

Abstract
Prolyl-4-hydroxylase domain (PHD) proteins are 2-oxoglutarate and dioxygen-dependent enzymes that mediate the rapid destruction of hypoxia-inducible factor α subunits. Whereas PHD1 and PHD3 proteolysis has been shown to be regulated by Siah2 ubiquitin E3 ligase-mediated polyubiquitylation and proteasomal destruction, protein regulation of the main oxygen sensor responsible for hypoxia-inducible factor α regulation, PHD2, remained unknown. We recently reported that the FK506-binding protein (FKBP) 38 specifically interacts with PHD2 and determines PHD2 protein stability in a peptidyl-prolyl cis-trans isomerase-independent manner. Using peptide array binding assays, fluorescence spectroscopy, and fluorescence resonance energy transfer analysis, we defined a minimal linear glutamate-rich PHD2 binding domain in the N-terminal part of FKBP38 and showed that this domain forms a high affinity complex with PHD2. Vice versa, PHD2 interacted with a non-linear N-terminal motif containing the MYND (myeloid, Nervy, and DEAF-1)-type Zn2+ finger domain with FKBP38. Biochemical fractionation and immunofluorescence analysis demonstrated that PHD2 subcellular localization overlapped with FKBP38 in the endoplasmic reticulum and mitochondria. An additional fraction of PHD2 was found in the cytoplasm. In cellulo PHD2/FKBP38 association, as well as regulation of PHD2 protein abundance by FKBP38, is dependent on membrane- anchored FKBP38 localization mediated by the C-terminal transmembrane domain. Mechanistically our data indicate that PHD2 protein stability is regulated by a ubiquitin-independent proteasomal pathway involving FKBP38 as adaptor protein that mediates proteasomal interaction. We hypothesize that FKBP38-bound PHD2 is constantly degraded whereas cytosolic PHD2 is stable and able to function as an active prolyl-4-hydroxylase.
The heterodimeric α/β transcription factor complexes of hypoxia-inducible factors (HIFs)4 are central regulators of the cellular, local, and systemic response to reduced oxygen partial pressure (pO2) (1, 2). Under normoxic conditions, two highly conserved prolyl residues within the oxygen-dependent degradation domain of HIFα subunits are hydroxylated by members of the prolyl-4-hydroxylase domain (PHD) family (also called egg laying-defective nine homolog (EGLN) or HIF prolyl hydroxylase) (3–5). Hydroxylated prolines are then bound by an E3 ubiquitin ligase complex containing the von Hippel-Lindau tumor suppressor protein (pVHL) as recognition subunit, mediating polyubiquitylation and proteasomal degradation of HIFα subunits (6–8). In addition, factor inhibiting HIF hydroxylates under normoxic conditions an asparaginyl residue in the C-terminal transcriptional transactivation domain of HIFα subunits, preventing the association with the CH1 domain of the p300 and cAMP-responsive element-binding protein-binding protein (CBP) co-activators (9, 10). PHDs and factor inhibiting HIF belong to the 2-oxoglutarate- and iron-dependent dioxygenase superfamily and act as cellular oxygen sensors by correlating the availability of oxygen with the regulation of HIF protein stability as well as transcriptional activity. Reduced tissue oxygenation leads to a concomitant decline in PHD and factor inhibiting HIF activity, resulting in the accumulation of HIFα subunits that translocate to the nucleus, heterodimerize with the constitutively expressed aryl hydrocarbon receptor nuclear translocator/HIFβ subunit, and function as active transcription factors (11, 12).
Higher metazoans have three PHD genes; whereas PHD2 mRNA levels are ubiquitously expressed, PHD1 mRNA is present at high levels in testes, and PHD3 mRNA is most abundant in the heart (13). Additionally a fourth enzyme (P4H) has been identified (14). P4H is membrane-anchored in the endoplasmic reticulum (ER); hence it is more closely related to the collagen prolyl hydroxylases but it can also hydroxylate HIFα subunits in vitro (15). Using short interfering RNAs, PHD2 was shown to be the main regulator of normoxic HIFα protein regulation (16). Genetic models have supported these findings; whereas Phd1−/− and Phd3−/− mice were apparently normal, Phd2−/− mice died between embryonic days 12.5 and 14.5 because of placental and heart defects (17). PHD2 was shown to be the main regulator of vascular growth (18) as well as renal erythropoiesis in adult mice (19), and somatic PHD2 inactivation resulted in polycythemia and dilated cardiomyopathy (20).
Transcript levels of PHD2 and PHD3, but not PHD1, are themselves regulated by HIF, and induction under hypoxic conditions leads to attenuated HIFα levels (21, 22). Initially it has been suggested that this feedback regulation would provide protection from reoxygenation (23, 24). We recently suggested that induced PHD levels compensate for the lack of oxygen also under hypoxic conditions and thereby define an adapted HIF threshold (25). In addition, not only increased PHD levels but also overactivation of the catalytic activity of the PHD isoforms by chronic hypoxia has been proposed (26). The catalytic activity of HIF hydroxylases can also be modulated by interfering with the different cofactors. Krebs cycle intermediates like fumarate and succinate inhibit PHDs, and genetic defects in fumarate hydratase or succinate dehydrogenases predispose to tumor formation involving accumulation of HIFα (27–29). In addition, also cellular availability of iron and ascorbate influences the catalytic activity of HIF hydroxylases. Iron and ascorbate supplementation suppressed HIF-1α accumulation in cancer cells, and decreased ascorbate levels resulted in HIF-1α stabilization (30, 31). Finally also reactive oxygen species as well as nitric oxide have been shown to interfere with HIFα stabilization by modulating PHD activity (32–34).
Apart from regulation of PHD mRNA levels and hydroxylase activity, differences in PHD protein abundance are directly linked to hydroxylation activity because HIF hydroxylation is a non-reversible process. Strikingly whereas PHD1 and PHD3 protein stability has been reported to be regulated by polyubiquitylation through the ubiquitin ligase Siah2, probably involving additional protein interfaces, the proteolytic regulation of the main oxygen sensor PHD2 remained unknown (35, 36).
FK506-binding protein 38 (FKBP38) is an immunophilin and belongs to the peptidyl-prolyl cis-trans isomerase protein family (37). Ca2+/calmodulin has been shown to bind and activate FKBP38, leading to modulation of Bcl-2 function (38–40). The FKBP38/Bcl-2 interaction was blocked by binding of Hsp90 to the tetratricopeptide repeat domain of FKBP38 (41, 42). In vivo, FKBP38 has been shown to control neural tube patterning. Genetic mouse models have suggested that FKBP38 functions as a negative regulator of the sonic hedgehog signaling pathway, thereby promoting bone morphogenic protein signaling (43, 44). Recently we reported that FKBP38 interacts with PHD2 and regulates PHD2 protein stability (45). Although this effect was independent of the peptidyl-prolyl cis-trans isomerase activity of FKBP38, the precise molecular mechanism remained unknown. Here we biochemically characterized the PHD2/FKBP38 interaction in more detail by defining the minimal FKBP38 fragment required for PHD2 binding and provide evidence that in cellulo interaction as well as functional regulation of PHD2 protein abundance depends on the membrane-associated localization of FKBP38. These findings imply that PHD2 is not diffusely distributed in the cytoplasm but localizes to defined intracellular structures that determine PHD2 protein stability and hydroxylase function.
EXPERIMENTAL PROCEDURES
Plasmids
If not indicated otherwise, cloning was carried out using Gateway technology (Invitrogen). Cloning of PCR fragments into entry vectors and recombination to destination vectors were described previously (45). All restriction enzymes were purchased from MBI Fermentas (Labforce, Nunnigen, Switzerland) or New England Biolabs (Bioconcept, Allschwil, Switzerland). pENTR4-PHD2 from residues 1 to 162 (pENTR4-PHD21–162), pENTR4-PHD21–31, pENTR4-PHD21–58, and pENTR4-PHD21–114 were obtained by amplifying the corresponding PHD2 cDNAs from the plasmid pENTR/D-PHD2 by PCR and cloning into XhoI/NcoI-digested pENTR4. pENTR/D-PHD221–426 and pENTR/D-PHD263–426 were obtained by digesting with MscI/ScaI or MscI/KasI followed by Klenow fill-in and religation. pENTR4-FKBP381–389 was generated by inserting a stop codon in pENTR4-FKBP381–412 by site-directed mutagenesis. The inserts of all entry vectors were verified by DNA sequencing (Microsynth, Balgach, Switzerland). The mammalian Matchmaker vectors pM and pVP16 (Clontech, Takara Bio Europe) were converted to destination vectors by ligation of the Gateway vector conversion cassette reading frame B (Invitrogen) into the EcoRI sites of pM and pVP16. The mammalian one-hybrid plasmid pM-HIF-1α370–429-VP16-AD and GST-HIF-1α530–826 were generated as described previously (45, 46). V5-tagged expression vectors were generated by recombination of entry vectors with pcDNA3.1/nV5-DEST. Fluorescent expression vectors pECFP-C1 and pEYFP-C1 (Clontech) were converted to destination vectors by ligation of the Gateway vector conversion cassette reading frame B (Invitrogen) into the SmaI sites of pECFP-C1 and pEYFP-C1 to generate pECFP-C1-DEST and pEYFP-C1-DEST, respectively.
