

Abstract
DC-SIGN is a C-type lectin receptor of dendritic cells and is involved in the early stages of numerous infectious diseases. DC-SIGN is organized into a tetramer enabling multivalent interaction with pathogens. Once formed, the DC-SIGN-pathogen complex can be internalized into compartments of increasing acidity. We have studied the pH dependence of the oligomerization state and conformation of the entire extracellular domain and neck region. We present evidence for equilibrium between the monomeric and tetrameric states of the extracellular domain, which exhibits a marked dependence with respect to both pH and ionic strength. Using solution x-ray scattering we have obtained a molecular envelope of the extracellular domain in which a model has been built. Our results highlight the central role of the neck domain in the pH-sensitive control of the oligomerization state, in the extended conformation of the protein, and in carbohydrate recognition domain organization and presentation. This work opens new insight into the molecular mechanism of ligand release and points to new avenues to block the first step of this important infection pathway.
Dendritic cells (DCs),2 which are found in peripheral tissues and act as sentinels against invading pathogens are considered to be the most efficient professional antigen presenting cells identified so far (1). A major subset of these DCs, dermal DCs, is characterized by the membrane expression of the DC-SIGN (dendritic cell-specific ICAM3 grabbing non-integrin) receptor (CD209). DC-SIGN is a calcium-dependent (C-type) lectin able to recognize highly glycosylated proteins. It is implicated in the early stages of many viral infections (2) and is spatially distributed in well defined membranous microdomains with an average diameter of 200 nm that act as docking platforms for pathogens and endogenous antigen attachment (3, 4). DC-SIGN binds to viral pathogens through their exposed glycoproteins such as HIV-1 envelope protein (gp120) (5), GP1 of Ebola (6), E1 and E2 of HCV (7, 8), and Dengue virus E glycoprotein (9). Apart from the viral world, DC-SIGN has also been implicated in infection processes involving fungi (10, 11), bacteria, such as Neisseria meningitidis (12) and Leishmania pifanoi (13), and parasites, such as Schistosoma mansoni (14). In addition, DC-SIGN mediates DC contacts with endothelial cells, naive T lymphocytes, and neutrophils by interacting with the endogenous adhesion molecules ICAM2 (15), ICAM3 (16), and the CD11b/CD18 integrin (17), respectively.
DC-SIGN is a type II membrane protein comprising three main domains: a cytoplasmic region, a transmembrane segment, and an extracellular domain (ECD). The ECD can be divided into two structurally and functionally distinct regions: a neck region involved in the tetramerization of the receptor and a calcium-dependent carbohydrate-recognition domain (CRD), which is at the heart of the molecular recognition processes mediated by DC-SIGN.
The cytoplasmic region contains recycling and internalization motifs important for targeting receptors, together with their associated ligands, to subcellular compartments as shown for DEC 205 and DC-SIGN itself (18, 19). Indeed, in some conditions, combined deletion of the triacidic cluster (EEE), the dileucine (LL), and the tyrosine-based (YXXL) internalization motifs induces the loss of DC capacity to enhance T-cell HIV-1 infection (19). The ability of DC-SIGN to promote T-cell infection by HIV-1 involves different pathways depending on the kinetics of the infection process. HIV transfers from DCs to T cells have been described to occur in two phases: a short-term phase (up to 24 h after viral exposure) and a long-term phase (from 24 to 72 h) (20, 21). The long-term phase is proposed to involve membrane fusion and subsequent DC infection and does not need to concern us further in this study. In contrast, short-term phase transfer involves virion internalization into DCs through vesicles. A pH dependence of HIV transfer has been suggested, based on the abrogation of the trans-enhancement of T-cell infection upon intracellular pH neutralization by concanamycin A (19). Indeed, internalization of HIV particles into low pH non-lysosomal compartments has been described in DC-SIGN-expressing cells (19). At the molecular level, a pH dependence of DC-SIGN binding to gp120 has been reported (22), as well as ligand release from DC-SIGN in pH conditions mimicking the acidic luminal environment of endosomes/lysosomes (23). However, no molecular explanation has yet been proposed for this phenomenon that might play a key role in the initiation mechanisms of DC-SIGN-mediated infectious processes. An effect of pH on the CRD binding properties has been reported (22) but no detailed molecular study of the pH dependence of the oligomerization properties of the neck domain, essential for the integrity of ECD, has yet been reported.
