

Abstract
Enzymes that modify DNA are faced with significant challenges in specificity for both substrate binding and catalysis. We describe how single hydrogen bonds between M.HhaI, a DNA cytosine methyltransferase, and its DNA substrate regulate the positioning of a peptide loop which is ∼28 Å away. Stopped-flow fluorescence measurements of a tryptophan inserted into the loop provide real-time observations of conformational rearrangements. These long-range interactions that correlate with substrate binding and critically, enzyme turnover, will have broad application to enzyme specificity and drug design for this medically relevant class of enzymes.
Sequence-specific modification of DNA is essential for nearly all forms of life and contributes to a myriad of biological processes including gene regulation, mismatch repair, host defense, DNA replication, and genetic imprinting. Methylation of cytosine and adenine bases is a key epigenetic process whereby phenotypic changes are inherited without altering the DNA sequence (1). The central role of the bacterial and mammalian S-adenosylmethionine (AdoMet)2-dependent DNA methyltransferases in virulence regulation and tumorigenesis, respectively, have led these enzymes to be validated targets for antibiotic and cancer therapies (2, 3). However, AdoMet-dependent enzymes catalyze diverse reactions, and the design of potent and selective DNA methyltransferase inhibitors is particularly challenging (4, 5). The design of drugs that bind outside the active site is a particularly attractive means of inhibition for enzymes with common cofactors like AdoMet because off-target inhibition often leads to toxicity (6). Unfortunately, robust methods to identify and characterize such critical binding sites distal from the active site have not been developed.
DNA methyltransferases bind to a particular DNA sequence, stabilize the target base into an extrahelical position within the enzyme active site, and transfer the methyl moiety from AdoMet to the DNA (7). During this process, dramatic changes in the DNA structure such as bending, base flipping, or the intercalation of residues into the recognition sequence are often accompanied by large scale protein rearrangements (8). Here we characterized a specific conformational rearrangement of M.HhaI, a model DNA cytosine C5 methyltransferase with a cognate recognition sequence of 5′-GCGC-3′. Many structures of M.HhaI are available at high resolution including an ensemble of complexes with either cognate or nonspecific DNA (9, 10). Reorganization of an essential catalytic loop (residues 80–100) is regulated by sequence-specific protein-DNA interactions that occur ∼28 Å away from the catalytic loop (Fig. 1). Our work quantifies the importance of such distal communication in sequence-specific DNA modification and provides plausible structural mechanisms.
FIGURE 1.

Loop interactions in M.HhaI. A, two superimposed structures of M.HhaI are shown with the catalytic loop highlighted. Enzymes are in blue (open conformer) and red (closed conformer) with the cofactor in orange, the flipped cytosine in green, and position 1 of the DNA recognition sequence colored by atom along with Cys-81, Ile-86, and Arg-240. The large blue arrow shows the long-range structural communication between Arg-240 and the catalytic loop. B, close-up of the interactions between Arg-240, Ile-86, and position 1 of the recognition sequence. The flipped cytosine is in green. Removal of N2 from the guanosine with an inosine base maintains near cognate loop motion while removal of O6 with a 2AP base has almost no loop motion.
DNA-dependent positioning of the catalytic loop in M.HhaI was first observed crystallographically; cognate DNA stabilizes the loop-closed conformer, while nonspecific DNA leaves the loop in the open conformer (9, 10). Correct positioning of this loop is essential for catalysis because C81, the active site nucleophile that attacks the target cytosine base at the C6 position (supplemental Fig. S1), is ∼9.6 Å away in the loop-open conformer (Fig. 1A). Populating the closed conformer of the loop is essential for tight DNA binding and stabilizing the target cytosine that is flipped out of the DNA duplex (11–13). Using stopped-flow fluorescence spectroscopy to monitor the environment of tryptophan (Trp) residues inserted into the catalytic loop, we recently observed reorganization of this loop upon DNA binding in the absence of cofactor using the M.HhaI mutants W41F, W41F/K91W, and W41F/E94W (12). Loop positioning and the interconversion between the open and closed conformers, as determined from the intensities and rates of change in fluorescence signal are highly dependent on DNA sequence and confirm that cognate DNA stabilizes the loop-closed conformer whereas nonspecific DNA stabilizes the open conformer.