Chemicals
Chemicals were purchased from the following companies: cycloheximide (CHX) (Sigma), E64 (Alexis Biochemicals, Lausen, Switzerland), 4-(2-aminoethyl)benzenesulfonyl fluoride (Alexis Biochemicals), MG132 (Sigma), N-acetyl-Leu-Leu-Met (Calbiochem, Merck Chemicals, VWR International, Switzerland), and Pepstatin A (Alexis Biochemicals). All other chemicals were purchased from Sigma.
Peptide Synthesis
Peptides were produced by solid-phase peptide synthesis with the robot Syro II (MultiSynTech, Witten, Germany) using 0.15 mmol of preloaded Fmoc-Asn(Trt)-Wang resin (Novabiochem, Läufelfingen, Switzerland). The synthesis was performed by Fmoc strategy and standard protocol as described previously (38). The purity of the peptides was evaluated by analytical HPLC, and the correct molecular masses were confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry.
Peptide Array Synthesis
Using the standard SPOT synthesis protocol (47), the peptides were synthesized stepwise by an Abimed Asp 222 synthesizer on a cellulose membrane derivatized with two β-Ala residues as linker.
Immunoblot Analysis of PHD2 Interaction with the Peptide Array
Before immunoblot screening, the dry peptide array membranes were rinsed for 10 min in methanol and three times for 20 min in TBS buffer (30 mm Tris/HCl, pH 7.6, 170 mm NaCl, 6.4 mm KCl). PHD2 variant solutions (100 nm) in TBS buffer were allowed to react with peptide array membranes for 4 h at 4 °C under gentle shaking. The membrane was subsequently washed three times with TBS buffer, and bound protein was blotted onto nitrocellulose membranes and analyzed using polyclonal rabbit anti-PHD2 antibodies (Novus Biologicals, Lubio Science, Lucerne, Switzerland).
Protein Binding Assay
Streptavin-agarose (Sigma) was saturated with biotin-EEEEEEEEEEDDLSELPPLE-NH2 peptide and washed three times with incubation buffer (25 mm Tris/HCl, pH 7.5, 200 mm NaCl, 1 mm dithiothreitol). PHD2 variants (1 μm) were incubated either in the presence or absence of Ac-EEEEEEEEEEDDLSELPPLE-NH2 peptide (20 μm) and FKBP38 (10 μm) with the affinity matrix for 1 h at 4 °C. Subsequently the samples were washed three times with incubation buffer and subjected to SDS-PAGE. Binding of PHD2 was analyzed using polyclonal rabbit anti-PHD2 antibodies (Novus Biologicals). For incubation with endogenous rat proteins, rat liver was decomposed in 50 mm Hepes buffer (4% Chaps, 1% dithiothreitol, 100 mm NaCl, 0.1% NaN3, pH 7.5) and centrifuged at 14,000 × g for 10 min at 4 °C. The supernatant was applied to the affinity matrix analogously to the PHD2 proteins.
Fluorescence Spectroscopy
Steady-state fluorescence spectra were recorded on a PerkinElmer Life Sciences FluoroMax2 fluorescence spectrometer using a 1 × 1-cm cuvette with an excitation wavelength of 280 nm and excitation and emission slit widths of 5 and 3 nm, respectively. To compensate for inner filter effects, the samples for fluorescence measurements were diluted to an optical density at 280 nm of 0.15. Protein samples were applied in 25 mm Tris/HCl, pH 7.5, 200 mm NaCl, 1 mm dithiothreitol. The binding constant (KD) was calculated from the fluorescence intensity by using the equation
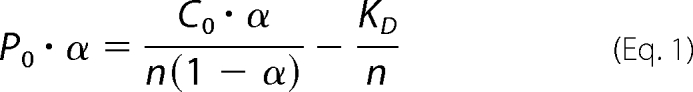 |
where P0 is total protein concentration, α is (Fmax − F)/(Fmax − F0), Fmax is fluorescence intensity at saturation, F0 is initial fluorescence intensity, n is the number of independent binding sites, C0 is total PHD2 concentration at each addition, and KD is the dissociation constant.
Protein Expression and Purification
GST and GST fusion proteins were expressed in Escherichia coli BL21-AI (Invitrogen) by induction with 0.02% arabinose for 4 h and purified using glutathione-Sepharose beads (GE Healthcare). Purification of GST-PHD2 from Sf9 insect cells was described previously (48). S4 and S2 were expressed in E. coli BL21(DE3)pLysS from pGEX-6P-PSMC1 and pGEX-6P-PSMD2, respectively.
In Vitro Transcription/Translation (IVTT) and GST Pulldown
IVTT reactions were carried out as described by the manufacturer (Promega, Dübendorf, Switzerland) using recombined destination vectors in the presence of [35S]Met (Hartmann Analytic, Braunschweig, Germany). Purified GST-tagged proteins (10 μg) were diluted in bead binding buffer (50 mm potassium phosphate, pH 7.5, 150 mm KCl, 1 mm MgCl2, 10% glycerol, 1% Triton X-100) and incubated with glutathione-Sepharose beads. For pulldown experiments, 20 μl of rabbit reticulocyte IVTT reactions or 10 μg of purified recombinant proteins were incubated for 2 h at 4 °C with bound GST fusion proteins in co-IP buffer (50 mm Tris/HCl, pH 7.6, 2 mm EDTA, 100 mm NaCl, 0.1% Triton X-100), washed four times with co-IP buffer, boiled in sample buffer (40 mm Tris/HCl, pH 6.8, 1% SDS, 50 mm β-mercaptoethanol) for 5 min, and separated by SDS-PAGE. Gels were stained with Coomassie Blue and dried, and radioactively labeled proteins were detected by phosphorimaging (Molecular Imager FX, Bio-Rad).
Cell Culture and Transient Transfection
HeLa cervical carcinoma, HEK293 embryonic kidney carcinoma, MCF-7 breast cancer, mouse ts20, and H38-5 cells were cultured in high glucose Dulbecco's modified Eagle's medium (Sigma) as described previously (46). Generation of stable RNAi-mediated down-regulation of FKBP38 was reported previously (45). Briefly HeLa cells were transfected by calcium phosphate coprecipitation with pSilencer2.1-U6 hygro (Ambion, Huntingdon, UK) containing either a control or FKBP38 targeting sequence, and single clones were selected by limited dilution. FKBP38 expression was analyzed by reverse transcription quantitative PCR and immunoblotting. Strong FKBP38 down-regulation was observed in stable clones 3D6 and 2G8. Transient transfections were performed with the polyethylenimine (Polysciences, Warrington, PA) method as reported before (25).
Immunoblotting
Total cell lysates were prepared using RIPA buffer (50 mm Tris/HCl, pH 8.0, 1 mm EDTA pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS). Protein concentration was determined using the BCA assay (Pierce, Perbio Science). Immunoblotting was performed as described previously (49). Briefly protein was separated by SDS-PAGE, electrotransferred onto nitrocellulose membranes (GE Healthcare), and incubated with antibodies. The following antibodies were used: rabbit anti-human PHD2 (Novus Biologicals), rabbit anti-mouse PHD2 (Novus Biologicals), rabbit anti-FKBP38 (39), mouse anti-β-actin (Sigma), mouse anti-protein-disulfide isomerase (Novus Biologicals), rabbit anti-mitofilin (Novus Biologicals), mouse anti-p53 (Santa Cruz Biotechnology, Labforce, Nunningen, Switzerland), mouse anti-V5 (Invitrogen), mouse anti-GST (Sigma), goat anti-S2 (Novus), and rabbit anti-S4 antibodies (Novus).