The neck region comprises seven complete and one incomplete 23-amino acid long repeat. These repeats exhibit a distinct pattern of hydrophobic residues, positioned at regular intervals along an α-helical stretch that could favor coiled-coil interaction between helices from the four chains within a tetramer (24, 25). Comparison of circular dichroism spectra from the entire ECD and the CRD fragment led to a similar suggestion (26). The neck domain, which is responsible for tetramer formation and stability of the whole ECD, is thereby at the onset of the avidity mechanism that endows the DC-SIGN receptor with a very high affinity toward its target as compared with the weak affinity of isolated CRD. Indeed, the dissociation constant of the complex with the HIV gp120 envelope protein is in the nanomolar range with the whole ECD, whereas it is in the millimolar range when using monomeric CRD (22). Moreover, DC-SIGN neck region variants, resulting from alternative splicing and genomic polymorphism, exhibit altered sugar binding (27). DC-SIGN neck polymorphism has recently been correlated with differential susceptibility to HIV-1 and other pathogens as a result of modified DC-SIGN multimerization on the cell surface (27). Beyond the avidity mechanism already mentioned, the association of the four chains within the neck largely constraints the CRD spatial arrangement that must bear on the ligand interaction and recognition properties. However, despite the importance of the DC-SIGN tetrameric state in molecular recognitions, structural information is scarce and mostly restricted to the CRD moiety. High-resolution structures of the CRD in complex with various carbohydrate ligands are available (28–31). The partial structure of a repeat has been obtained using a truncated dimeric form of the related homolog, DC-SIGNR (29, 32). However, structural information on the tetramer is still lacking although it is a prerequisite to a precise understanding of CRD presentation, pathogen recognition, and to rationalize emerging strategies for the design of DC-SIGN multivalent inhibitors (33–36).
Our study of the ECD, and more particularly of the neck domain, addresses two issues. First of all, we investigate the role of the neck domain in the structural organization of DC-SIGN ECD. We analyze its stability and dynamic as a function of pH conditions encountered in the various cell compartments. Our studies involve various constructs (CRD, ECD, and neck region) using several techniques: analytical ultracentrifugation, size exclusion chromatography (SEC), and circular dichroism (CD). We show that physiologically encountered pH variations significantly affect the protein oligomerization and structure. Slightly acidic pH destabilizes the tetramer as well as the CRD structure with possible consequences on the strength of ligand binding, which is strongly dependent on the multiplicity of interactions. In parallel, a molecular envelope of DC-SIGN ECD derived from small angle x-ray scattering (SAXS) measurements in solution highlights the extended organization of DC-SIGN and suggests a tighter arrangement of the four CRDs than hereto proposed.
EXPERIMENTAL PROCEDURES
Cloning
The cDNA coding for the DC-SIGN CRD (amino acids 254–404) was obtained by PCR and cloned into pET15b (Novagen). The protein was expressed in Escherichia coli as previously described (37).
The cDNA encoding DC-SIGN ECD (amino acids 66–404) was cloned into a pET30b plasmid (Novagen) resulting in expression vector pET30b-DC-SIGN ECD. Expression was carried out in E. coli as previously described (35).
