

Abstract
Wall teichoic acids are a chemically diverse group of anionic polymers that constitute up to 50% of the Gram-positive cell wall. These polymers play a pivotal role in virulence and have been implicated in a diverse range of physiological functions. The TagF-like family of enzymes has been shown to be responsible for wall teichoic acid priming and polymerization events. Although many such enzymes are well validated therapeutic targets, a mechanistic understanding of this enzyme family has remained elusive. TagF is the prototypical teichoic acid polymerase and uses CDP-glycerol to catalyze synthesis of the linear (1,3)-linked poly(glycerol phosphate) teichoic acid in Bacillus subtilis 168. Here we used a synthetic soluble analog of the natural substrate of the enzyme, Lipid ϕ, to conduct the first detailed mechanistic investigation of teichoic acid polymerization. Through the use of a new high pressure liquid chromatography-based assay to monitor single glycerol phosphate incorporations into the Lipid ϕ analog, we conducted a detailed analysis of reaction product formation patterns and unequivocally showed TagF to be non-processive in vitro. Furthermore by monitoring the kinetics of polymerization, we showed that Lipid ϕ analog species varying in size have the same Km value of 2.6 μm and validated use of Bi Bi velocity expressions to model the TagF enzyme system. Initial rate analysis showed that TagF catalyzes a sequential Bi Bi mechanism where both substrates are added to the enzyme prior to product release consistent with a single displacement chemical mechanism.
Wall teichoic acids are a group of phosphate-containing anionic carbohydrate polymers that constitute up to 50% of the dry weight of the Gram-positive cell wall (1). Teichoic acids play a pivotal role in virulence and have been implicated in a diverse range of physiological functions including cation homeostasis, nutrient trafficking, binding of envelope proteins, and regulation of autolysins (2–4). Our knowledge of wall teichoic acid synthesis largely stems from studies conducted in the model bacterium Bacillus subtilis 168, which expresses a linear (1,3)-linked poly(glycerol phosphate) teichoic acid (5–7). Through these studies, our group and others have identified the genetic requirements for poly(glycerol phosphate) synthesis; however, until recently, a biochemical understanding of priming and polymerization events have been confounded by the interfacial localization of these enzymatic steps (8, 9). Indeed interfacial localization has hindered the understanding of the synthesis of many other important cell wall components such as O-antigen, polysialic acid, lipoarabinomannan, oligomers for N-linked glycosylation, and others (10–13). A breakthrough in our ability to study the enzymes involved in the lipid-linked steps of wall teichoic acid synthesis was made by Ginsberg et al. (14) with the development of synthetic substrate analogs of lipid-linked wall teichoic acid intermediates. These substrate analogs have since facilitated the detailed mechanistic study of uncharacterized teichoic acid enzymes and were used to reconstitute all intracellular steps in Staphylococcus aureus wall teichoic acid synthesis in vitro (15, 16).
Genetic and biochemical studies have given rise to a model for the synthesis of poly(glycerol phosphate) wall teichoic acid in B. subtilis 168 in which polymer synthesis is carried out on the intracellular surface of the cytoplasmic membrane by stepwise additions of sugars to an undecaprenol phosphate lipid carrier via the tag (teichoic acid glycerol) gene products (7, 17). Polymer synthesis is initiated by TagO, which catalyzes the transfer of N-acetylglucosamine-1-phosphate from UDP-GlcNAc to undecaprenol phosphate to create Lipid α (18) (the new nomenclature for lipid-linked teichoic acid intermediates proposed by Pereira and Brown (17) is summarized in Table 1). N-Acetylmannosamine (ManNAc)2 is transferred to Lipid α from UDP-ManNAc by TagA, producing Lipid β that is “primed” with sn-glycerol-3-phosphate by TagB to create the polymerization substrate Lipid ϕ.1 (9, 14, 16). Some 30–50 glycerol phosphate residues are subsequently added to Lipid ϕ.1 by TagF, and the intracellular steps of teichoic acid synthesis are completed via polymer glucosylation by TagE (6, 8). Intracellular teichoic acid is then exported to the outer leaflet of the cytoplasmic membrane by the TagG/H ATP-binding cassette transport system and transferred to peptidoglycan by a currently unknown enzyme (19).
TABLE 1.
Recently proposed nomenclature for wall teichoic acid intermediates
Shown is the nomenclature proposed for wall teichoic acid biosynthetic intermediates (17). Intermediates were named according to the enzyme utilizing the molecule as a substrate. Lipid α is the substrate for TagA, Lipid β is the substrate for TagB. Lipid ϕ.n species are substrates for TagF where n indicates the number of glycerol phosphate residues in the molecule. For example, Lipid ϕ.1 is the product of the TagB-catalyzed priming reaction where a single glycerol phosphate residue is added. und, undecaprenol; P, phosphate; GroP, sn-glycerol-3-phosphate.