Bioluminescence Assays for Proteasomal Activity
Assays were performed as described by the manufacturer (Proteasome-GloTM assay system, Promega). Briefly HeLa cells were lysed in a buffer containing 20 mm Tris/HCl, pH 7.2, 150 mm NaCl, 0.1 mm EDTA, 1 mm 2-mercaptoethanol, 5 mm ATP, 20% glycerol, 0.1% Triton X-100. The lysate was centrifuged at 800 × g for 5 min at 4 °C to pellet the nuclei. The supernatant was centrifuged at 100,000 × g for 1 h at 4 °C, and the resulting supernatant was saved as the cytosolic fraction. The pellet was further lysed in the same lysis buffer as described above except that Triton X-100 was 0.5% and centrifuged at 16,000 × g for 15 min at 4 °C. The resulting supernatant was saved as the membrane fraction. Lysates (5 μg in 10 μl of 10 mm Hepes, pH 7.6) were incubated at room temperature with 10 μl of Suc-LLVY-GloTM (chymotrypsin-like), Z-LRR-GloTM (trypsin-like), or Z-nLPnLD-GloTM (where nL is norleucine) (caspase-like) reagent for 30 min before hydrolysis of the peptide was measured (Berthold Technologies, Regensdorf, Switzerland).
Subcellular Fractionation
Cells were incubated in hypotonic buffer (10 mm Hepes, pH 7.5, 1 mm EGTA, 25 mm KCl) on ice for 20 min and afterward Dounce homogenized in homogenization buffer (10 mm Hepes, pH 7.5, 1 mm EDTA, 250 mm sucrose). Cell lysates were centrifuged at 3,000 × g for 15 min at 4 °C to pellet nuclei and non-lysed cells. To separate the membrane from the cytosolic fraction, the supernatant was centrifuged at 100,000 × g for 1 h at 4 °C. The membrane fraction was resuspended in homogenization buffer, overlaid on a 10–30% iodixanol gradient, and centrifuged at 150,000 × g for 18 h to separate the membrane fractions. 1-ml fractions were obtained by puncture of the centrifugation tubes and analyzed by immunoblotting.
Fluorescence Microscopy and Fluorescence Resonance Energy Transfer (FRET) Analysis
Cells were cultivated on microscope coverslips, washed twice with ice-cold phosphate-buffered saline, fixed on ice for 30 min with 4% paraformaldehyde, and permeabilized with 0.1% saponin in phosphate-buffered saline. Endogenous as well as transfected proteins were detected using the indicated antibodies. Cellular organelles were stained with MitoTracker (Molecular Probes, Invitrogen), wheat germ agglutinin-Alexa Fluor 594 (Molecular Probes, Invitrogen) and with antibodies against protein-disulfide isomerase or calreticulin (Novus Biologicals). Immune complexes were visualized with goat anti-rabbit Alexa Fluor 488, goat anti-mouse Alexa Fluor 488, goat anti-rabbit Alexa Fluor 568, or goat anti-mouse Alexa Fluor 568 (Molecular Probes, Invitrogen), respectively. Nuclei were stained with 4′,6-diamidino-2-phenylindole (Sigma) for 30 min. After extensive washings with phosphate-buffered saline, the microscope slides were embedded in Mowiol and analyzed by confocal laser scanning microscopy (SP1, Leica Microsystems). For FRET analysis, HEK293 cells were transfected with the indicated plasmids, and FRET was monitored as described previously (50). FRET signals were analyzed using the sensitized FRET method (51).
Mammalian One- and Two-hybrid Assays
Mammalian one- and two-hybrid analyses were performed using the mammalian Matchmaker system (Clontech) as described previously (45). HeLa cells were transiently co-transfected with 10 ng of one-hybrid or 1.5 μg of Gal4 DNA binding domain (DBD) and 1.5 μg of Gal4 activation domain (AD) fusion protein vectors together with 500 ng of firefly luciferase reporter vector pGRE5xE1b and 20 ng of pRL-SV40. Luciferase reporter gene activity was determined using the Dual-Luciferase reporter assay system according to the manufacturer's instructions (Promega).
RESULTS
Biochemical Characterization of the PHD2/FKBP38 Interaction
Based on our previous discovery of the interaction between PHD2 and the N-terminal domain of FKBP38, we performed a peptide scan to identify the actual interaction sites. Thereto we incubated purified recombinant PHD2 with a peptide array of 13-amino acid peptides corresponding to the sequence of FKBP38, consecutively shifted forward by 1 amino acid. PHD2 bound to a cluster of peptides corresponding to the FKBP38 residues 37–56 in the N-terminal extension preceding the catalytic domain (Fig. 1, A and B, upper panel). PHD2 did not bind to the inverted sequence of this motif in the peptide array, demonstrating stereospecificity of the identified interaction (Fig. 1B, lower panel). Analogous experiments investigating the binding of recombinant FKBP38 to a PHD2 peptide array did not provide a distinct binding pattern indicating a binding site that could not be mimicked by the 13-mer peptides (data not shown).
FIGURE 1.

Analysis of the PHD2 interaction domain in FKBP38. A, domain architecture of FKBP381–412. PPIase, peptidyl-prolyl cis-trans isomerase; TPR, tetratricopeptide repeats; CaM, calmodulin-binding site; TM, TM domain. B, an array of 13-mer peptides spanning the FKBP38 sequence was synthesized with forward shifts by 1 amino acid. PHD2 interaction with the peptide array of the FKBP38 forward sequence (upper panel) and reverse sequence (lower panel) was analyzed by immunoblotting. The PHD2 interaction pattern is displayed in the framed region. The respective binding motif comprised the peptides b13–24, which correspond to FKBP3837–56. The reverse sequence comprising the same residues in the reverse order in the peptides f1–12 was not found to interact with PHD2 (lower panel). C, immunoblot analysis of the interaction between two different PHD2 variants and a biotin-labeled FKBP3837–56 peptide-bound streptavidin matrix using anti-PHD2 antibodies. The streptavidin matrix alone served as control. D, endogenous proteins from rat liver lysates were incubated with an FKBP3837–56 affinity matrix, and PHD2 binding was detected by immunoblotting. The streptavidin matrix alone served as binding control. Representative values in the lower column diagrams are average relative band intensities with S.E. of several independent experiments. p values were obtained by unpaired t tests (*, p < 0.05; **, p < 0.01). E, fluorescence measurements at an excitation wavelength of 278 nm with 1 μm FKBP38 (—), 1 μm PHD2 (····), and a 1:1 mixture of both proteins (- - - -). The calculated spectrum (_ _ _) represents the sum of the individual protein spectra as it should appear when the components do not interact. F, titration curve resulting from fluorescence measurements at 340 nm (excitation at 278 nm) of a sample containing 1 μm FKBP38/PHD2 and various concentrations of a peptide corresponding to FKBP3837–56. The straight line represents the fit according to the equation under “Experimental Procedures.” rel., relative; cps, counts/s.
Based on these results, the FKBP3837–56 peptide biotinyl-EEEEEEEEEEDDLSELPPLE-NH2, which corresponds to the PHD2-interacting motif, was immobilized on streptavidin beads and analyzed for binding to purified PHD2 protein variants. PHD21–426 strongly bound to the affinity matrix, and binding was significantly diminished in the presence of the FKBP3837–56 peptide or FKBP38 itself (Fig. 1C), indicating competition between the matrix and soluble FKBP38 variants for PHD2 binding. In contrast, PHD2170–426 bound to the affinity matrix only very weakly, suggesting specific binding between the N-terminal PHD2 domain and FKBP3837–56. Furthermore endogenous rat PHD2 from crude liver lysates bound only to the affinity matrix, and binding was efficiently decreased by addition of the FKBP3837–56 peptide (Fig. 1D). The identity of bound PHD2 was confirmed by mass spectrometry analysis (data not shown).