In this study, W41F/K91W and W41F/E94W M.HhaI were preincubated with cognate (COG), non-cognate (NC), or nonspecific (NS) DNA and mixed with cofactor or cofactor product, AdoMet and S-adenosylhomocysteine (AdoHcy), respectively, in a stopped-flow apparatus. Differences in observed fluorescence intensity are indicative of shifts in the populations of the various loop conformers; no observable fluorescence change suggests no significant change in population and thus, essentially no loop positioning to the closed conformer. Non-cognate and cognate sequences are nearly identical but differ by a single base change, whereas the nonspecific DNA substrate has no similarity to the cognate sequence. As a methyltransferase searches for the cognate site within a genome, it must encounter both non-cognate and nonspecific DNA sequences and be able to distinguish these from the cognate sequence. We examine both binding, using the cofactor product AdoHcy, and catalysis, using the AdoMet cofactor of M.HhaI, with these various DNA substrates.
EXPERIMENTAL PROCEDURES
Mutant Construction
Four mutants of WT M.HhaI were constructed, R240A, W41F, W41F/K91W, and W41F/E94W as previously reported (12). Briefly, PCR mutagenesis was done using the QuikChange kit (Stratagene). Sequenced plasmids were transformed and the enzymes purified using a His6 tag and a nickel column. Enzymes were further purified using a second cation exchange column. The protein concentration was determined by Bradford, SDS-PAGE densitometry, and absorbance at 280 nm.
Substrate Design and Preparation
DNA substrates used for kinetic, thermodynamic, and fluorescence studies were all synthesized by Midlands DNA (Midland, TX) and HPLC-purified. The following single strand oligonucleotide sequences were used, COGTOP: 5′-GGGAATTCATGGCGCAGTGGGTGGATCCTG-3′, COGBOT: 3′-CCCTTAAGTACCGCGTCACCCACCTAGGAC-5′, MCOGTOP: 5′-GGGAATTCATGGMGCAGTGGGTGGATCCAG-3′, MCOGBOT: 3′-CCCTTAAGTACCGMGTCACCCACCTAGGAC-5′, NC-1TOP: 5′-GGGAATTCATGACGCAGTGGGTGGATCCAG-3′, NC-1BOT: 3′-CCCTTAAGTACTGMGTCACCCACCTAGGAC-5′, NC-PURTOP: 5′-GGGAATTCATGNCGCAGTGGGTGGATCCTG-3′, NC-2APTOP: 5′-GGGAATTCATGPCGCAGTGGGTGGATCCTG-3′, NC-INOTOP: 5′-GGGAATTCATGICGCAGTGGGTGGATCCTG-3′, NSTOP: 5′-CAACAACTTCTTCTTCTTCT TCTTCTTCTTCTTCAACAAC-3′, NSBOT: 3′- GTTGTTGAAGAAGAAGAAGAAGAAGAAGAAGAAGTTGTTG-5′, and ABTOP: 5′-GGGAATTCATGGBGCAGTGG GTGGATCCTG-3′. Recognition sites are underlined, and target bases are in bold where M is 5-methyl cytosine, P is 2-aminopurine (2AP), I is inosine, B is abasic, and N is nebularine (purine). Duplex substrates were annealed to form UCOG DNA from COGTOP and COGBOT, COG DNA from COGTOP and MCOGBOT, MCOG DNA from MCOGTOP and MCOGBOT, NC-1 DNA from NC-1TOP and NC-1BOT, NC-PUR DNA from NC-PURTOP and MCOGBOT, NC-2AP DNA from NC-2APTOP and MCOGBOT, NC-INO DNA from NC-INOTOP and MCOGBOT, NS DNA from NSTOP and NSBOT, and AB DNA from ABTOP and MCOGBOT. All substrates, except UCOG, MCOG, and NS DNA, are hemi-methylated to force bound enzymes to a single orientation. AdoMet and AdoHcy were purchased from Sigma. For DNA KD measurements, DNA substrates were radiolabeled by using [γ-32P]ATP (Amersham Biosciences) and T4 polynucleotide kinase (New England Biolabs). All substrate concentrations were spectrophotometrically determined using Beer's Law and calculated extinction coefficients (14).