| Enzyme | Substrate | Chemical composition |
|---|---|---|
| TagA | Lipid α | GlcNAc-1-P-P-und |
| TagB | Lipid β | ManNAc-β(1–4)-GlcNAc-1-P-P-und |
| TagF | Lipid ϕ.n | (GroP)n-ManNAc-β(1–4)-GlcNAc-1-P-P-und |
| TagF | Lipid ϕ.n analog | (GroP)n-ManNAc-β(1–4)-GlcNAc-1-P-P-tridecane |
Based on sequence identity and crude mechanistic studies of B. subtilis 168 TagB and TagF enzymes, teichoic acid primases, oligomerases, and polymerases have been grouped into the TagF-like enzyme family that share a conserved ≈300-residue C-terminal catalytic domain and a basic N-terminal domain of variable size (9, 20). In efforts to expand our mechanistic understanding of this unique enzyme family, soluble substrate analogs have been used to study wall teichoic acid priming and polymerization events in B. subtilis 168 and S. aureus (14, 15, 21). Through these studies, the Lipid ϕ analog has been validated as a suitable substrate for kinetic investigation of B. subtilis 168 TagF (21). To firmly establish the processivity of the prototypical TagF enzyme, herein we developed a robust HPLC-based assay that allowed us to monitor single glycerol phosphate incorporations into a radiolabeled Lipid ϕ analog. We analyzed patterns of product accumulation to determine enzyme processivity and showed unequivocally that soluble TagF utilizes a non-processive polymerization mechanism. Further we took advantage of this finding to validate application of Bi Bi initial rate expressions to the TagF system. We conducted the first detailed steady state kinetic mechanistic study of wall teichoic acid polymerization and showed that poly(glycerol phosphate) synthesis is mediated via a sequential Bi Bi mechanism. We posit a single displacement active site mechanism where Lipid ϕ directly attacks the pyrophosphate linkage of CDP-glycerol.
EXPERIMENTAL PROCEDURES
General Methods
Chemicals and enzymes were purchased from Sigma with the following exceptions: dibasic potassium phosphate was from EMD Biosciences (Gibbstown, NJ); imidazole, Tris, and urea were from Bioshop (Burlington, Ontario, Canada); alkaline phosphatase and Complete Mini protease inhibitor mixture were from Roche Applied Science; [γ-32P]ATP, [α-32P]CTP, and Ultima-Flo M scintillation fluid were from PerkinElmer Life Sciences; and sn-[U-14C]glycerol-3-phosphate was purchased from Amersham Biosciences. Ni2+-chelating columns and Superdex 200 columns were from Amersham Biosciences. Chromatography was performed on either an Amersham Biosciences ATKA fast protein liquid chromatography system or a Waters HPLC system. All enzyme assays were conducted with N-terminally hexahistidine-tagged TagF from B. subtilis 168 prepared as described previously (8).
CDP-glycerol Synthesis
CDP-glycerol and CDP-[U-14C]glycerol were synthesized using TarD from S. aureus and methods described previously (22). [α-32P]CDP-glycerol was synthesized similarly with the addition of 125 μCi of [α-32P]CTP (3000 Ci/mmol) to the reaction. [β-32P]CDP-glycerol was synthesized in two enzymatic steps. sn-Glycerol-3-[32P]phosphate was synthesized using 4 units/μl glycerol kinase (from bakers' yeast), 250 μCi of [γ-32P]ATP (3000 Ci/mmol), 90 μm ATP, and 90 μm glycerol in a reaction buffer that contained 50 mm HEPES, pH = 8, 20 mm MgCl2. The reaction was permitted to go to completion before being filtered with a 5000 molecular weight cutoff centrifugal filter (Millipore). The filtrate was used in the place of sn-glycerol-3-phosphate to synthesize [β-32P]CDP-glycerol as described previously (22). sn-Glycerol-3-[32P]phosphate synthesis was monitored by following the conversion of [γ-32P]ATP to glycerol-3-[32P]phosphate using a paired ion exchange (PIC)-HPLC separation method. Samples were loaded onto an Inertsil ODS-3 (4 × 150-mm, 5-μm) column with buffer Pic A (15 mm dibasic potassium phosphate, 10 mm tetrabutylammonium hydrogen sulfate, pH = 7) and eluted with a linear 3-min gradient to 100% buffer Pic B (15 mm dibasic potassium phosphate, 10 mm tetrabutylammonium hydrogen sulfate, 30% (v/v) acetonitrile, pH = 7). Analytes were visualized by in-line scintillation counting.
Lipid ϕ.1 Analog Synthesis
The Lipid ϕ.1 analog was chemoenzymatically synthesized as described previously (21). The [14C]Lipid ϕ.1 analog was synthesized using the same method but substituting CDP-[U-14C]glycerol for CDP-glycerol. The [32P]Lipid ϕ.1 analog was synthesized using the same method but substituting [β-32P]CDP-glycerol for CDP-glycerol.