To further analyze the interaction between FKBP38 and PHD2, measurements of protein fluorescence of both interaction partners were performed. The fluorescence spectrum of the mixed proteins was blue-shifted by 2 nm, and the amplitude was increased by 12% compared with the calculated sum of the individual protein spectra (Fig. 1E). In comparison, no changes in the protein fluorescence were observed when PHD2170–426 was added to FKBP38 (supplemental Fig. S1), confirming the requirement of the N-terminal PHD2 domain for the interaction with FKBP38. The increase in the fluorescence signal that occurs upon FKBP38/PHD2 interaction was reduced in the presence of the FKBP3837–56 peptide in a concentration-dependent manner (Fig. 1F). The measurements resulted in a KD value of 1.48 ± 0.15 μm. Isothermal titration calorimetry measurements characterized the interaction between PHD2 and the N-terminal extension of FKBP38 as a 1:1 complex with a KD of 895 ± 148 nm (data not shown). The interaction between PHD2 and FKBP3837–56 was measured in parallel by isothermal titration calorimetry resulting in a KD of 1.28 ± 0.19 μm, which is similar to the result of the fluorescence titration curve.
Mapping the FKBP38 Interaction Domain in PHD2
The PHD2 protein contains an N-terminal MYND (myeloid, Nervy, and DEAF-1)-type Zn2+ finger domain from residues 21 to 58 and a prolyl-4-hydroxylase catalytic domain from residues 205 to 391 (Fig. 2A). We have shown previously that the N-terminal region of PHD2 between residues 1 and 169 interacts with FKBP38 (45). To characterize the interaction domain in more detail, we generated several PHD2 N-terminal deletion constructs and analyzed the interaction by GST pulldown experiments. GST-FKBP38 strongly interacted with IVTT 35S-labeled full-length PHD2 and PHD21–162. No interaction was observed with any of the N-terminal deletions, PHD221–426, PHD263–426, and PHD2170–426 (Fig. 2B). GST alone did not bind to the different PHD2 fragments. Equal input of recombinant proteins was confirmed by Coomassie Blue staining (data not shown). PHD2 C-terminal deletion fragments were purified as GST fusion proteins from bacteria, and the in vitro interaction was tested with IVTT 35S-labeled FKBP38 (Fig. 2, C and D). FKBP38 only bound to GST-PHD21–426 and GST-PHD21–114 but not to GST-PHD21–31 and GST-PHD21–58. Taken together, these data suggest a non-linear region of PHD2, comprising the residues 1–20 and 59–114, as the interaction domain with FKBP38.
FIGURE 2.

Mapping the interaction domain of PHD2. A, schematic representation of the PHD2 domain architecture and the PHD2 constructs used. B, IVTT 35S-labeled PHD2 variants were incubated with GST-FKBP38 or GST alone. Protein complexes were pulled down with glutathione-Sepharose beads, separated by SDS-PAGE, and visualized by phosphorimaging. C and D, IVTT 35S-labeled FKBP38 was incubated with recombinant GST-PHD21–426, GST-PHD2170–426, GST-PHD21–31, GST-PHD21–58, GST-PHD21–114, or GST alone. Protein complexes were pulled down and visualized as described above.
Ubiquitin-independent Proteasomal Degradation of PHD2
To explore the molecular mechanism of FKBP38-mediated PHD2 protein regulation, we first sought to investigate the proteolytic regulation of PHD2. MCF-7 cells were cultivated under normoxic or hypoxic conditions and reoxygenated in the presence of the cysteine protease inhibitor E64, the serine protease inhibitor 4-(2-aminoethyl)benzenesulfonyl fluoride, the proteasomal inhibitor MG132, the calpain and cathepsin inhibitor N-acetyl-Leu-Leu-Met, the aspartyl protease inhibitor Pepstatin A, or solvent controls. PHD2 protein levels remained unaffected by the various protease inhibitors except for MG132, which slightly increased PHD2 protein levels (data not shown). To further investigate the degradation pathway of PHD2, we induced PHD2 protein levels by hypoxia and analyzed PHD2 protein abundance under reoxygenation conditions in the presence or absence of MG132 and/or CHX. Overall PHD2 protein abundance as well as PHD2 protein half-life increased in the presence of MG132 (Fig. 3, A and B, respectively). Of note, addition of hydrogen peroxide from 1 to 250 μm altered neither PHD2 nor FKBP38 protein levels (data not shown).
FIGURE 3.

Proteolytic regulation of PHD2. Cellular extracts were prepared, separated by SDS-PAGE, and analyzed by immunoblotting. A, MCF-7 cells were preincubated for 24 h at 0.2% O2 and then reoxygenated for 20 or 40 h in the presence of solvent control, MG132 (5 μm), and/or CHX (50 μm). B, MCF-7 cells were cultivated for 24 h under 0.2% O2 before CHX (50 μm) and solvent control or CHX and MG132 (5 μm) were added for the indicated time periods. PHD2 and β-actin protein levels were analyzed by immunoblotting. C, mouse ts20 cells were cultivated at either 34 or 39 °C for 24, 32, or 48 h, and cellular extracts were analyzed by immunoblotting. D, mouse ts20 cells reconstituted with a wild-type E1 gene (H38-5) were incubated under the same conditions as described in C, and cellular proteins were analyzed by immunoblotting. reox, reoxygenation; ctrl, control; rel., relative. *, p < 0.05; **, p < 0.01.
To investigate whether ubiquitylation is required for proteolytic PHD2 regulation, we made use of mouse ts20 cells that harbor a temperature-sensitive E1 ubiquitin-activating enzyme (52). Whereas wild-type p53 accumulated because of decreased ubiquitin-dependent proteasomal degradation when ts20 cells were cultivated at restrictive 39 °C, PHD2 protein abundance was not affected (Fig. 3C). As control, incubation of ts20 cells stably transfected with a wild-type E1 gene (H38-5) at 34 or 39 °C neither resulted in p53 nor PHD2 accumulation (Fig. 3D). In addition, FKBP38 protein levels were not regulated by ubiquitin-mediated proteasomal degradation (data not shown). Furthermore we analyzed whether PHD2 protein stability is altered under hypoxic conditions. PHD2 protein levels were determined in cells treated with CHX and cultivated for different time points under normoxic or hypoxic conditions. PHD2 protein stability was not affected by the differences in pO2 (supplemental Fig. S2).
FKBP38 Interacts Simultaneously with Proteasomal Subunits as well as PHD2 and Modulates Membrane-associated Proteasomal Activity
Nakagawa et al. (53) reported that FKBP38 interacts with its tetratricopeptide repeat domains (residues 228–339) with subunits of the 26 S proteasome and proposed that anchoring proteasomal activity to organellar membranes is mediated by FKBP38. To further elaborate the mechanism by which PHD2 protein stability is regulated by FKBP38, we used GST pulldown assays and confirmed that FKBP38 interacts with the proteasomal subunits S2 and S4 (Fig. 4, A and B). Furthermore binding of S2 or S4 to FKBP38 did not influence simultaneous binding of PHD2 corroborating that FKBP38 interacts with different domains with these proteins. FRET methodology was used to analyze whether S4 also interacts in cells with FKBP38. FRET signals were detected when CFP-S4 and YFP-FKBP38 were expressed in HEK293 cells (Fig. 4C). To investigate whether cellular proteasomal activity is modulated by FKBP38 expression, we measured chymotrypsin-, trypsin-, and caspase-like activities in parental as well as FKBP38-down-regulated HeLa cells. Whereas cytosolic proteasomal activities were increased by FKBP38 suppression, membrane-associated proteolytic activities were reduced compared with parental cells (Fig. 4D). Generation and analysis of cells harboring stable RNAi-mediated down-regulation of FKBP38 was reported previously (45) (see also “Experimental Procedures”). These data indicate that FKBP38 anchors the 26 S proteasome to intracellular membranes and might thereby regulate proteasomal PHD2 degradation.
FIGURE 4.

FKBP38 regulates proteasomal activity. A and B, recombinant GST or GST-FKBP38 proteins were incubated with recombinant S2 or S4 and PHD2 proteins, and protein complexes were pulled down with glutathione-Sepharose beads, separated by SDS-PAGE, and visualized by immunoblotting. C, HEK293 cells were transiently transfected with CFP-S4 and YFP-FKBP38 plasmids, and FRET analysis was performed. Subcellular distribution of FRET efficiency signals ranging from 0 to 60% was visualized in false color mode as indicated by the color bar (black, 0%; white, 60%). FRET efficiencies of single cells were averaged and plotted to the acceptor/donor fluorescence ratio. D, cytosolic or membrane fractions of parental or FKBP38-down-regulated 2G8 HeLa cells were incubated with Suc-LLVY-, Z-LRR-, or Z-nLPnLD-aminoluciferin. Results are mean values ±S.E. of n = 3 independent experiments performed in triplicates. wt, wild type; RLU, relative luciferase units.