Determination of kcat
Steady state product formation over time was monitored at 22 °C using a filter binding assay previously described (14) to determine observed kcat values (supplemental Table S1). These rate constants are only observed kcat values because substrate concentrations were not fully saturating; however, all enzymes were measured under identical conditions. Enzyme (1.9 nm) was added to 3H-labeled AdoMet (6.0 μm) and COG DNA (9.5 μm) in methyltransferase reaction buffer (MRB, 100 mm Tris, pH 8.0, 10 mm EDTA, 1 mm β-mercaptoethanol). Aliquots were spotted onto 2.3-cm DE-81 filters at 10-min intervals. Washed filters were counted for 3H, and the data were fit to a straight line using SigmaPlot. Samples were analyzed with a Beckman Coulter LS6500 MultiPurpose Scintillation Counter.
Determination of kchem
Kinetic constants for the rate of methyl transfer (kchem) were measured at 22 °C using a single turnover filter binding assay, which was previously described (14) (supplemental Table 1). Enzyme (1.0 μm) was added to 3H-labeled AdoMet (4.0 μm) and DNA (500 nm) in MRB with time points taken over 300 s. DNA substrates measured were COG DNA, NC-PUR DNA, NC-2AP DNA, and NC-INO DNA. To quench the reaction, 12 μl of reaction mixture was added to 6 μl of 0.4% SDS with 0.5 m HCl and then neutralized with 6 μl of 0.5 m Tris base, and 20 μl were spotted onto DE-81 filters. To catch time points for WT with COG DNA, a KinTek Corp. RQF-3 rapid quench instrument was required. Quenched reactions from the rapid quench instrument were spotted on filters. Washed filters were counted for 3H, and data were fit to exponential curves using SigmaPlot (Systat).
Determination of KD DNA
12% non-denaturing PAGE mobility shifts were used to measure KD values for all DNA substrates with the addition of AdoHcy and AdoMet (supplemental Table S1) as previously described (14). For all shifts, 32P-labeled DNA (∼20 pm) was used with cofactor either AdoHcy or AdoMet (500 μm) in MRB. Enzyme was titrated from 1 pm to 13 μm at 22 °C. Gels were scanned on a Storm 840 densitometer (Amersham Biosciences), and bottom band densitometry was determined using ImageQuant (Amersham Biosciences). Data were fit to a rectangular hyperbola using SigmaPlot (Systat).
Determination of Km AdoMet
Michaelis-Menten constants for AdoMet were determined for W41F, W41F/K91W, and W41F/E94W using a filter binding assay as previously reported (14). Enzyme (500 pm) was mixed with COG DNA (200 nm) followed by AdoMet titrations (50–2600 nm) in MRB at 22 °C. A single time point was taken at 45 min in duplicate and spotted onto DE-81 filters. Washed filters were counted for 3H, and data were fit to linear equations to determine the rates, which were then plotted versus AdoMet concentration. This data were fit to exponential curves using SigmaPlot (Systat) to give reported values.
Stopped-flow Fluorescence
Time-resolved measurements were made at 22 °C with excitation at 290 nm and a cut-off filter at 295 nm similar to protocols previously reported (12). Briefly, 4-mm slit widths were used on an Applied Photophysics SX.18MV stopped-flow reaction analyzer equipped with a single channel emission photomultiplier tube positioned 90° from the excitation beam. All measurements were done in FLB (fluorescence buffer, 100 mm Tris, pH 8.0, 100 mm NaCl, and 10 mm EDTA), and data were fit using SigmaPlot (Systat). At least ten shots were averaged for each data point, which was then repeated in at least duplicate. Trp fluorescent measurements were made using AdoHcy, AdoMet, UCOG, COG, MCOG, NC-1, NC-PUR, NC-2AP, NC-INO, and NS DNA. For all measurements enzyme (500 nm) was preincubated with DNA (750 nm) and mixed in the stopped-flow apparatus with cofactor (500 μm). Traces were made over 50 s in FLB. Drift corrections were made in Excel (Microsoft), and corrected data were plotted in SigmaPlot (Systat).