Enzymatic Assays
Enzyme assays were conducted in duplicate at 30 °C with 2.5 nm TagF, 50 mm Tris, 30 mm MgCl2, and the indicated concentrations of CDP-glycerol and the Lipid ϕ.1 analog and quenched with the addition of an equal volume of freshly prepared 8 m urea. Reaction progress was determined by one of two stopped HPLC-based assays that allowed for quantification of the conversion of the radiolabeled Lipid ϕ.1 analog to Lipid ϕ.2 or quantification of the conversion of CDP-glycerol to CMP. Conversion of the [14C]Lipid ϕ.1 analog or [32P]Lipid ϕ.1 analog to Lipid ϕ.2 was followed by separation of radioactive species using an anion exchange column (J. T. Baker Inc. WP Quat, 4.6 × 50 mm, 5 μm). Samples were eluted with a continuous 4-min gradient from 0.25 to 6% (v/v) triethylamine bicarbonate in double distilled H2O and visualized with in-line scintillation counting. Conversion of CDP-glycerol to CMP was followed with the PIC-HPLC protocol described above but with isocratic sample loading and elution at 15% Pic B followed by flushing of the column with 100% Pic B. Analytes were visualized by UV absorbance at 271 nm or in-line scintillation counting. Fitting of data to initial rate expressions was done using a non-linear sum of least squares regression method with either Sigma Plot 8.0 or the Enzyme Kinetics Module 1.1 available for Sigma Plot 8.0 (SPSS, Inc., Chicago, IL).
RESULTS
TagF Processivity
To investigate the processivity of TagF, we examined patterns of product accumulation under initial rate conditions, an approach similar to those used to establish the processivity of polysialic acid, polygalacturonate, and type 3 capsular polysaccharide polymerases in Escherichia coli, Petunia axillaris, and Streptococcus pneumonia, respectively (23–26). In the TagF system, a processive mechanism would yield a small number of polymeric Lipid ϕ analog product molecules equivalent to the concentration of free enzyme provided the polymer did not reach full length. Furthermore the ratio of consumption of the donor substrate, CDP-glycerol, to the amount of polymeric Lipid ϕ analog produced would be greater than 1. If the TagF system was non-processive, the enzyme would dissociate from both products following catalysis (26, 27). In the non-processive case and under initial rate conditions, probability dictates that in the presence of a large excess of substrates the enzyme would interact with any particular substrate molecule only once. Thus, the most abundant product species would contain a single glycerol phosphate addition, and the amount of CDP-glycerol consumed would be equivalent to the amount of product.
To monitor incorporation of glycerol phosphate units into the TagF substrate, the Lipid ϕ.1 analog, we developed a stopped assay in which the radiolabeled Lipid ϕ analog reaction products were separated by anion exchange HPLC with in-line radioactivity detection following termination of the reaction with urea. Reactions were conducted with equimolar amounts of CDP-glycerol and the [14C]Lipid ϕ.1 analog (200 μm), and substrate turnover was limited to 15%. The quenched reaction mixture was separated into two aliquots for quantification of the Lipid ϕ.1 and Lipid ϕ.n analogs as well as CDP-glycerol turnover. Fig. 1A shows an anion exchange separation of radiolabeled Lipid ϕ analog species from a quenched reaction in which the Lipid ϕ.2 analog was the only radioactive product (species identity was determined by mass spectroscopy; data not shown). Quantification of these peaks indicated the production of 29 μm Lipid ϕ.2 analog. The enzyme concentration in this experiment was 2.5 nm. Exclusive production of the Lipid ϕ.2 analog in an amount over 10,000 times larger than the enzyme concentration would only be possible by non-processive glycerol phosphate additions. To confirm that Lipid ϕ.2 analog species were generated by non-processive additions of glycerol phosphate, CDP-glycerol turnover was quantified by PIC-HPLC and determined to be 29 μm (Fig. 1B). The equivalent consumption of both substrates confirmed that TagF utilized a non-processive mechanism and suggested that initial velocity expressions developed for non-polymerase Bi Bi enzyme systems could be used to describe the TagF polymerase system.
FIGURE 1.

TagF is non-processive. Reactions were conducted with equimolar amounts of CDP-glycerol, the [14C]Lipid ϕ.1 analog (200 μm), and 2.5 nm TagF. A, Lipid ϕ analog species were separated following reaction with TagF by an anion exchange HPLC assay and visualized with in-line scintillation counting. Integration of peaks indicated 29 μm Lipid ϕ.2 analog was produced. Peak identities were confirmed by liquid chromatography-mass spectrometry (data not shown). B, CMP was quantified following the reaction by separation of CDP-glycerol and CMP via PIC-HPLC and visualized by absorbance at 271 nm. Integration of peaks indicated the formation of 29 μm CMP.