FKBP38 and PHD2 Co-localize in Mitochondrial and ER Fractions
FKBP38 contains a C-terminal transmembrane domain (Fig. 1A) and has been reported to be an integral ER and mitochondrial membrane protein with a topology in which the protein is exposed to the cytoplasm (39, 43). Using indirect immunofluorescence, we found co-localization of endogenous FKBP38 with ER (calnexin) and mitochondria (MitoTracker) markers but not with the Golgi apparatus (wheat germ agglutinin), confirming the published results (supplemental Fig. S3). Endogenous PHD2 was found to localize in the cytoplasm and mitochondria and to a lesser extent in the ER but not in the Golgi region (supplemental Fig. S3). Next we analyzed the subcellular localization of PHD2 by biochemical fractionation in parental (wild-type) as well as FKBP38 down-regulated (2G8) HeLa cells cultivated under normoxic or hypoxic conditions (Fig. 5). Overall PHD2 protein abundance was elevated in FKBP38-silenced (Fig. 5, B and D) compared with parental (Fig. 5, A and C) HeLa cells as reported previously. FKBP38 protein was present in fractions containing mitochondria (mitofilin) as well as ER membranes (protein-disulfide isomerase) as expected. PHD2 also was mainly found in mitochondria and ER fractions but also was found in the high density fraction 10 and in the cytosolic fraction (Fig. 5E). In FKBP38-down-regulated cells (2G8), higher PHD2 protein levels were observed in ER fractions under hypoxic (Fig. 5D) compared with normoxic (Fig. 5B) conditions. Taken together, PHD2 co-localized with FKBP38 in fractions containing ER and mitochondrial membranes and was also found in membrane-free cytosolic fractions in which FKBP38 was not detectable.
FIGURE 5.

Analysis of FKBP38 and PHD2 protein levels by biochemical fractionation. Parental HeLa (wild type (wt)) or FKBP38-silenced (2G8) HeLa cells were cultivated at 20% O2 (A and B) or at 0.2% O2 (C and D). Cellular membranes were separated from cytosolic fractions by differential centrifugation and then separated in a 10–30% iodixanol gradient. 1-ml fractions were collected and analyzed by immunoblotting. Mitofilin served as a mitochondria marker, and protein-disulfide isomerase (PDI) served as an ER marker. E, cytosolic fractions of parental HeLa and HeLa 2G8 cells were analyzed by immunoblotting.
FKBP38 Membrane-associated Localization Is Required for the Interaction with PHD2 within Cells
FRET technology was applied to investigate whether membrane insertion of FKBP38 is required for PHD2 interaction. Strong FRET signals were observed when CFP-FKBP38 and YFP-PHD2 were expressed in HEK293 cells but not when the PHD2 interaction domain within FKBP38 was deleted (CFP-FKBP3899–412; Fig. 6A). Surprisingly no FRET signal could be recorded when an FKBP38 mutant lacking the transmembrane (TM) domain (CFP-FKBP381–389) was co-expressed with PHD2 despite the presence of the N-terminal interaction domain (Fig. 6A and supplemental Fig. S4). Whereas YFP-labeled FKBP38 and FKBP3899–412 co-localized with both of the subcellular localization vectors CFP-ER and CFP-Mito (data not shown), deletion of the TM domain of FKBP38 led to a more homogeneous distribution throughout the cytoplasm and to some nuclear localization (Fig. 6A). However, in in vitro GST pulldown experiments FKBP38 interacted with PHD2 independently of the FKBP38 TM domain (Fig. 6B). To confirm the requirement of the FKBP38 TM domain for cellular PHD2 interaction, we applied mammalian two-hybrid technology. The activity of a co-transfected luciferase reporter gene construct was greatly enhanced when DBD-PHD2 and AD-FKBP381–412 were co-transfected in HeLa cells, but consistent with the FRET data, no luciferase reporter gene activity was measured when the TM domain was deleted in FKBP38 (FKBP381–389; Fig. 6C). These data suggest that the TM domain of FKBP38 is required for the binding to PHD2 in a cellular context.
FIGURE 6.

FKBP38 transmembrane domain is required for PHD2 interaction. A, HEK293 cells were transiently transfected with the indicated CFP or YFP plasmids, and FRET analysis was performed. B, recombinant GST, GST-FKBP38, or GST-FKBP381–389 proteins were incubated with IVTT 35S-labeled PHD2, and protein complexes were pulled down with glutathione-Sepharose beads, separated by SDS-PAGE, and visualized by phosphorimaging. C, HeLa cells were transiently transfected with Gal4 DBD and Gal4 activation domain (VP16-AD) fusion protein vectors, Gal4 response element-driven firefly luciferase reporter, and a Renilla luciferase control vector. Following transfection, the cells were incubated under normoxic (20% O2) or hypoxic (0.2% O2) conditions, and luciferase reporter gene activities were determined 16 h later. Firefly to Renilla luciferase activity ratios were normalized to the normoxic negative control DBD-p53/AD-CP (CP) (VP3 polyoma virus coat protein) co-transfection that was arbitrarily defined as 1. DBD-p53/AD-LT (LT) (large T antigen) served as a positive control. Mean values ±S.E. are shown of n = 3 independent experiments performed in triplicates.
FKBP38 Transmembrane Domain Is Necessary for Regulation of PHD2 Protein Abundance
To investigate whether the FKBP38 TM domain is required for functional regulation of PHD2, we transfected cells with full-length FKBP38, the peptidyl-prolyl cis-trans isomerase domain-lacking mutant FKBP38Δ98–257, or the TM domain mutant FKBP381–389. FKBP38 as well as FKBP38Δ98–257 localized to the ER and mitochondria as expected, whereas deletion of the TM domain in FKBP381–389 resulted in localization throughout the cytoplasm (supplemental Fig. S4). We reported previously that short interfering RNA-mediated FKBP38 down-regulation led to increased PHD2 protein levels and reduced HIF-1α protein accumulation. On the other hand and in agreement with the previous results, expression of full-length FKBP38 and FKBPΔ98–257 enhanced the activity of a luciferase HIF-1α one-hybrid reporter construct under normoxic as well as hypoxic conditions (Fig. 7A). However, FKBP381–389 had no effect on the HIF-1α stability reporter and was comparable to the mock-transfected control (Fig. 7A), indicating that the TM domain of FKBP38 is not only required for in cellulo interaction but also for functional PHD2 regulation. Similar results were obtained in cells stably transfected with a control or FKBP38 short interfering RNA plasmid (3D6 and 2G8). Generation of these cells was described previously (45). Expression of full-length FKBP38 increased HIF-1α370–429 one-hybrid stability, whereas FKBP381–389 had no effect (Fig. 7B). Note that the activity of the HIF-1α one-hybrid reporter was decreased in mock-transfected FKBP38 knockdown cells compared with control cells due to increased PHD2 protein abundance. Next we investigated the effect of FKBP38 or FKBP381–389 overexpression on endogenous PHD2 protein levels. PHD2 protein levels were increased in FKBP38-down-regulated cells (3D6 and 2G8) compared with control cells, and PHD2 was attenuated when FKBP38 expression was reconstituted (Fig. 7C). Expression of FKBP381–389 did not revert elevated PHD2 protein levels, confirming that functional PHD2 protein regulation requires membrane-associated subcellular localization of FKBP38.
FIGURE 7.

