membrane environment. We also used COILS (15) and Paircoil (16) to predict whether the human HO-1 TM helix (residues 264–288) folds as a coiled coil. Both methods predicted heptad repeats indicated by the a-g positions, as shown in Fig. 4A. The amino acids at the “a” and “d” positions were hydrophobic, as in a typical coiled coil. When these residues were placed on the helical wheel generated by GCG (Accelrys Inc., San Diego), Leu-267, Leu-274, and Val-281 were found to be in the “a” position, whereas Trp-270, Leu-277, and Gly-284 were in the “d” position (Fig. 4B). We propose that these residues form the interface between two HO-1 TM α-helices, as shown in the manually docked model (Fig. 4C). The TM segment (residues 267–285) in each molecule was modeled as a right-handed α-helix by the InsightII program (Accelrys Inc. San Diego).
FIGURE 3.

The CD spectra of the human HO-1 TMS. A, sequence alignment of the last 25 amino acids in the C-terminal of vertebrate HO-1s. The red box indicates the predicted TM α-helix. B, CD spectra of a 20 μm solution of synthetic human HO-1 C-terminal 25 residue peptide in 10 mm SDS (solid line) or DPC (dashed line) at 25 °C in 20 mm phosphate buffer (pH 7.0).
FIGURE 4.

Prediction of a heptad repeat in the TM helix (residues 267–285) of HO-1. Using the COILS and Paircoil2 methods, a heptad repeat (a-g positions) in the TM helix was predicted, as shown in the sequence (A) or on a helical wheel (B). The amino acids at the “a” and “d” positions are hydrophobic, as in a typical coiled coil. C, proposed interface between two HO-1 TM α-helices.
The Role of Trp-270 Residue in HO-1 Oligomerization
Because Trp-270, an aromatic amino acid conserved in vertebrate HO-1s, may contribute a strong thermodynamic force in the interface, we proposed that mutation of Trp-270 might have an impact on the oligomeric state of HO-1. To test this, two HO-1 mutants, YFP-HO-1-W270A and YFP-HO-1-W270N, were constructed and their expression, function, and subcellular localization in HEK293 cells examined. The W270A mutation did not significantly affect the subcellular localization of HO-1 (Fig. 5A). Although the activity of W270A HO-1 was slightly lower than that of wild type HO-1, the difference was not statistically significant (p = 0.102) (Fig. 5C). In contrast, YFP-HO-1-W270N was localized not only in the ER, but also in the cytoplasm and nucleus (Fig. 5A), suggesting that this mutant is susceptible to proteolytic cleavage. The subcellular localization of the W270N mutant was confirmed by subcellular fractionation (Fig. 5B). In addition, HO enzymatic activity in the microsomal fraction of cells expressing the W270N mutant was significantly lower than that in cells expressing wild type HO-1 (Fig. 5C). To examine whether the different Trp-270 mutations have differential effects on the TMS α-helical structure, we performed additional CD experiments with the synthetic C-terminal peptides containing these mutations. Fig. 6 shows the CD spectra of these peptides in the presence of SDS. We then used three different methods, SELCON3, CONTIN/LL, and CDSSTR, to perform the quantitative analysis of the secondary structure content of each peptide (12). The results were summarized in Table 1. It was noted that the averaged α-helix contents of wild-type. W270A, and W270N peptides were about 60–70%, indicating that the α-helix remained as a main structure for the mutated peptides. This observation suggests that the effect of W270N mutation on HO-1 stability and enzymatic activity was not due to the impact on the α-helix of HO-1 TMS. To examine whether the Trp-270 mutations affected HO-1 intermolecular interactions, FRET was performed on the YFP- and CFP-fused mutant proteins. Fig. 7A shows the epi-fluorescence images obtained. Fig. 7B shows that the FR and FE in the granular or perinuclear ER in cells expressing the W270A mutant were not significantly different from those in cells expressing wild type HO-1. In contrast, the FRET signal detected in the ER of cells expressing the W270N mutant was significantly lower than that in cells expressing wild type HO-1 (Fig. 7B). Moreover, the FR observed in the cytoplasm of cells expressing the W270N mutant was close to 1 (Fig. 7B).
FIGURE 5.

The W270N mutation renders HO-1 susceptible to proteolysis. A, confocal images of HEK293 cells co-transfected with YFP-HO-1-W270A or YFP-HO-1-W270N and ER-DsRed. B, subcellular localization of HO-1 examined by Western blot analysis of different fractions isolated from HEK293 cells 24 h after transfection with plasmids coding for YFP-HO-1-FL, YFP-HO-1-W270N, or YFP-HO-1-CΔ24. C, HEK293 cells were transfected with plasmids coding for YFP-HO-1-FL, YFP-HO-1-W270A, or YFP-HO-1-W270N, then, 24 h later, the microsomal fractions were prepared and HO activities measured. The activity was normalized to the corresponding microsomal HO-1 protein level determined by Western blot analysis. The specific activity in cells expressing YFP-HO-1-FL was taken as 100%. *, p < 0.01 versus cells expressing YFP-HO-1-FL. n = 5 in each group.