Apparent Kinetic Constants for the Lipid ϕ Analog
We recently completed a preliminary kinetic investigation of TagF using the same synthetic Lipid ϕ analog used in this work. Our previous study indicated that the Lipid ϕ analog could be a suitable substrate for elucidation of the steady state kinetic mechanism of TagF. In that study, we found a low apparent Km of 2.6 ± 0.2 μm for the Lipid ϕ analog and a rapid apparent turnover number of 27 s−1 (21). The low Km for the Lipid ϕ analog presented a technical challenge in assaying TagF activity within the conventional range of 10–15% substrate turnover that is generally accepted for steady state kinetic experiments (28). For this reason, we explored the possibility of working outside the range of 10–15% turnover for the Lipid ϕ analog by investigating whether TagF differentiated between the analog in different degrees of polymerization. To determine whether TagF distinguished between Lipid ϕ analog species as the degree of polymerization increased, we measured the apparent kinetic parameters Km and kcat/Km for the Lipid ϕ.1 analog by monitoring the conversion of the [32P]Lipid ϕ.1 analog to Lipid ϕ.2 analog by anion exchange HPLC with in-line radioactivity detection. TagF (25 pm) was incubated with a fixed, saturating concentration of CDP-glycerol (700 μm) and 1–32 μm Lipid ϕ.1 analog. Initial rate data were fit to Equation 1. The apparent kinetic parameters Km and kcat/Km were observed to be 3.4 ± 0.6 μm and 9.4 × 106 m−1 s−1, respectively.
 |
Interestingly the observed Km and kcat/Km values obtained when conversion of the Lipid ϕ.1 analog to the Lipid ϕ.2 analog was measured were roughly equivalent to those we have measured previously (Km = 2.6 ± 0.2 μm and kcat/Km = 1.1 × 107 m−1 s−1) in an assay where multiple additions of glycerol phosphate occurred for each Lipid ϕ.1 analog; oligomers of approximately eight glycerol phosphate residues were produced (the degree of Lipid ϕ analog polymerization was approximated by the ratio of CDP-glycerol consumed to the amount of the Lipid ϕ.1 analog initially present as described previously (21)). Roughly equivalent kinetic parameters generated using these two assay systems suggested that TagF did not differentiate between the Lipid ϕ.1 analog and Lipid ϕ analogs with degrees of polymerization up to 8.
Although TagF did not seem to differentiate between the Lipid ϕ.1 analog and a Lipid ϕ analog with up to eight additions of glycerol phosphate, it was possible that a change in kinetic properties might occur at a higher degree of polymerization as has been observed in studies using synthetic polymerization substrates for GlfT, a polygalactofuranosyltransferaserase from Mycobacterium tuberculosis, and polysialyltransferases from both E. coli and Neisseria meningitidis (29, 30). To investigate the possibility of a changing Km value for the substrate as length increased we took the following approach. We reasoned that measurement of the reaction rate during polymerization could be used to detect a changing Michaelis constant provided that the system was kept within initial rate conditions with respect to CDP-glycerol. Fig. 2, inset, depicts model product versus time plots for each of three possibilities: increasing, decreasing, or no change in Km. A large change in the slope of this plot results from even a modest 2-fold change in Km, confirming that measurement of the rate of product accumulation is a sensitive method to detect a changing Michaelis constant. Reactions with 2 mm CDP-glycerol and 2 μm [14C]Lipid ϕ.1 analog were incubated for up to 20 min prior to reaction termination. Product accumulation was determined by conversion of CDP-glycerol to CMP and plotted as a function of time in Fig. 2. The accumulation of polymeric Lipid ϕ analog was measured at each reaction end point by gel filtration as described previously (data not shown) (8). Interestingly product accumulation remained linear with time as the Lipid ϕ analog was polymerized to 76 glycerol phosphate units in length, a size much larger than the approximate 30–50-unit poly(glycerol phosphate) wall teichoic acid found in vivo. Our results indicated that species of the Lipid ϕ analog with at least 76 glycerol phosphate units serve a functionally equivalent role in the TagF polymerase assay system and could therefore be described with the same term in an initial velocity expression.
FIGURE 2.

Poly(glycerol phosphate) synthesis is linear with time and independent of polymer size. Reactions were conducted with 2 mm CDP-glycerol, 2 μm Lipid ϕ.1 analog, and 2.5 nm TagF, and reaction progress was determined by CMP formation via PIC-HPLC. The degree of Lipid ϕ analog polymerization at each time point was approximated as reported previously by the ratio of CDP-glycerol consumption to the initial concentration of the Lipid ϕ.1 analog (21). Inset, calculated data are shown. A 2-fold increase (- - -) and 2-fold decrease (· · ·) in Km are compared with a substrate with a constant (—) Km. The simulated data were generated using reaction conditions equivalent to those used in the experiment in which the substrate concentration was equivalent to the Km.
Initial Rate Analysis for TagF
Having demonstrated that the TagF polymerase system could be accurately described with non-polymerase Bi Bi velocity expressions, we took advantage of conventional approaches in steady state enzymology to investigate TagF reaction kinetics (28). We conducted an initial rate analysis of TagF in the absence of products to gain insight into catalytic mechanism and determine true kinetic parameters for the reaction. A 5 × 5 matrix of reactant concentrations was explored, and reaction progress was monitored by following the conversion of CDP-glycerol to CMP. Initial rate data were fit to all rate expressions in the Enzyme Kinetics Module 1.1 for Sigma Plot 8.0 and found to be best described by the sequential random Bi Bi mechanism (Equation 2). Best fit was determined using the Akaike information criterion corrected for small sample size that uses estimates of accuracy and precision to quantitatively rank the suitability of models for describing a data set (31).