Simulations
Fitting of fluorescence traces was done with Scientist (Micro Math). Enzyme turnover was under single turnover conditions with enzyme (1 μm) preincubated with DNA (750 nm) and mixed in the stopped-flow apparatus with cofactor (500 μm). Traces were made over 2, 20, and 200 s in FLB, which were tabulated as one file in Excel. The fit to the data are shown in Equations 1–8,
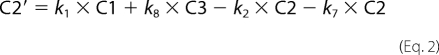 |
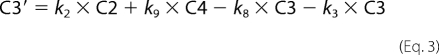 |
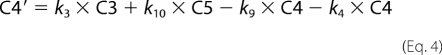 |
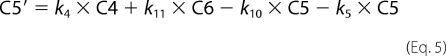 |
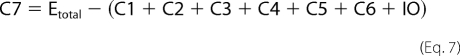 |
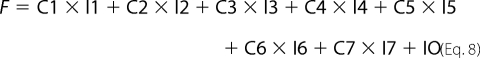 |
where C1–C7 are the concentrations of the various intermediate enzyme conformers having fluorescence intensities I1–I7, respectively, and k1–k11 are the respective forward and reverse rate constants, IO is the normalization factor, Etotal is the total enzyme concentration, and F is the total fluorescent signal measured.
RESULTS AND DISCUSSION
Controlling the Loop Position from 28 Å Away
When mixed with NS DNA and either AdoMet or AdoHcy, no loop closure was observed in M.HhaI (Fig. 2B) (12). Similar results were observed with NC-1 DNA (Fig. 2, A and B) and other non-cognate DNA substrates previously used (NC-2 to NC-6: TCGC, CCGC, GCGA, GCGT, GCGG; data not shown) (15). DNA recognition elements contacting either position one or four of the cognate sequence must contribute significantly to loop positioning because none of the non-cognate DNA substrates induce loop closure. Thus, although spatially removed by ∼28 Å, our data show that the recognition domain (residues 191–303) and the catalytic loop (residues 80–100) are conformationally linked (Fig. 1). Inspection of the first base pair of the recognition sequence in the 1.96 Å cocrystal structure (16) reveals no protein interactions to the cytosine base whereas three can be seen to the guanosine base, two from Arg-240 via the major groove to guanosine O6 and N7 and one from Ile-86 via the minor groove to guanosine N2 (Fig. 1B). When compared with its cognate DNA substrate, M.HhaI discriminates against NC-1 ∼2,800-fold through a combination of a 70-fold change in binding and a 40-fold change in the rate of catalysis (15). To further elucidate the exact protein-DNA interactions controlling loop rearrangement and DNA specificity, we used a series of non-cognate DNA substrates, which have base pair substitutions only at the first position of the cognate recognition sequence 5′-GCGC-3′, namely NC-1 (ACGC) (15), NC-INO (ICGC), NC-2AP (2CGC), and NC-PUR (PCGC) (Fig. 2A).
FIGURE 2.

Stopped-flow fluorescence of the catalytic loop in M.HhaI. All plots have enzyme (500 nm) preincubated with DNA (750 nm) and mixed with cofactor (500 μm). A, key of DNA substrates showing the structure of bases used, recognition sequence where the target base is raised, and position 1 is underlined in bold, DNA name, and observed kchem values. N.D. is not determined. B, W41F/E94W traces with various DNA substrates and AdoHcy, NS (gray), NC-1 (black), NC-2AP (pink), NC-PUR (orange), NC-INO (red), and COG (cyan). Only NC-INO shows loop motion similar to COG DNA. C, W41F/E94W traces with COG and MCOG DNA mixed with cofactors AdoMet and AdoHcy. NS and AdoHcy (black), MCOG with AdoMet (dark green), MCOG with AdoHcy (light green), COG with AdoHcy (cyan), COG with AdoMet (blue), and COG with both AdoHcy and AdoMet (500 μm each; purple). The catalytic loop cannot remain closed with methylated DNA and AdoMet, loop opening correlates with cofactor release, and loop opening contributes to kcat. D, COG data from C over 2 s. E, W41F/E94W catalysis with non-cognate DNA substrates and AdoMet. NS (gray), NC-2AP (pink), NC-PUR (orange), NC-INO (red), and COG (blue) are shown. Loop motion with NC-INO is nearly identical to COG DNA while NC-PUR and NC-2AP have highly perturbed motion. F, data from E over 2 s. G, W41F/K91W traces show nearly identical loop motion to W41F/E94W but with increasing signal. NS DNA and AdoHcy (black), NC-2AP with AdoHcy (pink), NC-PUR with AdoHcy (orange), NC-INO with AdoHcy (red), MCOG with AdoMet (dark green), MCOG with AdoHcy (light green), COG with AdoHcy (cyan), and COG with AdoMet (blue). All loop rearrangements are nearly identical for both W41F/K91W and W41F/E94W validating our analysis and interpretations.