FKBP38 transmembrane domain is necessary for functional regulation of PHD2. A, MCF-7 cells were transiently transfected with Gal4-DBD-HIF-1α370–429-VP16-AD expression vector, Gal4 response element-driven firefly luciferase reporter, and a Renilla luciferase control vector and either co-transfected with V5-FKBP38, V5-FKBP38Δ98–357, V5-FKBP381–389, or a mock plasmid (pcDNA3.1-LacZ). 24 h post-transfection, cells were either cultured under normoxic or hypoxic conditions for an additional 16 h before relative luciferase activities were determined. Results are presented as mean values of relative luciferase activities ±S.E. of n = 4 independent experiments performed in triplicates. p values were obtained by paired t tests (**, p < 0.01; *, p < 0.05). Expression of the transfected vectors was verified by immunoblotting against the V5 tag. B, HeLa FKBP38 RNAi control cells (ctrl) and FKBP38 RNAi-depleted cells (3D6 and 2G8) were transiently transfected with the indicated plasmids as described in A. Results are mean values ±S.E. of n = 7 independent experiments performed in triplicates. C, parental HeLa as well as RNAi control (ctrl) and FKBP38 down-regulated (3D6 and 2G8) cells were transiently transfected with the indicated plasmids, and PHD2, V5, and β-actin were detected by immunoblotting.
DISCUSSION
HIFα protein stability is tightly controlled by O2-dependent hydroxylation of specific prolyl residues within the oxygen-dependent degradation domain by the HIF prolyl-4-hydroxylases. In addition to the transcriptional feedback regulation of PHD2 and PHD3 by HIF itself, the catalytic activity of the PHDs is influenced by numerous factors such as Krebs cycle intermediates, availability and oxidation state of iron, intracellular ascorbate levels, reactive oxygen species, and nitric oxide. Whereas the protein stability of PHD1 and PHD3 is regulated by polyubiquitylation and proteasomal degradation, the molecular mechanism of proteolytic PHD2 regulation remained enigmatic. We recently reported that PHD2 protein stability is modulated by the peptidyl-prolyl cis-trans isomerase FKBP38 in a peptidyl-prolyl cis-trans isomerase-independent manner. In the present study, we biochemically characterized the interaction of PHD2 with FKBP38 in more detail, providing evidence that PHD2 is degraded in the proteasome via an ubiquitin-independent mechanism and showing that the membrane-associated subcellular localization of FKBP38 is important for functional regulation of PHD2.
The motif required for PHD2 binding comprised a minimal linear glutamate-rich binding domain from the FKBP38 residues 37–56 (Fig. 1). Measurements of the protein fluorescence and isothermal titration calorimetry resulted in a KD value of about 1 μm for the interaction between FKBP38 and PHD2, providing evidence for a high affinity complex. In addition, FRET efficiency for the PHD2/FKBP38 interaction was very high, indicating a short distance between the two molecules and thus close interaction (Fig. 6). When compared with FRET data recently obtained for the HIF-1α/HIF-1β heterodimer, PHD2 and FKBP38 may be closer to each other than the subunits of the HIF-1 dimer (50). Although so far no data are available to correlate distances between proteins within the HIF system with function, our data may also define the range of protein-protein distances that is acceptable for active protein complexes. Larger distances may in fact be indicative of additional proteins/co-activators contributing to the active complex (54).
FKBP38 interacts with a non-linear motif in PHD2 located between residues 1 and 114 (Fig. 2). This region contains a MYND-type Zn2+ finger domain that has been shown to constitute a protein/protein interaction domain implicated in transcriptional repression and has been found among others also in histone methyltransferases (55–57). Although addition of the Zn2+ chelator N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine increased the hydroxylation activity of full-length PHD2 but not that of a MYND deletion mutant (58), it did not influence the interaction of PHD2 with FKBP38 in vitro (data not shown). Previously the ubiquitin E3 ligase Siah2 has been proposed to target PHD1 and PHD3 for proteasome-dependent degradation, whereas PHD2 protein levels remained unchanged (35, 36). Interestingly the MYND domain was also implicated in proteolytic regulation (59) because N-terminal MYND domain deletion rendered PHD2 susceptible to Siah2-mediated polyubiquitylation and proteasomal degradation (60). Hence the MYND domain, which is among PHDs only found in PHD2, might be involved in alternative proteolytic regulation of PHD2 compared with PHD1 and PHD3. Because crystallization of full-length PHD2 was unsuccessful and a structure was only obtained for the catalytic domain of PHD2 (61), identification of the PHD2/FKBP38 interaction domains might assist computer-based modeling and development of small molecules that could interfere with the PHD2/FKBP38 interaction, resulting in modulated PHD2 protein abundance and HIFα stability.
The mechanism of proteolytic PHD2 regulation is still unknown. Here we show that inhibition of the proteasomal degradation pathway increased PHD2 protein stability, although blocking the ubiquitin-activating enzyme E1 did not lead to the accumulation of PHD2 (Fig. 3). This suggests that polyubiquitylation is not required for proteasomal PHD2 degradation. Interestingly a recent approach of measuring global protein stability in mammalian cells and determination of proteins whose stability was changed in response to proteasomal inhibition identified PHD2 as proteasome substrate (62). Ubiquitin-independent proteasomal degradation has been shown for a number of proteins (63–68), and accumulating evidence underscores the importance of this pathway in addition to classic ubiquitin-dependent degradation in the proteasome (69). How selectivity of substrate recognition is achieved in ubiquitin-independent proteasomal degradation is still a matter of debate. Several mechanisms such as protein oxidation (70, 71) and indirect substrate recognition through adaptor molecules (72, 73) have been proposed. Intriguingly we confirmed data reported by Nakagawa et al. (53) and showed that FKBP38 interacts with proteasomal subunits in vitro as well as in cellulo (Fig. 4). Simultaneous binding of PHD2 and proteasomal subunits S2 or S4 to FKBP38 suggests that FKBP38 might act as an adaptor protein, mediating proteasomal interaction and subsequent degradation of PHD2. Similarly to what has been described for Fkbp38−/− mice (53), biochemical fractionation revealed reduced proteasomal activity in membrane fractions derived from FKBP38-down-regulated cells (Fig. 4D), providing an explanation for the increased PHD2 protein stability in our FKBP38 knockdown clones.
Although the presence of the N-terminal PHD2 interaction domain of FKBP38 is sufficient to confer in vitro interaction, our data suggest that functional regulation of PHD2 protein abundance depends on the endogenous, membrane-associated subcellular localization of FKBP38 (Figs. 6 and 7). Independent reports have indicated that PHD2 can localize to different subcellular compartments. Initial overexpression data indicated that PHD2 is mainly located in the cytoplasm (74, 75), and analysis of endogenous PHD2 protein localization confirmed these data, although also nuclear staining was observed in both normal and neoplastic tissues (76) as well as various cell lines (77). Recently also mitochondrial and peroxisomal PHD2 localization was reported (78). We observed mainly cytoplasmic PHD2 localization partially overlapping with a mitochondrial marker and to a lesser extent also with an ER marker (Fig. 5 and supplemental Fig. S3). Although PHD2 and FKBP38 form a high affinity complex, PHD2 does not completely co-localize with FKBP38, and no shift in subcellular PHD2 localization was observed in FKBP38-depleted cells. Therefore, we hypothesize that a fraction of PHD2 binds to FKBP38, which serves as an adaptor molecule and mediates ubiquitin-independent proteasomal degradation, whereas cytosolic PHD2 is stable and able to function as an active prolyl-4-hydroxylase under optimal enzymatic conditions. Clearly future research is needed to clarify which factors regulate the association of PHD2 with FKBP38 and how proteasomal recognition of non-ubiquitylated PHD2 occurs.
In summary, we biochemically identified the interacting regions of PHD2 and FKBP38 and provide evidence that PHD2 protein stability is regulated by ubiquitin-independent proteasomal proteolysis, coherent with the observation that membrane-associated subcellular localization of FKBP38 is required for functional regulation of PHD2 protein levels. Whereas PHD1 and PHD3 have been shown to undergo Siah2-mediated polyubiquitylation and proteasomal degradation, this is the first report about the molecular mechanism regulating PHD2 protein stability. Specific pharmacological disruption of the FKBP38/PHD2 interaction might be a promising approach to modulate PHD2 protein abundance and HIFα protein stability.
Supplementary Material
Acknowledgments
We thank C. Aerni and P. Spielmann for excellent technical assistance and Dr. M. Malesevic for peptide and peptide array synthesis. The mouse ts20 cells harboring a temperature-sensitive E1 ubiquitin-activating enzyme and the reconstituted H38-5 cells were kindly provided by Dr. C. Borner (Institute for Molecular Medicine, University of Freiburg, Germany). The plasmids pGEX-6P-PSMC1 (S4) and pGEX-6P-PSMD2 (S2) were kindly provided by Dr. M. Shirane and Dr. K. I. Nakayama.