A and B are either CDP-glycerol or Lipid ϕ analog, αKA and αKB are the Michaelis constants for A and B, respectively, and α is a constant that modifies the dissociation constant for substrate binding a transitory enzyme complex (28). Initial rate data are shown in double reciprocal form in Fig. 3. Km values, summarized in Table 2, were calculated to be 2.6 ± 1.4 μm for the Lipid ϕ analog and 180 ± 77 μm for CDP-glycerol. The secondary rate constants kcat/Km for the Lipid ϕ analog and CDP-glycerol were determined to be 3.4 × 107 and 4.8 × 105 s−1 m−1, respectively.
FIGURE 3.

TagF catalyzes a sequential Bi Bi mechanism. Initial velocity data are presented in double reciprocal plots of 1/velocity versus 1/[substrate]. A, CDP-glycerol was varied from 100 to 1600 μm while the Lipid ϕ analog was held constant at 0.5 (▼), 1 (○), 2 (●), 4 (□), and 8 (■) μm. B, the Lipid ϕ analog was varied from 0.5 to 8 μm while CDP-glycerol was held constant at 100 (▼), 200 (○), 400 (●), 800 (□), and 1600 (■) μm. Experiments were conducted with 2.5 nm TagF, and initial rates were determined by monitoring the conversion of CDP-glycerol to CMP via PIC-HPLC. Initial rate data were fit to all initial velocity expressions in the Enzyme Kinetics Module 1.1 for Sigma Plot 8.0 using a non-linear sum of least squares regression method and were found to be best described by the expression for a sequential random mechanism (Equation 2). Error bars indicate S.E.
TABLE 2.
Summary of kinetic constants
| Substrate | Km | kcat | kcat/Km | αa |
|---|---|---|---|---|
| μm | s−1 | s−1m−1 | ||
| Lipid ϕ analog | 2.6 ± 1.4 | 87 ± 8.4 | 3.4 × 107 | 0.60 ± 0.46 |
| CDP-glycerol | 180 ± 77 | 4.8 × 105 |
a α is a constant in Equation 2 that modifies the dissociation constant for substrate binding to a transitory enzyme complex (28).
Product Inhibition
Initial velocity studies were able to differentiate between sequential and non-sequential kinetic mechanisms; however, to gain insight into substrate binding order, experiments needed to be conducted in the presence of reaction products (32). The inhibitory effect of 0.3–3 mm CMP with either CDP-glycerol or the Lipid ϕ analog as the varied substrate (co-substrate was held constant at Km concentration) was determined. Rate data were tested against all inhibition models in the Enzyme Kinetics Module 1.1 in Sigma Plot 8.0 and ranked by the Akaike information criterion corrected for small sample size. When CDP-glycerol was the varied substrate, velocity data were found to be best described by the competitive inhibition model (Equation 3), whereas the partial non-competitive inhibition model best described the data when the Lipid ϕ analog was the varied substrate (Equation 4 where β is the constant modifying the rate of product formation when the inhibitor is bound to the enzyme·substrate complex (28)).
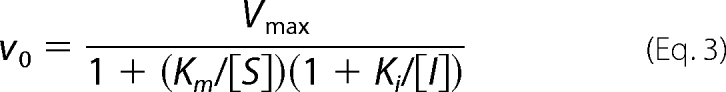 |
Product inhibition data are presented in double reciprocal form in Fig. 4 and summarized in Table 3. The competitive inhibition observed with CMP while CDP-glycerol was the varied substrate suggested that both molecules bound the same enzyme form, either the apoenzyme or a transitory TagF·Lipid ϕ analog complex where the latter describes a Theorell-Chance mechanism (28, 33). The partial non-competitive inhibition we observed with CMP and the Lipid ϕ analog when the latter was the varied substrate ruled out a rapid equilibrium random kinetic mechanism (28).
FIGURE 4.

Inhibition of TagF by CMP. Inhibition of TagF by the reaction product CMP is presented in double reciprocal plots of 1/velocity versus 1/[substrate]. A, CMP was held constant at 0 (■), 300 (□), 1000 (●), and 3000 (○) μm while CDP-glycerol was varied between 100 and 1600 μm, and Lipid ϕ analog was fixed at 2 μm. B, CMP was held constant at 0 (■), 100 (□), 300 (●), and 1000 (○) μm while the Lipid ϕ analog was varied from 1 to 16 μm with CDP-glycerol fixed at 180 μm. Experiments were conducted with 2.5 nm TagF, and initial rates were determined by monitoring the conversion of [α-32P]CDP-glycerol to [α-32P]CMP via PIC-HPLC. Rate data were fit to all inhibition models in the Enzyme Kinetics Module 1.1 for Sigma Plot 8.0 using a non-linear sum of least squares regression method. When CDP-glycerol was the varied substrate the data were best described by the competitive inhibition model (Equation 3). With the Lipid ϕ analog as the varied substrate the data were best described by the non-competitive (partial) inhibition model (Equation 4). Error bars indicate S.E.