Replacement of the guanosine at position one with purine (NC-PUR) removes both the O6 and N2 moieties and nearly eliminated loop closure as determined by the fluorescence intensity at equilibrium (Fig. 2B). P:C base pairs are largely unperturbed in critical helical parameters when compared with G:C base pairs (17), suggesting that the lack of loop closure is due to the loss of direct protein-DNA interactions. The backbone carbonyl of Ile-86 in the catalytic loop interacts with the guanosine N2 (Fig. 1B) and was previously implicated as being important for sequence recognition (18). Surprisingly, substitution of 2AP at position one (NC-2AP) does not recover the loop positioning observed with COG DNA even though the amino moiety of 2AP directly contacts the catalytic loop (Fig. 2B). Importantly, the kinetic contributions leading to this difference in equilibrium population between NC-2AP and COG DNA appear to reflect altered rate constants of closing shown by slower observed rate constants for both NC-2AP and NC-PUR when compared with COG (supplemental Table S2). The side chain of Arg-240 in the small domain interacts with the guanosine O6; substitution of inosine for purine (NC-INO) at this position results in complete recovery of the equilibrium and kinetic fluorescence changes to cognate levels (Fig. 2B and supplemental Table S2). Thus, the interaction between Arg-240 and guanosine O6 is a predominant determinant of loop closure in the presence of AdoHcy.
Loop Motion during Catalysis with Cognate DNA under Various Methylation States
To further elucidate the importance of loop closure, especially those involving specific protein-DNA interactions such as that between Arg-240 and the guanosine at position one (Fig. 1B), we monitored loop reorganization during catalytic turnover. Unlike proteins which are limited to binding, enzymes catalyze chemical reactions which must be coupled to the recognition process. Until now, all experiments have used AdoHcy, the cofactor product, and examined only aspects of substrate binding. To examine the role of the catalytic loop during catalysis, experiments were run with AdoMet and allowed to proceed through complete enzymatic turnover. Cognate DNA substrates were examined under a variety of methylation states (supplemental Table S2), either unmethylated (UCOG; data not shown), hemi-methylated (COG; Fig. 2), or fully methylated (MCOG; Fig. 2, C and G). UCOG DNA was measured but was indiscernible from COG substrates with both AdoMet and AdoHcy. Although COG DNA substrates were predominantly studied, MCOG data provide a basis for interpretation of other DNA substrates and is presented first.
When mixed with enzyme and AdoMet, MCOG DNA gave very little observable change in fluorescence signal, substantially different from the AdoHcy trace, which itself was roughly half as intense as AdoHcy and COG DNA (Fig. 2C). Structural examination of the WT protein-DNA complex suggests that a steric clash between the methyl groups of AdoMet and 5-methyl cytosine could prevent the loop from closing properly.
Loop motion during catalysis with COG DNA and AdoMet is different from COG DNA and AdoHcy in predominantly three ways: (i) with AdoMet, the loop shows an opening motion after the initial closure (Fig. 2C), (ii) the initial rate constants for loop closure are much faster with AdoMet (Fig. 2D and supplemental Table S2), and (iii) there is a lag with AdoMet before the loop reopens (Fig. 2, C and D). Traces of loop motion during catalysis fit to single or double exponentials during initial loop closure (decrease in signal for E94W and increase in signal for K91W) and to single exponentials for loop reopening (supplemental Table S2). In contrast, simulations run on the overall trace which includes the lag phase, revealed that the proper determination of intermediates and kinetic constants is extremely complicated, requiring multiple intermediates, each with different fluorescence intensity and a potential reverse rate. Although extensive population studies would need to be devised to fully separate each intermediate, these catalytic traces provide a real-time examination of conformational rearrangements responsible for enzyme catalysis.