This work was supported by grants from the Forschungskredit der Universität Zürich (to G. C.), the Sassella-Stiftung (to G. C. and S. B.), the Hartmann Müller-Stiftung (to G. C.), the Krebsliga des Kanton Zürich (to G. C.), the Olga Mayenfisch Stiftung (to G. C.), 6th Framework Programme of the European Commission/State Secretariat for Education and Research (SER) Grant EUROXY LSHC-CT-2003-502932/SBF 03.0647-2 (to R. H. W.), Swiss National Science Foundation Grant 3100A0-104219 (to R. H. W. and G. C.), and Deutsche Forschungsgemeinschaft Grant FA 225/19-3 (to J. F.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- HIF
- hypoxia-inducible factor
- ER
- endoplasmic reticulum
- FKBP
- FK506-binding protein
- FRET
- fluorescence resonance energy transfer
- GST
- glutathione S-transferase
- IVTT
- in vitro transcription/translation
- KD
- dissociation constant
- PHD
- prolyl-4-hydroxylase domain
- E1
- ubiquitin-activating enzyme
- E3
- ubiquitin-protein isopeptide ligase
- MYND
- myeloid, Nervy, and DEAF-1
- CHX
- cycloheximide
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- Trt
- trityl
- Chaps
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- RNAi
- RNA interference
- Suc
- succinyl
- Z
- benzyloxycarbonyl
- DBD
- DNA binding domain
- AD
- activation domain
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- TM
- transmembrane.
REFERENCES
- 1.Semenza G. L. (2007) Sci. STKE 2007,cm8. [DOI] [PubMed] [Google Scholar]
- 2.Wenger R. H., Stiehl D. P., Camenisch G. (2005) Sci. STKE 2005,re12. [DOI] [PubMed] [Google Scholar]
- 3.Bruick R. K., McKnight S. L. (2001) Science 294,1337–1340 [DOI] [PubMed] [Google Scholar]
- 4.Epstein A. C., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y. M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P. H., Pugh C. W., Schofield C. J., Ratcliffe P. J. (2001) Cell 107,43–54 [DOI] [PubMed] [Google Scholar]
- 5.Ivan M., Haberberger T., Gervasi D. C., Michelson K. S., Günzler V., Kondo K., Yang H., Sorokina I., Conaway R. C., Conaway J. W., Kaelin W. G., Jr. (2002) Proc. Natl. Acad. Sci. U.S.A. 99,13459–13464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., Ratcliffe P. J. (1999) Nature 399,271–275 [DOI] [PubMed] [Google Scholar]
- 7.Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J. M., Lane W. S., Kaelin W. G., Jr. (2001) Science 292,464–468 [DOI] [PubMed] [Google Scholar]
- 8.Jaakkola P., Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., von Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Science 292,468–472 [DOI] [PubMed] [Google Scholar]
- 9.Mahon P. C., Hirota K., Semenza G. L. (2001) Genes Dev. 15,2675–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lando D., Peet D. J., Whelan D. A., Gorman J. J., Whitelaw M. L. (2002) Science 295,858–861 [DOI] [PubMed] [Google Scholar]
- 11.Schofield C. J., Ratcliffe P. J. (2004) Nat. Rev. Mol. Cell Biol. 5,343–354 [DOI] [PubMed] [Google Scholar]
- 12.Kaelin W. G., Jr., Ratcliffe P. J. (2008) Mol. Cell 30,393–402 [DOI] [PubMed] [Google Scholar]
- 13.Lieb M. E., Menzies K., Moschella M. C., Ni R., Taubman M. B. (2002) Biochem. Cell Biol. 80,421–426 [DOI] [PubMed] [Google Scholar]
- 14.Oehme F., Ellinghaus P., Kolkhof P., Smith T. J., Ramakrishnan S., Hütter J., Schramm M., Flamme I. (2002) Biochem. Biophys. Res. Commun. 296,343–349 [DOI] [PubMed] [Google Scholar]
- 15.Koivunen P., Tiainen P., Hyvärinen J., Williams K. E., Sormunen R., Klaus S. J., Kivirikko K. I., Myllyharju J. (2007) J. Biol. Chem. 282,30544–30552 [DOI] [PubMed] [Google Scholar]
- 16.Berra E., Benizri E., Ginouvès A., Volmat V., Roux D., Pouysségur J. (2003) EMBO J. 22,4082–4090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeda K., Ho V. C., Takeda H., Duan L. J., Nagy A., Fong G. H. (2006) Mol. Cell Biol. 26,8336–8346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeda K., Cowan A., Fong G. H. (2007) Circulation 116,774–781 [DOI] [PubMed] [Google Scholar]
- 19.Takeda K., Aguila H. L., Parikh N. S., Li X., Lamothe K., Duan L. J., Takeda H., Lee F. S., Fong G. H. (2008) Blood 111,3229–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minamishima Y. A., Moslehi J., Bardeesy N., Cullen D., Bronson R. T., Kaelin W. G., Jr. (2008) Blood 111,3236–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Metzen E., Stiehl D. P., Doege K., Marxsen J. H., Hellwig-Bürgel T., Jelkmann W. (2005) Biochem. J. 387,711–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pescador N., Cuevas Y., Naranjo S., Alcaide M., Villar D., Landázuri M. O., Del Peso L. (2005) Biochem. J. 390,189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marxsen J. H., Stengel P., Doege K., Heikkinen P., Jokilehto T., Wagner T., Jelkmann W., Jaakkola P., Metzen E. (2004) Biochem. J. 381,761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Appelhoff R. J., Tian Y. M., Raval R. R., Turley H., Harris A. L., Pugh C. W., Ratcliffe P. J., Gleadle J. M. (2004) J. Biol. Chem. 279,38458–38465 [DOI] [PubMed] [Google Scholar]
- 25.Stiehl D. P., Wirthner R., Köditz J., Spielmann P., Camenisch G., Wenger R. H. (2006) J. Biol. Chem. 281,23482–23491 [DOI] [PubMed] [Google Scholar]
- 26.Ginouvès A., Ilc K., Macías N., Pouysségur J., Berra E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105,4745–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isaacs J. S., Jung Y. J., Mole D. R., Lee S., Torres-Cabala C., Chung Y. L., Merino M., Trepel J., Zbar B., Toro J., Ratcliffe P. J., Linehan W. M., Neckers L. (2005) Cancer Cell 8,143–153 [DOI] [PubMed] [Google Scholar]
- 28.Selak M. A., Armour S. M., MacKenzie E. D., Boulahbel H., Watson D. G., Mansfield K. D., Pan Y., Simon M. C., Thompson C. B., Gottlieb E. (2005) Cancer Cell 7,77–85 [DOI] [PubMed] [Google Scholar]
- 29.Pollard P. J., Brière J. J., Alam N. A., Barwell J., Barclay E., Wortham N. C., Hunt T., Mitchell M., Olpin S., Moat S. J., Hargreaves I. P., Heales S. J., Chung Y. L., Griffiths J. R., Dalgleish A., McGrath J. A., Gleeson M. J., Hodgson S. V., Poulsom R., Rustin P., Tomlinson I. P. (2005) Hum. Mol. Genet. 14,2231–2239 [DOI] [PubMed] [Google Scholar]
- 30.Knowles H. J., Raval R. R., Harris A. L., Ratcliffe P. J. (2003) Cancer Res. 63,1764–1768 [PubMed] [Google Scholar]
- 31.Pagé E. L., Chan D. A., Giaccia A. J., Levine M., Richard D. E. (2008) Mol. Biol. Cell 19,86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mansfield K. D., Guzy R. D., Pan Y., Young R. M., Cash T. P., Schumacker P. T., Simon M. C. (2005) Cell Metab. 1,393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagen T., Taylor C. T., Lam F., Moncada S. (2003) Science 302,1975–1978 [DOI] [PubMed] [Google Scholar]