TABLE 3.
Summary of inhibition constants
| Inhibitor | Varied substrate | Fixed substrate | Type of inhibition | Ki | βa |
|---|---|---|---|---|---|
| μm | |||||
| CMP | CDP-glycerol | Lipid ϕ analog | Competitive | 182 ± 35 | |
| CMP | Lipid ϕ analog | CDP-glycerol | Non-competitive (partial) | 156 ± 30 | 0.23 ± 0.04 |
a β is a constant in Equation 4 that modifies the rate of product formation when inhibitor is bound to the enzyme·substrate complex (28).
As we have shown above, the Lipid ϕ.n analog reaction products functioned as substrates in a manner indistinguishable from the Lipid ϕ.1 analog, making these reaction products unsuitable for product inhibition studies. Our investigation of product inhibition on TagF was therefore limited to CMP and indicated that TagF utilized a sequential mechanism where CDP-glycerol can (or must) be added to TagF first or a Theorell-Chance mechanism where the Lipid ϕ analog could be the first substrate to bind.
DISCUSSION
A mechanistic understanding of wall teichoic acid priming and polymerization and regulation of these events has remained elusive. In this study, we have begun to shed light on these elements by conducting the first detailed mechanistic investigation of wall teichoic acid polymerization. Using a new HPLC-based assay to monitor incorporation of single glycerol phosphate residues into the synthetic Lipid ϕ analog polymerization substrate, we unequivocally showed that TagF is non-processive in this assay system (illustrated in Fig. 5A). We used this discovery as a foundation for the use of non-polymerase initial velocity expressions to describe TagF and to enable a steady state kinetic investigation of TagF-catalyzed teichoic acid polymerization.
FIGURE 5.

TagF is a non-processive enzyme with a proposed single displacement mechanism. The data presented herein are consistent with the following models. A, TagF binds CDP-glycerol and Lipid ϕ to form a ternary complex. The ternary complex dissociates to liberate free enzyme and product following each catalytic event. B, an active site base (B) deprotonates the terminal hydroxyl of Lipid ϕ to facilitate nucleophilic attack on the β-phosphate of CDP-glycerol.
In our kinetic investigation of TagF we found that, surprisingly, Lipid ϕ analog species with up to 76 glycerol phosphate residues functioned equivalently in the reaction system, indicative of an unchanging Km value for the Lipid ϕ analog regardless of the degree of polymerization. In a previous study exploring acceptor substrate specificity for TagF, we have shown that CDP-glycerol can be used as an acceptor substrate albeit at a much slower rate (34). Together these results are intriguing and suggest a model where TagF does not distinguish among Lipid ϕ species in varying degrees of polymerization. Indeed these findings suggest that TagF may interact largely with the terminal glycerol phosphate residue of Lipid ϕ. However, the reaction rate with CDP-glycerol as the acceptor substrate for polymerization was far slower (kcat of 27 min−1) than that seen here (34). These findings strongly suggest that chemical matter present in the Lipid ϕ analog and absent in CDP-glycerol facilitates far more efficient catalysis. Detailed examinations of acceptor substrate specificity and structural studies of the TagF·Lipid ϕ complex will be needed to define the chemical requirements for efficient recognition of the acceptor substrate by TagF.
Our investigation of kinetic parameters for Lipid ϕ analog species of varying length also gives clues into possible mechanisms of poly(glycerol phosphate) length regulation. Previous work on TagF had ruled out a mechanism of length regulation via accessory proteins such as Wzz-dependent length regulation of Wzy-dependent O-antigen polymerization (8, 12). This suggested that TagF may utilize an intrinsic mechanism of regulation, such as the mechanism of product inhibition used by the Campylobacter jejuni N-linked glycosylation polymerase PglH, or a membrane-dependent mechanism (13). The results we have presented in this study have shown that the Lipid ϕ analog could be polymerized at a constant velocity, indicative of an equivalent Km value for polymers of varying lengths, suggesting that regulation of polymer length is not achieved by a mechanism intrinsic to the enzyme in solution. As we have suggested previously, a growing body of evidence is pointing toward a mechanism of wall teichoic acid length regulation that is, at least in part, mediated by association of TagF with the membrane (34). Indeed intracellular localization studies using green fluorescent protein-Tag enzyme fusion proteins have shown that wall teichoic acid enzymes, including TagF, are localized to the inner surface of the cytoplasmic membrane (9, 35, 36). With the demonstrated localization of TagF to the membrane and the implication of membrane association as an essential component of polymer length regulation, it is possible that membrane association may effect a physical change on TagF that is required for length regulation.