Loop reopening after the methylation of COG DNA, which forms MCOG DNA, is driven by the cofactor under single turnover conditions. To reopen the loop after initial closure and methyltransfer, the excess AdoMet in solution (500 μm) displaces the negligible amounts of product AdoHcy (750 nm) and creates the steric clash that prevented loop closure with AdoMet and MCOG DNA (Fig. 2C). To test this hypothesis, COG DNA methylation was performed under conditions where AdoMet and AdoHcy were at equivalent concentrations (500 μm each). Critically, initial intensities of closure were nearly identical for both AdoMet and AdoMet/AdoHcy additions confirming roughly equivalent populations of the closed conformer (Fig. 2D). When equivalent amounts of AdoHcy and AdoMet were present in solution after methyltransfer, roughly equivalent amounts of enzyme associated to each cofactor, AdoMet and AdoHcy, leading to different ternary complexes. The equilibrium intensity when both cofactors are present (purple) indicates a greater amount of loop-closed conformers when compared with just AdoMet (Fig. 2C, blue). Thus, it appears that some enzymes have bound AdoHcy and MCOG DNA and have populated the closed conformer greater than with AdoMet and MCOG DNA. These intensities are very close to the respective MCOG DNA traces (Fig. 2C, green). Finally, rate constants of initial loop closure are also consistent with this model (supplemental Table S2). With MCOG DNA, AdoHcy binding to M.HhaI leads to the closed conformer whereas AdoMet binding leads to the open conformer.
Although the cofactor alone has no impact on loop positioning (12), rate constants of loop closure with AdoMet were faster than those with AdoHcy when in the presence of COG DNA, (Fig. 2D and supplemental Table S2). This suggests that loop positioning in the ternary complex is highly dependent on the cofactor, especially for initial closure, and that a ternary complex is briefly formed with the loop in the open conformer since DNA binding is concentration-dependent while loop repositioning is not (19). Whereas absolute cofactor concentration-dependent loop rearrangements could not be accurately measured due to the large amounts of AdoMet needed to keep all enzymes bound, no dependence was observed for either DNA or cofactor under the conditions used.
Lag Phase of Loop Motion during Catalysis
The lag period between initial loop closure and loop reopening increases with DNA concentration; however, a substantial lag (∼2–3 s; data not shown) remains under single-turnover conditions with limiting DNA. Because the enzymes are saturated and fully bound to the DNA substrates throughout these studies (supplemental Table S1), the lag must be due to a sum of multiple kinetic steps with nearly equivalent rate constants, implying the existence of several intermediates. Furthermore, the half-life derived from our observed kchem values (∼6 s, Fig. 2A) suggests that immediately after methylation, the loop reopens thereby releasing the cofactor. At ∼6 s in the stopped-flow trace, the loop appears to be well past initial closure, through the lag phase, and into loop reopening when half the DNA is methylated. This mechanism accounts for the decreased maximal intensity of COG DNA with AdoMet when compared with AdoHcy (Fig. 2C); the closed conformer was not as populated with AdoMet because the loops in some protein-DNA complexes are reopening after methyl transfer. Thus, methylation, or more specifically the conversion of AdoMet to AdoHcy, controls loop reopening which appears to follow rapid cofactor diffusion from the active site, as suggested by others (19).
Under multiple turnover conditions, with excess DNA, the lag phase is extended and represents the steady state population of loop motion during catalysis. Loop reopening does not begin until enzymes are bound to MCOG DNA after methyl transfer; hypothetically, if the concentration of COG DNA were kept constant while MCOG DNA was removed, the loop would never fully populate the open state. Also, because loop reopening is so much slower than methyltransfer, the transient species with COG DNA and AdoMet with a closed loop accumulates, leading to an increased lag period with the loop predominantly closed as determined by the maximum amplitude of fluorescence signal. If the intermediate were bound to MCOG DNA and AdoHcy or AdoMet, the lag would occur at a much lower intensity (Fig. 2, C and G). Loop reopening kinetics (after 20 s) fit well to single exponential curves with rate constants of ∼0.08 s−1 (supplemental Table S2) suggesting that this step contributes to kcat (∼0.05 s−1, supplemental Table S1) (18).
Loop Motion with Non-cognate DNA Substrates
Catalysis using AdoMet was also monitored with non-cognate DNA substrates to determine if loop positioning plays an active role in specificity and catalysis. Fluorescence traces with NC-PUR DNA and AdoMet show that the population of the closed conformer is significantly decreased when compared with COG DNA as determined by the maximum intensities (Fig. 2, E and F). This most likely accounts for the decrease in observed kchem because without proper loop closure, C81 is not in proximity to attack the flipped cytosine base (Fig. 2A and supplemental Fig. S1).