FIGURE 6.

The CD spectra of the HO-1 TMS containing Trp-270 mutations. The CD spectra of 20 μm of synthetic HO-1 C-terminal peptides with wild-type sequence (solid line), W270A (dashed line), or W270N (dotted line) mutation in 20 mm phosphate buffer (pH 7.0) containing 200 mm SDS were recorded at 25 °C.
TABLE 1.
Secondary structure contents of wild type and mutated TMS peptides estimated from CD spectra acquired at pH 7.0 and 25 °C using three different methods as described under “Experimental Procedures”
| TMS | Method | H(r)a | H(d)b | S(r)c | S(d)d | Trne | Unrdf | α-Helix content (%) |
|---|---|---|---|---|---|---|---|---|
| Wild type | SELCON3 | 0.473 | 0.220 | 0.003 | 0.014 | 0.057 | 0.274 | 69.3 |
| CONTIN/LL | 0.481 | 0.194 | 0.005 | 0.028 | 0.063 | 0.229 | 67.5 | |
| CDSSTR | 0.574 | 0.199 | 0.052 | 0.010 | 0.034 | 0.133 | 77.3 | |
| W270A | SELCON3 | 0.510 | 0.229 | 0.000 | 0.006 | 0.047 | 0.248 | 73.9 |
| CONTIN/LL | 0.488 | 0.131 | 0.043 | 0.033 | 0.075 | 0.230 | 61.9 | |
| CDSSTR | 0.433 | 0.194 | 0.051 | 0.071 | 0.077 | 0.176 | 62.7 | |
| W270N | SELCON3 | 0.392 | 0.200 | 0.015 | 0.031 | 0.096 | 0.299 | 59.2 |
| CONTIN/LL | 0.435 | 0.167 | 0.008 | 0.037 | 0.090 | 0.262 | 60.2 | |
| CDSSTR | 0.476 | 0.196 | 0.054 | 0.045 | 0.046 | 0.186 | 67.2 |
a H(r), regular α-helix.
b H(d), distorted α-helix.
c S(r), regular β-strand.
d S(d), distorted β-strand.
e Trn, turns.
f Unrd, unordered.
FIGURE 7.

The W270N mutation decreases HO-1 oligomerization. A, epi-fluorescent images of HEK293 cells 24 h after transfection with plasmids coding for CFP-HO-1-FL and YFP-HO-1-FL (HO-1-FL), CFP-HO-1-W270A and YFP-HO-1-W270A (HO-1-W270A), or CFP-HO-1-W270N and YFP-HO-1-W270N (HO-1-W270N). The granular ER is indicated by the stars in the insets and the perinuclear ER by the arrows. B, average FR and FE values in the granular and perinuclear ER or the cytoplasm. The number of cells quantified in each group is shown in parentheses. *, p < 0.05; ***, p < 0.005 versus the corresponding compartment in cells expressing wild type HO-1. †, p < 0.05 versus the granular ER of cells expressing HO-W270N.
DISCUSSION
The present study demonstrates, for the first time, that HO-1 can form dimers and oligomers in the ER membrane. Although oligomerization is a common feature of many proteins containing a single TMS, whether HO-1 could undergo oligomerization was not known. To test this possibility, we first performed chemical cross-linking on intact cells overexpressing HO-1 using a membrane permeable cross-linker, DSP. SDS-PAGE and Western blot analysis revealed the formation of HO-1 dimers and higher oligomers, which was not influenced by the protein concentration. The co-immunoprecipitation of Flag-HO-1 and YFP-HO-1 from lysates of cells co-expressing these two proteins provided support for HO-1 protein-protein interactions. In addition to the biochemical assessments, we also performed FRET analysis to examine protein-protein interactions in live cells. Consistent with the biochemical results, the FRET data provided convincing evidence to support the existence of HO-1 as preformed oligomers in the ER. Moreover, no oligomerization was observed for the truncated HO-1 lacking the TMS, indicating that the protein-protein interactions are driven by the TMS. A recent study demonstrated that the TMS is essential for the maximal catalytic activity of a recombinant human HO-1 in an in vitro assay (17). It has been shown that the TMS can increase the binding of HO-1 to NADPH-cytochrome P450 reductase (17). The present finding demonstrates a new role of the TMS in the formation of the quaternary structure of HO-1 in native membranes.