The construct allowing DC-SIGN S-Neck expression has been generated in two steps. In a first step, the sequence encoding the whole DC-SIGN ECD region was obtained by PCR using pET30b-DC-SIGN ECD vector as DNA matrix and the primers EC5′ (5′-GCA TTA GGT CTC TGC GCT CCA TAA GTC AGG AAC AAT C-3′) and EC3′ (5′-GCA GCA GGT CTC TTA TCA CTA CGC AGG AGG GGG G-3′). Addition of a Strep-Tag II at the N terminus of the construct was achieved by cloning the PCR product into a pASK6 vector (IBA GmbH). The vector and the PCR product were both digested by BsaI before ligation using the DNA Rapid Ligation Kit (Roche) to lead to the pASK6-DC-SIGN ECD plasmid. In a second step, the Strep-tagged DC-SIGN Neck encoding fragment was obtained by changing, within the pASK6-DC-SIGN ECD plasmid, the His254 codon by a stop codon immediately downstream of the neck sequence. This site-directed mutagenesis was performed by PCR using the following primers: Neck forward (5′-GTG GAA CGC CTG TGA CAC CCC TGT CC-3′, underlined: stop codon) and Neck reverse (5′-GG ACA GGG GTG TCA CAG GCG TTC CAC-3′, underlined, stop codon). This PCR product and the pET20b plasmid (Novagen) were digested with XbaI and HindIII before ligation. The resulting plasmid was termed pET20b-DC-SIGN S-Neck.
Protein Expression and Purification
Upon expression, DC-SIGN CRD and ECD formed inclusion bodies and were refolded as previously described (26). Purification of functional proteins was achieved by affinity chromatography on a mannan-agarose column (Sigma) equilibrated with 25 mm Tris-HCl, pH 8, 150 mm NaCl, 4 mm CaCl2 (buffer A). After loading, DC-SIGN CRD was purified as a delayed fraction, whereas in the case of DC-SIGN ECD the protein was tightly bound to the column and eluted in the same buffer without CaCl2 but supplemented with 10 mm EDTA (buffer B). This step was followed by SEC, using a Superose 6 column equilibrated with buffer A.
DC-SIGN-S-Neck was expressed in E. coli BL21(DE3) in 1 liter of LB culture supplemented with 100 μg/ml ampicillin at 37 °C. Expression was induced by addition of 0.1 mm isopropyl 1-thio-β-d-galactopyranoside when the culture had reached an A600 of 0.6 and maintained for 4 h. The protein was expressed in the cytoplasm in a soluble form. Cells were harvested by a 20-min centrifugation at 5000 × g at 4 °C. The pellet was resuspended in 25 ml of a solution containing 0.2 m NaCl, 20 mm Tris-HCl, pH 8, 10 mm MgCl2, DNase I, and one anti-protease mixture tablet (Complete EDTA free, Roche). Cells were disrupted by sonication and cell debris eliminated by centrifugation at 100,000 × g for 45 min at 4 °C. The supernatant was loaded onto a Strep-Tactin Superflow column (IBA GmbH) at 20 °C. Unbound proteins were washed away with buffer C (150 mm NaCl, 25 mm Tris-HCl, pH 8, 1 mm EDTA) before DC-SIGN S-Neck was eluted with buffer D (150 mm NaCl, 25 mm Tris-HCl, pH 8, 1 mm EDTA, 2.5 mm desthiobiotine). Eluted fractions were analyzed by SDS-PAGE (12%) and the DC-SIGN S-Neck containing fractions were pooled and concentrated to 4 mg/ml by ultrafiltration (YM10 membrane from Amicon). The Strep-Tag II was cleaved off using restriction grade factor Xa (Qiagen) at a ratio of 0.1 units/mg of Strep-tagged protein for a 30-min incubation at 4 °C. Elimination of the cleaved off Strep-Tag II was performed by SEC. The factor Xa-cleaved sample was loaded onto a Superdex 200 column (Amersham Biosciences) previously equilibrated with 150 mm NaCl and 25 mm Tris-HCl, pH 8. The protein-containing fractions were pooled and concentrated. The resulting protein, after tag cleavage, was termed DC-SIGN Neck. The molecular mass of each construct was confirmed by mass spectroscopy. Each construct was also checked by N-terminal sequencing.