NC-INO DNA and COG DNA traces with AdoMet were nearly identical (Fig. 2, E and F), further supporting the notion that the interaction between Arg-240 and guanosine O6 is a major sequence determinant, which controls loop closure. Replacement of O6 into position one of the recognition sequence with an inosine base completely recovers loop motion when compared with NC-PUR/AdoMet traces. Because Arg-240 is the only protein moiety in contact with the O6, it strongly suggests that this residue communicates with and controls the positioning of the catalytic loop. NC-2AP DNA was measured with AdoMet but was not as definitive (Fig. 2, E and F and supplemental Discussion).
The robustness of this fluorescence-based approach is supported by our observation that the W41F/K91W and W41F/E94W mutants have nearly identical loop rearrangements and are highly functional, with catalytic parameters very close to WT. In addition, loop closure with W41F/K91W (Fig. 2G) results in an increase in signal, the exact opposite to observations made with the W41F/E94W mutant (Fig. 2, B and C). However, the differential quenching can be structurally justified; in the WT structure, the loop-open conformer has Lys-91 buried while Glu-94 is exposed (10). In the closed conformer, Lys-91 is exposed while Glu-94 is buried (9). W41F/K91W showed nearly identical catalytic parameters (supplemental Table S1) to WT and W41F/E94W in addition to similar kinetics of loop motion (supplemental Table S2). This second mutant validates this fluorescence method of analysis and bolsters our conclusions about loop motion in this class of enzymes.
R240A M.HhaI
To further examine the Arg-240 interaction with the guanosine O6, we removed the guanidinium side chain by constructing the R240A mutant of M.HhaI. The binding of R240A M.HhaI to its cognate DNA was decreased ∼23,000-fold from WT values and represents almost complete loss of affinity with values nearly identical to WT and NS DNA (supplemental Table S3). Interestingly, disruption of the Arg-240 to guanosine O6 interaction through either protein mutagenesis or with modified DNA substrates have similar impacts on affinity (WT/NC-2AP KD of 18 nm and R240A/COG KD of 394 nm versus WT/COG KD of 17 pm, supplemental Tables S1 and S3). This loss of binding energy (∼6–7 kcal/mol) is much larger than what is typically observed for individual protein-DNA contacts associated with binding, suggesting this interaction controls the formation of other contacts, perhaps through loop rearrangements (20, 21). The complete recovery of this binding energy with a DNA substrate lacking the target base (abasic substrate, AB DNA, supplemental Table S3) suggests that Arg-240 is involved in base flipping which itself could account for improper loop closure if base flipping and loop positioning are fully coupled (11). Interestingly, R240A shows the same 8-fold decrease in kchem with COG DNA as WT does with NC-2AP DNA (60 ms−1 and 74 ms−1, respectively, supplemental Table S3) further supporting the notion that removal of the Arg-240 to guanosine O6 interaction through modified DNA bases or mutant enzymes gives equivalent impacts on catalysis. Finally, the R240A mutant is unable to distinguish between the various non-cognate substrates, all giving a kchem of ∼50 ms−1. Thus, the side chain of Arg-240 and its contact with the DNA is essential for sequence discrimination. The combined data strongly support the critical interaction between Arg-240 and guanosine O6 as being a predominant determinant of loop closure, binding affinity, catalysis, and DNA discrimination.
We have measured catalytic loop reorganization in M.HhaI and provided a structural basis for sequence-specific DNA discrimination during both binding and catalysis. Although mechanisms posited to account for the extraordinary specificity achieved by DNA-modifying enzymes are scarce, our results provide real-time observations of conformational rearrangements responsible for substrate specificity, which lead to controlled catalysis. Elements in the recognition domain, especially the single hydrogen bond between Arg-240 of the enzyme and O6 of the guanosine at the first position in the recognition sequence, contribute significantly to the correct positioning of the catalytic loop. Arg-240 and the recognition domain are ∼28 Å away from the catalytic loop in the open conformer and indirectly determine both loop positioning and kinetics. Loop transitions correlate well with observed kchem and kcat rate constants and are heavily modulated by cofactors. Steric clashes in the active site most likely disrupt base flipping, prevent loop closure, and drive loop reopening post-catalysis. These long-range interactions that correlate with substrate binding and critically, enzyme turnover, will have broad application to enzyme specificity and drug design for this medically relevant class of enzymes.