The crystal structures of the soluble human and rat HO-1 lacking the TMS have been determined (18, 19), but nothing is known about the tertiary and quaternary structures of the TMS in HO-1. Structural studies on membrane proteins have revealed that the TMS is often α-helical (6, 7). Using CD spectroscopy and molecular modeling, we predicted that the HO-1 TMS folds as an α-helix. Transmembrane helix-helix interactions have been reported for several proteins with single transmembrane α-helices (6, 7), but information regarding the sequence motifs required for the interactions is limited. The human erythrocyte glycophorin A is the most characterized membrane protein where the dimerization has been shown to be mediated through a GXXXG motif in the transmembrane helices (20, 21). Studies on a membrane-helix model have revealed that the hydrogen bonds formed by polar residues, such as Asn, Asp, or Glu, can drive the self-association of membrane helices (22–26). Moreover, a heptad repeat sequence with a leucine zipper motif can also contribute to the lateral interactions of TMS through van der Waals interactions (27). The HO-1 TMS does not contain these typical interaction motifs. However, computational modeling predicted a heptad repeat in which the residues at the “a” and “d” positions consisted of three Leu (Leu-267, Leu-274, and Leu-277), one Val (Val-281), one Trp (Trp-270), and one Gly (Gly-284), which may serve as the interface for the helix-helix interaction through aliphatic side-chain packing. Because Trp has been shown to stabilize the self-association of helices through aromatic ring π-π interactions (28, 29), we proposed that Trp-270 may have an impact on the lateral interactions between TMSs. The W270A mutation did not significantly affect HO-1 ER localization and oligomerization, indicating that the aromatic π interaction of Trp was not required for the TMS-TMS interaction. Although Asn residues in two transmembrane α-helices can theoretically form interhelical hydrogen bonds, which might contribute to dimer formation (22, 23), the W270N mutation resulted in weaker oligomerization, as revealed by the lower FRET efficiency. Because W270N mutation did not affect the α-helix structure as shown in CD experiment, we speculated that the polar interactions introduced by Asn in the HO-1 TMS somehow decreased the packing effects of other interfacial residues. More studies are required to clarify this issue. Interestingly, the W270N mutation significantly increased the susceptibility of HO-1 to proteolysis without affecting its integration in the ER membrane. These observations clearly support the importance of oligomerization in the proper organization and stabilization of HO-1 in the ER membrane. Moreover, the activity of microsomal W270N HO-1 was significantly lower than that of the wild type HO-1, suggesting that the oligomeric state may have an impact on the catalytic function of HO-1 in the ER. A previous study showed that NADPH-cytochrome P450 reductase and biliverdin reductase bind to soluble human HO-1 at sites, which are partially overlapping or allosterically modulated (30, 31). It is conceivable that oligomerization may allow the simultaneous binding of these two proteins on different HO-1 monomers and facilitate the cooperative reaction needed for heme degradation and subsequent bilirubin production. We noticed that the activity of W270A HO-1 was also slightly lower than that of wild type HO-1, although the difference was not statistically significant. The possibility that Trp-270 mutations to other amino acids might affect to various extents the interaction between HO-1 TMS and NADPH-cytochrome P450 reductase (17) cannot be completely ruled out in the present study. Further work is required to clarify this issue.
In conclusion, the present study demonstrates that the TMS of HO-1 not only functions as a membrane anchor, but also drives the self-association which is crucial for the stabilization and function of HO-1 in the ER. Given the importance of HO-1 in various pathophysiological states associated with cellular stress, genetic variations affecting TMS-TMS interactions may have a significant impact on disease progression. Moreover, whether the oligomeric state can be altered under specific cellular contexts, such as oxidative stress and hypoxia, remains to be explored. Although our data support the importance of Trp-270 in the TMS-TMS interaction, the roles of other residues require further investigation. To gain further insights into the structural framework of helix-helix interactions, studies are currently in progress to determine the structure of HO-1 TMS-TMS dimers in detergent micelles by high resolution nuclear magnetic resonance.
This work was supported in parts by grants from the National Science Council of Taiwan (to R.-C. S., NSC-95-2320-B-001-028-MY3; to L.-Y. C., NSC-95-2320-B-001-008), and an Academia Sinica Investigatorship (to L.-Y. C.).
- HO-1
- heme oxygenase-1
- YFP
- yellow fluorescence protein
- CFP
- cyan fluorescence protein
- FRET
- fluorescence resonance energy transfer
- TMS
- transmembrane segment
- ER
- endoplasmic reticulum
- DSP
- dithiobis[succinimidylpropionate]
- ER
- endoplasmic reticulum.