Oligomerization State Analysis by Size Exclusion Chromatography
We used a calibrated Superdex 200 column to study the pH and salt effects on the protein oligomerization state. The column, kept at a constant temperature of 20 °C was first equilibrated with the appropriate buffer at varying pH (pH range 5.1 to 8.9). For pH variation studies, all buffers were used at a 25 mm concentration (acetate for pH 5.1, potassium phosphate for pH 5.9–7.5, and Tris for pH 8.9), supplemented with 150 mm NaCl. For experiments based on ionic strength variations, a 25 mm phosphate buffer, pH 7.4, was supplemented with an increasing amount of NaCl yielding the final concentrations series: 0, 0.05, 0.15, 0.3, 0.6, 1, 1.5, 2, 2.5, 3, 3.5, and 4 m. DC-SIGN EC and DC-SIGN Neck stock solutions (32 mg/ml) were diluted to 2.5 mg/ml in the appropriate buffer and then centrifuged to eliminate potential aggregates. Each sample (250 μl) was then loaded onto the previously equilibrated column and eluted in the same buffer. The collected fractions were analyzed by 12% SDS-PAGE. Analysis of the oligomerization state distribution was performed in parallel to the analytical ultracentrifugation.
Analytical Ultracentrifugation
Sedimentation velocity experiments were performed in a Beckman XL-I analytical ultracentrifuge using an AN-60 TI rotor (Beckman instruments), at 20 °C. Samples were handled in a buffer with appropriate pH. The molar mass and partial specific volume of DC-SIGN Neck and ECD chains were estimated from the amino acid composition using the SEDNTREP software: 38706 Da and 0.733 ml/g for the whole ECD, and 21228 Da and 0.748 ml/g for the neck.
Sedimentation velocity experiments were carried out at 42,000 rpm, using 400-μl protein samples, loaded into the two channel 1.2-cm path length centerpieces equipped with sapphire windows, and monitored at 5-min intervals using absorbance and interference optics. Absorbance scans were recorded overnight at 279 nm with a radial step size of 30 μm. Sedimentation velocity profiles were analyzed using the size distribution analysis from the program SEDFIT that provides a continuous distribution of sedimentation coefficients, c(s) (38). Typically, 20 regularly spaced experimental profiles obtained over a total 6-h sedimentation were globally modeled. The continuous distribution of sedimentation coefficients (c(s)) analysis was performed considering 200 particles on a grid of 300 radial points, and fitting the value of the frictional ratio f/f0. For the regularization procedure a confidence level of 0.7 was used.
The interpretation in terms of oligomer use the following formula,
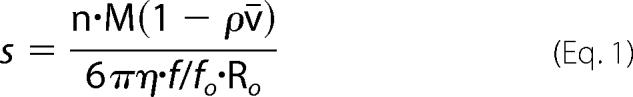 |
with s the sedimentation coefficient, n the number of monomers per complex, M the monomer molecular mass, R0 the radius of a spherical particle of density 1/v̄ and mass n.M, f/f0 the frictional ratio, v̄ the partial specific volume of the protein, η and ρ the viscosity and the density of the solvent, respectively. All s values have been corrected for solvent density and viscosity and are therefore given as s20,w values.
Sedimentation Coefficient from Models
The value of the sedimentation coefficient was calculated from the model of DC-SIGN using the program HYDROPRO (39). The solvent viscosity was 0.010 poise, the solvent density was 0.998 g/ml, and the temperature was 293 K. The effect of hydration is included in an effective atomic radius of 3.2 Å for all non-hydrogen atoms in the primary hydrodynamic model.
Circular Dichroism
Circular dichroism spectra were measured on a Jobin Yvon CD6 spectropolarimeter operating at room temperature. Quartz cells with a 1-mm path length were used for recording spectra in the UV region (180–260 nm). DC-SIGN Neck and DC-SIGN ECD were diluted to a final concentration of 0.2 mg/ml in the buffered solutions at each pH value and centrifuged to eliminate aggregates. These buffered solutions were prepared by mixing the required amounts of acidic and basic potassium phosphate at a final concentration of 25 mm with 150 mm NaCl.