Supplementary Material
Acknowledgments
We thank Dr. Matthew Purdy for the His-tagged M.HhaI construct, assistance in analyzing kinetic results, and critical review of this manuscript. We also thank Dr. Ben Youngblood and Dr. Stephanie Coffin for insightful discussions, Dr. John Perona and Dr. Fredrick Dahlquist for critical review of this manuscript, and Dr. Stanley Parsons for kinetic analysis and discussions. A special thanks to Dr. Bill Russ and Dr. Rama Ranganathan for assistance and insights with the statistical coupling analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant GM053763. This work was also supported by National Science Foundation Grant MCB-9983125.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Discussion, Tables S1–S3, Figs. S1–S3, and Movie S1.
- AdoMet
- S-adenosylmethionine
- AdoHcy
- S-adenosylhomocysteine
- WT
- wild type
- COG
- cognate
- NC
- non-cognate
- NS
- nonspecific.
REFERENCES
- 1.Russo V. E. A., Martienssen R. A., Riggs A. D. (eds) (1996) Epigenetic Mechanisms of Gene Regulation, pp. 1–69, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 2.Egger G., Liang G., Aparicio A., Jones P. A. (2004) Nature 429, 457–463 [DOI] [PubMed] [Google Scholar]
- 3.Wahnon D. C., Shier V. K., Benkovic S. J. (2001) J. Am. Chem. Soc. 123, 976–977 [DOI] [PubMed] [Google Scholar]
- 4.Lee B. H., Yegnasubramanian S., Lin X., Nelson W. G. (2005) J. Biol. Chem. 280, 40749–40756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stresemann C., Lyko F. (2008) Int. J. Cancer 123, 8–13 [DOI] [PubMed] [Google Scholar]
- 6.Christopoulos A. (2002) Nat. Rev. Drug Disc. 1, 198–210 [DOI] [PubMed] [Google Scholar]
- 7.Bheemanaik S., Reddy Y. V., Rao D. N. (2006) Biochem. J. 399, 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai Y. C., Johnson K. A. (2006) Biochemistry 45, 9675–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klimasauskas S., Kumar S., Roberts R. J., Cheng X. D. (1994) Cell 76, 357–369 [DOI] [PubMed] [Google Scholar]
- 10.O'Gara M., Zhang X., Roberts R. J., Cheng X. D. (1999) J. Mol. Biol. 287, 201–209 [DOI] [PubMed] [Google Scholar]
- 11.Estabrook R. A., Lipson R., Hopkins B., Reich N. (2004) J. Biol. Chem. 279, 31419–31428 [DOI] [PubMed] [Google Scholar]
- 12.Estabrook R. A., Reich N. (2006) J. Biol. Chem. 281, 37205–37214 [DOI] [PubMed] [Google Scholar]
- 13.Youngblood B., Shieh F. K., de Los Rios S., Perona J. J., Reich N. O. (2006) J. Mol. Biol. 362, 334–346 [DOI] [PubMed] [Google Scholar]
- 14.Lindstrom W. M., Jr., Flynn J., Reich N. O. (2000) J. Biol. Chem. 275, 4912–4919 [DOI] [PubMed] [Google Scholar]
- 15.Youngblood B., Buller F., Reich N. O. (2006) Biochemistry 45, 15563–15572 [DOI] [PubMed] [Google Scholar]
- 16.Shieh F. K., Youngblood B., Reich N. O. (2006) J. Mol. Biol. 362, 516–527 [DOI] [PubMed] [Google Scholar]
- 17.Li Y., Zon G., Wilson W. D. (1991) Biochemistry 30, 7566–7572 [DOI] [PubMed] [Google Scholar]
- 18.Svedruzić Z. M., Reich N. O. (2004) Biochemistry 43, 11460–11473 [DOI] [PubMed] [Google Scholar]
- 19.Merkiene E., Klimasauskas S. (2005) Nucleic Acids Res. 33, 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin A. M., Sam M. D., Reich N. O., Perona J. J. (1999) Nat. Struct. Biol. 6, 269–277 [DOI] [PubMed] [Google Scholar]
- 21.Parry D., Moon S. A., Liu H. H., Heslop P., Connolly B. A. (2003) J. Mol. Biol. 331, 1005–1016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.