In this study we validated the use of Bi Bi velocity expressions to model the TagF polymerase system and in doing so were able to conduct the first detailed steady state kinetic mechanistic study of the prototypical teichoic acid polymerase, TagF. Initial rate experiments were consistent with a sequential (ternary complex) mechanism, and our investigation of product inhibition indicated that CDP-glycerol and CMP bind to the same enzyme form. These steady state investigations indicate that TagF utilizes either a sequential mechanism where CDP-glycerol can (or must) be added to the enzyme first or a Theorell-Chance mechanism where Lipid ϕ could be added to the enzyme first (28). It is tempting to speculate based on the similar enzymatic activities of glycosyltransferases utilizing undecaprenol-linked acceptors, such as TagA and MurG, that TagF will use an ordered Bi Bi mechanism where the sugar donor binds first (16, 37). It must be noted, however, that there is mechanistic precedence from the C. jejuni polymerase PglH for an ordered Bi Bi mechanism where the undecaprenol-linked acceptor is the first substrate to bind the enzyme (13). Therefore, a Lipid ϕ product analog that can bind to the same enzyme form as Lipid ϕ and that can inhibit TagF will be needed to differentiate between these possible mechanisms. With our discovery of a sequential Bi Bi mechanism for TagF and our previous report showing the dependence of TagF catalysis on two putative active site histidine residues (20), we propose a single displacement catalytic mechanism for TagF (illustrated in Fig. 5B) in which an active site base deprotonates the terminal hydroxyl of Lipid ϕ to activate the acceptor nucleophile for direct attack on the β-phosphate of CDP-glycerol.
The assay system used in this study allowed us to establish processivity and kinetic properties for TagF in a soluble, minimal environment that can be used as a reference to compare the properties of TagF in more physiological systems. In addition to the putative role that association of TagF with the membrane may play in length regulation, it is possible that membrane association may influence other aspects of enzyme activity such as processivity, substrate dissociation constants, or catalytic efficiency. Establishment of reference kinetic properties for TagF in this study will aid in elucidating the functional role played by the membrane and other components with which TagF may interact in vivo as assay systems are developed that reconstitute the true physiological environment of TagF.
This study has provided detailed insight into the mechanism of glycosyl transfer by TagF and the conserved TagF-like family of enzymes responsible for priming and polymerization events in wall teichoic acid synthesis. Many members of the TagF-like enzyme family are well validated yet unexploited therapeutic targets (3, 38, 39). We hope that the new mechanistic understanding of this unique enzyme family and the facile assay for primase/polymerase activity developed in this work will help in the development of new antibacterial compounds targeting wall teichoic acid polymerization.
Acknowledgment
We thank Dr. Kalinka Koteva (Department of Biochemistry and Biomedical Sciences and the Michael G. DeGroote Institute for Infectious Disease Research, McMaster University) for assistance with mass spectroscopy.
This work was supported by Canadian Institutes of Health Research Grant MOP-15496 and a Canada Research Chair (held by E. D. B.).
- ManNAc
- N-acetylmannosamine
- GlcNAc
- N-acetylglucosamine
- HPLC
- high pressure liquid chromatography
- PIC
- paired ion exchange.
REFERENCES
- 1.Hancock I. C. (1997) Biochem. Soc. Trans. 25, 183–187 [DOI] [PubMed] [Google Scholar]
- 2.Weidenmaier C., Peschel A. (2008) Nat. Rev. Microbiol. 6, 276–287 [DOI] [PubMed] [Google Scholar]
- 3.Weidenmaier C., Peschel A., Xiong Y. Q., Kristian S. A., Dietz K., Yeaman M. R., Bayer A. S. (2005) J. Infect. Dis. 191, 1771–1777 [DOI] [PubMed] [Google Scholar]
- 4.Neuhaus F. C., Baddiley J. (2003) Microbiol. Mol. Biol. Rev. 67, 686–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mauël C., Young M., Margot P., Karamata D. (1989) Mol. Gen. Genet. 215, 388–394 [DOI] [PubMed] [Google Scholar]
- 6.Mauël C., Young M., Karamata D. (1991) J. Gen. Microbiol. 137, 929–941 [DOI] [PubMed] [Google Scholar]
- 7.Bhavsar A. P., Brown E. D. (2006) Mol. Microbiol. 60, 1077–1090 [DOI] [PubMed] [Google Scholar]