REFERENCES
- 1.Maines M. D. (1988) FASEB J. 2, 2557–2568 [PubMed] [Google Scholar]
- 2.Abraham N. G., Kappas A. (2008) Pharmacol. Rev. 60, 79–127 [DOI] [PubMed] [Google Scholar]
- 3.Yoshida T., Sato M. (1989) Biochem. Biophys. Res. Commun. 163, 1086–1092 [DOI] [PubMed] [Google Scholar]
- 4.Yoshida T., Ishikawa K., Sato M. (1991) Eur. J. Biochem. 199, 729–733 [DOI] [PubMed] [Google Scholar]
- 5.Lin Q., Weis S., Yang G., Weng Y. H., Helston R., Rish K., Smith A., Bordner J., Polte T., Gaunitz F., Dennery P. A. (2007) J. Biol. Chem. 282, 20621–20633 [DOI] [PubMed] [Google Scholar]
- 6.Arkin I. T. (2002) Biochim. Biophys. Acta 1565, 347–363 [DOI] [PubMed] [Google Scholar]
- 7.Mackenzie K. R. (2006) Chem. Rev. 106, 1931–1977 [DOI] [PubMed] [Google Scholar]
- 8.Marianayagam N. J., Sunde M., Matthews J. M. (2004) Trends Biochem. Sci. 29, 618–625 [DOI] [PubMed] [Google Scholar]
- 9.Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 10.Lin P. H., Chiang M. T., Chau L. Y. (2008) Biochim. Biophys. Acta 1783, 1826–1834 [DOI] [PubMed] [Google Scholar]
- 11.Erickson M. G., Alseikhan B. A., Peterson B. Z., Yue D. T. (2001) Neuron 31, 973–985 [DOI] [PubMed] [Google Scholar]
- 12.Sreerama N., Woody R. W. (2000) Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 13.Yang N. C., Lu L. H., Kao Y. H., Chau L. Y. (2004) J. Biomed. Sci. 11, 799–809 [DOI] [PubMed] [Google Scholar]
- 14.Lakowicz J. R. (1999). Principles of Fluorescence Spectroscopy, 2nd Ed., pp. 367–394, Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 15.Lupas A., Van Dyke M., Stock J. (1991) Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]
- 16.McDonnell A. V., Jiang T., Keating A. E., Berger B. (2006) Bioinformatics 22, 356–358 [DOI] [PubMed] [Google Scholar]
- 17.Huber W. J., 3rd, Backes W. L. (2007) Biochemistry 46, 12212–12219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugishima M., Omata Y., Kakuta Y., Sakamoto H., Noguchi M., Fukuyama K. (2000) FEBS Lett. 471, 61–66 [DOI] [PubMed] [Google Scholar]
- 19.Schuller D. J., Wilks A., Ortiz de Montellano P. R., Poulos T. L. (1999) Nat. Struct. Biol. 6, 860–867 [DOI] [PubMed] [Google Scholar]
- 20.Lemmon M. A., Treutlein H. R., Adams P. D., Brünger A. T., Engelman D. M. (1994) Nat. Struct. Biol. 1, 157–163 [DOI] [PubMed] [Google Scholar]
- 21.Russ W. P., Engelman D. M. (2000) J. Mol. Biol. 296, 911–919 [DOI] [PubMed] [Google Scholar]
- 22.Zhou F. X, Cocco M. J., Russ W. P., Brunger A. T., Engelman D. M. (2000) Nat. Struct. Biol. 7, 154–160 [DOI] [PubMed] [Google Scholar]
- 23.Choma C., Gratkowski H., Lear J. D., DeGrado W. F. (2000) Nat. Struct. Biol. 7, 161–166 [DOI] [PubMed] [Google Scholar]
- 24.Zhou F. X., Merianos H. J., Brunger A. T., Engelman D. M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 2250–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gratkowski H., Lear J. D., DeGrado W. F. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dawson J. P., Weinger J. S., Engelman D. M. (2002) J. Mol. Biol. 316, 799–805 [DOI] [PubMed] [Google Scholar]
- 27.Gurezka R., Laage R., Brosig B., Langosch D. (1999) J. Biol. Chem. 274, 9265–9270 [DOI] [PubMed] [Google Scholar]
- 28.Ridder A., Skupjen P., Unterreitmeier S., Langosch D. (2005) J. Mol. Biol. 354, 894–902 [DOI] [PubMed] [Google Scholar]
- 29.Sal-Man N., Gerber D., Bloch I., Shai Y. (2007) J. Biol. Chem. 282, 19753–19761 [DOI] [PubMed] [Google Scholar]
- 30.Wang J., de Montellano P. R. (2003) J. Biol. Chem. 278, 20069–20076 [DOI] [PubMed] [Google Scholar]
- 31.Higashimoto Y., Sugishima M., Sato H., Sakamoto H., Fukuyama K., Palmer G., Noguchi M. (2008) Biochem. Biophys. Res. Commun. 367, 852–858 [DOI] [PMC free article] [PubMed] [Google Scholar]