- 34.Berchner-Pfannschmidt U., Yamac H., Trinidad B., Fandrey J. (2007) J. Biol. Chem. 282,1788–1796 [DOI] [PubMed] [Google Scholar]
- 35.Nakayama K., Frew I. J., Hagensen M., Skals M., Habelhah H., Bhoumik A., Kadoya T., Erdjument-Bromage H., Tempst P., Frappell P. B., Bowtell D. D., Ronai Z. (2004) Cell 117,941–952 [DOI] [PubMed] [Google Scholar]
- 36.Möller A., House C. M., Wong C. S., Scanlon D. B., Liu M. C., Ronai Z., Bowtell D. D. (2009) Oncogene 28,289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer G., Wittmann-Liebold B., Lang K., Kiefhaber T., Schmid F. X. (1989) Nature 337,476–478 [DOI] [PubMed] [Google Scholar]
- 38.Edlich F., Maestre-Martínez M., Jarczowski F., Weiwad M., Moutty M. C., Maleseviæ M., Jahreis G., Fischer G., Lücke C. (2007) J. Biol. Chem. 282,36496–36504 [DOI] [PubMed] [Google Scholar]
- 39.Edlich F., Weiwad M., Erdmann F., Fanghänel J., Jarczowski F., Rahfeld J. U., Fischer G. (2005) EMBO J. 24,2688–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirane M., Nakayama K. I. (2003) Nat. Cell Biol. 5,28–37 [DOI] [PubMed] [Google Scholar]
- 41.Edlich F., Erdmann F., Jarczowski F., Moutty M. C., Weiwad M., Fischer G. (2007) J. Biol. Chem. 282,15341–15348 [DOI] [PubMed] [Google Scholar]
- 42.Okamoto T., Nishimura Y., Ichimura T., Suzuki K., Miyamura T., Suzuki T., Moriishi K., Matsuura Y. (2006) EMBO J. 25,5015–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bulgakov O. V., Eggenschwiler J. T., Hong D. H., Anderson K. V., Li T. (2004) Development 131,2149–2159 [DOI] [PubMed] [Google Scholar]
- 44.Cho A., Ko H. W., Eggenschwiler J. T. (2008) Dev. Biol. 321,27–39 [DOI] [PubMed] [Google Scholar]
- 45.Barth S., Nesper J., Hasgall P. A., Wirthner R., Nytko K. J., Edlich F., Katschinski D. M., Stiehl D. P., Wenger R. H., Camenisch G. (2007) Mol. Cell Biol. 27,3758–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Camenisch G., Tini M., Chilov D., Kvietikova I., Srinivas V., Caro J., Spielmann P., Wenger R. H., Gassmann M. (1999) FASEB J. 13,81–88 [DOI] [PubMed] [Google Scholar]
- 47.Frank R. (1992) Tetrahedron 48,9217–9232 [Google Scholar]
- 48.Wirthner R., Balamurugan K., Stiehl D. P., Barth S., Spielmann P., Oehme F., Flamme I., Katschinski D. M., Wenger R. H., Camenisch G. (2007) Methods Enzymol. 435,43–60 [DOI] [PubMed] [Google Scholar]
- 49.Martin F., Linden T., Katschinski D. M., Oehme F., Flamme I., Mukhopadhyay C. K., Eckhardt K., Tröger J., Barth S., Camenisch G., Wenger R. H. (2005) Blood 105,4613–4619 [DOI] [PubMed] [Google Scholar]
- 50.Wotzlaw C., Otto T., Berchner-Pfannschmidt U., Metzen E., Acker H., Fandrey J. (2007) FASEB J. 21,700–707 [DOI] [PubMed] [Google Scholar]
- 51.Feige J. N., Sage D., Wahli W., Desvergne B., Gelman L. (2005) Microsc. Res. Tech. 68,51–58 [DOI] [PubMed] [Google Scholar]
- 52.Chowdary D. R., Dermody J. J., Jha K. K., Ozer H. L. (1994) Mol. Cell Biol. 14,1997–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakagawa T., Shirane M., Iemura S., Natsume T., Nakayama K. I. (2007) Genes Cells 12,709–719 [DOI] [PubMed] [Google Scholar]
- 54.Berchner-Pfannschmidt U., Frede S., Wotzlaw C., Fandrey J. (2008) Eur. Respir. J. 32,210–217 [DOI] [PubMed] [Google Scholar]
- 55.Ladendorff N. E., Wu S., Lipsick J. S. (2001) Oncogene 20,125–132 [DOI] [PubMed] [Google Scholar]
- 56.Lausen J., Cho S., Liu S., Werner M. H. (2004) J. Biol. Chem. 279,49281–49288 [DOI] [PubMed] [Google Scholar]
- 57.Brown M. A., Sims R. J., 3rd, Gottlieb P. D., Tucker P. W. (2006) Mol. Cancer 5,26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi K. O., Lee T., Lee N., Kim J. H., Yang E. G., Yoon J. M., Kim J. H., Lee T. G., Park H. (2005) Mol. Pharmacol. 68,1803–1809 [DOI] [PubMed] [Google Scholar]
- 59.Ju D., Wang X., Xu H., Xie Y. (2008) Mol. Cell Biol. 28,1404–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakayama K., Gazdoiu S., Abraham R., Pan Z. Q., Ronai Z. (2007) Biochem. J. 401,217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McDonough M. A., Li V., Flashman E., Chowdhury R., Mohr C., Liénard B. M., Zondlo J., Oldham N. J., Clifton I. J., Lewis J., McNeill L. A., Kurzeja R. J., Hewitson K. S., Yang E., Jordan S., Syed R. S., Schofield C. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103,9814–9819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yen H. C., Xu Q., Chou D. M., Zhao Z., Elledge S. J. (2008) Science 322,918–923 [DOI] [PubMed] [Google Scholar]
- 63.Murakami Y., Matsufuji S., Kameji T., Hayashi S., Igarashi K., Tamura T., Tanaka K., Ichihara A. (1992) Nature 360,597–599 [DOI] [PubMed] [Google Scholar]
- 64.Tarcsa E., Szymanska G., Lecker S., O'Connor C. M., Goldberg A. L. (2000) J. Biol. Chem. 275,20295–20301 [DOI] [PubMed] [Google Scholar]
- 65.Chen X., Chi Y., Bloecher A., Aebersold R., Clurman B. E., Roberts J. M. (2004) Mol. Cell 16,839–847 [DOI] [PubMed] [Google Scholar]
- 66.Bossis G., Ferrara P., Acquaviva C., Jariel-Encontre I., Piechaczyk M. (2003) Mol. Cell. Biol. 23,7425–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asher G., Shaul Y. (2005) Cell Cycle 4,1015–1018 [DOI] [PubMed] [Google Scholar]
- 68.Kalejta R. F., Shenk T. (2003) Proc. Natl. Acad. Sci. U.S.A. 100,3263–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jariel-Encontre I., Bossis G., Piechaczyk M. (2008) Biochim. Biophys. Acta 1786,153–177 [DOI] [PubMed] [Google Scholar]
- 70.Bader N., Grune T. (2006) Biol. Chem. 387,1351–1355 [DOI] [PubMed] [Google Scholar]
- 71.Prakash S., Tian L., Ratliff K. S., Lehotzky R. E., Matouschek A. (2004) Nat. Struct. Mol. Biol. 11,830–837 [DOI] [PubMed] [Google Scholar]
- 72.Zhang M., Pickart C. M., Coffino P. (2003) EMBO J. 22,1488–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li X., Lonard D. M., Jung S. Y., Malovannaya A., Feng Q., Qin J., Tsai S. Y., Tsai M. J., O'Malley B. W. (2006) Cell 124,381–392 [DOI] [PubMed] [Google Scholar]
- 74.Huang J., Zhao Q., Mooney S. M., Lee F. S. (2002) J. Biol. Chem. 277,39792–39800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Metzen E., Berchner-Pfannschmidt U., Stengel P., Marxsen J. H., Stolze I., Klinger M., Huang W. Q., Wotzlaw C., Hellwig-Bürgel T., Jelkmann W., Acker H., Fandrey J. (2003) J. Cell Sci. 116,1319–1326 [DOI] [PubMed] [Google Scholar]
- 76.Soilleux E. J., Turley H., Tian Y. M., Pugh C. W., Gatter K. C., Harris A. L. (2005) Histopathology 47,602–610 [DOI] [PubMed] [Google Scholar]
- 77.Berchner-Pfannschmidt U., Tug S., Trinidad B., Oehme F., Yamac H., Wotzlaw C., Flamme I., Fandrey J. (2008) J. Biol. Chem. 283,31745–31753 [DOI] [PubMed] [Google Scholar]
- 78.Khan Z., Michalopoulos G. K., Stolz D. B. (2006) Am. J. Pathol. 169,1251–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.