- 8.Schertzer J. W., Brown E. D. (2003) J. Biol. Chem. 278, 18002–18007 [DOI] [PubMed] [Google Scholar]
- 9.Bhavsar A. P., Truant R., Brown E. D. (2005) J. Biol. Chem. 280, 36691–36700 [DOI] [PubMed] [Google Scholar]
- 10.Freiberger F., Claus H., Günzel A., Oltmann-Norden I., Vionnet J., Mühlenhoff M., Vogel U., Vann W. F., Gerardy-Schahn R., Stummeyer K. (2007) Mol. Microbiol. 65, 1258–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berg S., Kaur D., Jackson M., Brennan P. J. (2007) Glycobiology 17, 35–56R [DOI] [PubMed] [Google Scholar]
- 12.Whitfield C. (2006) Annu. Rev. Biochem. 75, 39–68 [DOI] [PubMed] [Google Scholar]
- 13.Imperiali B., Troutman J. (2009) Biochemistry 48, 2807–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ginsberg C., Zhang Y. H., Yuan Y., Walker S. (2006) ACS Chem. Biol. 1, 25–28 [DOI] [PubMed] [Google Scholar]
- 15.Brown S., Zhang Y. H., Walker S. (2008) Chem. Biol. 15, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y. H., Ginsberg C., Yuan Y., Walker S. (2006) Biochemistry 45, 10895–10904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pereira M. P., Brown E. D. (2008) in Microbial Glycobiology: Structures, Relevance and Applications (Moran A. B., Hoist P., Itzstein O. eds) Academic Press, San Diego, CA, in press [Google Scholar]
- 18.Soldo B., Lazarevic V., Karamata D. (2002) Microbiology 148, 2079–2087 [DOI] [PubMed] [Google Scholar]
- 19.Lazarevic V., Karamata D. (1995) Mol. Microbiol. 16, 345–355 [DOI] [PubMed] [Google Scholar]
- 20.Schertzer J. W., Bhavsar A. P., Brown E. D. (2005) J. Biol. Chem. 280, 36683–36690 [DOI] [PubMed] [Google Scholar]
- 21.Pereira M. P., Schertzer J. W., D'Elia M. A., Koteva K. P., Hughes D. W., Wright G. D., Brown E. D. (2008) Chembiochem 9, 1385–1390 [DOI] [PubMed] [Google Scholar]
- 22.Badurina D. S., Zolli-Juran M., Brown E. D. (2003) Biochim. Biophys. Acta 1646, 196–206 [DOI] [PubMed] [Google Scholar]
- 23.Akita K., Ishimizu T., Tsukamoto T., Ando T., Hase S. (2002) Plant Physiol. 130, 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forsee W. T., Cartee R. T., Yother J. (2006) J. Biol. Chem. 281, 6283–6289 [DOI] [PubMed] [Google Scholar]
- 25.Guillaumie F., Sterling J. D., Jensen K. J., Thomas O. R., Mohnen D. (2003) Carbohydr. Res. 338, 1951–1960 [DOI] [PubMed] [Google Scholar]
- 26.Vionnet J., Vann W. F. (2007) Glycobiology 17, 735–743 [DOI] [PubMed] [Google Scholar]
- 27.Alderwick L. J., Birch H. L., Mishra A. K., Eggeling L., Besra G. S. (2007) Biochem. Soc. Trans. 35, 1325–1328 [DOI] [PubMed] [Google Scholar]
- 28.Segal I. H. (1993) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, pp. 1–15, 18,–97, 161,–224, 274,–344, 506–623; Wiley Interscience, New York [Google Scholar]
- 29.Alderwick L. J., Dover L. G., Veerapen N., Gurcha S. S., Kremer L., Roper D. L., Pathak A. K., Reynolds R. C., Besra G. S. (2008) Protein Expr. Purif. 58, 332–341 [DOI] [PubMed] [Google Scholar]
- 30.Willis L. M., Gilbert M., Karwaski M. F., Blanchard M. C., Wakarchuk W. W. (2008) Glycobiology 18, 177–186 [DOI] [PubMed] [Google Scholar]
- 31.Saffron C. M., Park J. H., Dale B. E., Voice T. C. (2006) Environ. Sci. Technol. 40, 7662–7667 [DOI] [PubMed] [Google Scholar]
- 32.Cook P. F., Cleland W. W. (2007) Enzyme Kinetics and Mechanism, pp. 121–204, Garland Science Publishing, New York [Google Scholar]
- 33.Pereira M. P., Brown E. D. (2004) Biochemistry 43, 11802–11812 [DOI] [PubMed] [Google Scholar]
- 34.Schertzer J. W., Brown E. D. (2008) J. Bacteriol. 190, 6940–6947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhavsar A. P., D'Elia M. A., Sahakian T. D., Brown E. D. (2007) J. Bacteriol. 189, 6816–6823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Formstone A., Carballido-López R., Noirot P., Errington J., Scheffers D. J. (2008) J. Bacteriol. 190, 1812–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ha S., Chang E., Lo M., Men M., Park P., Ge M., Walker S. (1999) J. Am. Chem. Soc. 121, 8415–8426 [Google Scholar]
- 38.Weidenmaier C., Kokai-Kun J. F., Kristian S. A., Chanturiya T., Kalbacher H., Gross M., Nicholson G., Neumeister B., Mond J. J., Peschel A. (2004) Nat. Med. 10, 243–245 [DOI] [PubMed] [Google Scholar]
- 39.D'Elia M. A., Pereira M. P., Chung Y. S., Zhao W., Chau A., Kenney T. J., Sulavik M. C., Black T. A., Brown E. D. (2006) J. Bacteriol. 188, 4183–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]