Small Angle X-ray Scattering
SAXS data were collected at the X33 beamline at EMBL/DESY, Hamburg (40). The sample to detector distance was 2.3 m covering the range of scattering vector (q) from 0.008 to 0.47 Å−1 (q = 4πsinθ/λ, with 2θ being the scattering angle and λ = 1.5 Å the wavelength of the x-rays. The detector used was the online image plate reader MAR345 from MarResearch. The samples were prepared at concentrations of 1 and 2 mg/ml in 25 mm Tris, pH 8, 60 mm NaCl, 4 mm CaCl2, 5% (v/v) glycerol. Two successive frames of 3 min each were recorded for proteins and buffers. No radiation damage was detected. SAXS data were normalized to the intensity of the incident beam, averaged, and background subtracted using the program package PRIMUS (41). The curves recorded at 1 and 2 mg/ml were identical and therefore free from intermolecular interactions. A few test patterns were also recorded during commissioning time on the Swing SAXS beamline at the SOLEIL storage ring and were found essentially identical to the EMBL data.
In the case of a very elongated particle the Guinier range is very narrow and, in the case of DC-SIGN, restricted to such small angles as to be mostly inaccessible (42). Consequently, the radius of gyration (Rg) and the intensity at the origin I(0) were derived from the distance distribution function P(r) computed using the program GNOM (43).
The conformation in solution of DC-SIGN was determined ab initio from the scattering curve using the programs DAMMIN (44) and GASBOR (45). The two programs generate a volume filled with beads or a compact chain of dummy residues, respectively, whose scattering pattern fits the experimental scattering curve as measured by the discrepancy χ,
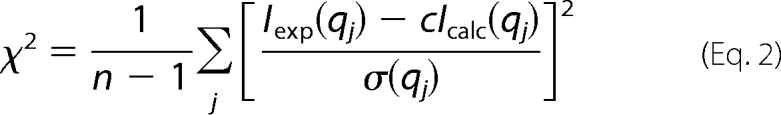 |
where n is the number of experimental points, c is a scaling factor, and Icalc(qj) and σ(qj) are the calculated intensity and experimental error, respectively. Models obtained from repeated calculations are superimposed and compared using the program suite DAMAVER (46). Their similarity is quantified using a normalized spatial discrepancy (47). Similar DAMMIN models for globular objects have normalized spatial discrepancy values of the order of 0.7, whereas corresponding GASBOR derived models exhibit values in the range of 1.0–1.4.
RESULTS
DC-SIGN Constructs, Production, and Purification
The truncated forms of DC-SIGN used in this study are presented in Fig. 1. DC-SIGN CRD and DC-SIGN ECD were overexpressed as inclusion bodies in E. coli BL21(DE3) strain. The two protein purification schemes started with the refolding procedure previously described (30). DC-SIGN CRD and ECD were produced without affinity tag and we used the physiological interaction of DC-SIGN with carbohydrate ligands for purification purposes. Oligomeric ECD tightly bound to the mannan-agarose column through an avidity based mechanism. Monomeric CRD, in contrast, with a low affinity for its natural ligands, did not firmly bind to the mannan-agarose column and was only delayed as previously described for other C-type lectin CRDs (48, 49). However, following a prior concentration to minimize the sample volume, monomeric CRD could be separated from the flow-through on the mannan-agarose column. DC SIGN-ECD and CRD were obtained to homogeneity (data not shown).
FIGURE 1.

Representations of DC-SIGN domain organization and the constructs used in these studies. CD, cytoplasmic domain; TM, transmembrane region; ND, neck domain required for oligomerization; S, Strep Tag; Xa, factor Xa cleavage site.
The DC-SIGN S-Neck protein was expressed as a soluble form in the E. coli cytoplasm. Thanks to the Strep-Tag II, the protein could be purified to homogeneity in a single affinity chromatography step. Following tag cleavage using factor Xa, DC-SIGN Neck was finally separated from the cleaved Strep-Tag II on a SEC column. Remarkably, purified DC-SIGN Neck protein eluted as two separate peaks of equivalent purity as judged from SDS-PAGE (Fig. 2, A and B). In what follows, the protein eluting within the peak of higher molecular mass is referred to as “form A,” that under the second one as “form B.”
FIGURE 2.

Analysis of the interconversion between the two distinct forms of DC-SIGN Neck. A, elution profile obtained for DC-SIGN Neck loaded onto the Superdex 200 column. B, SDS-PAGE analysis of the two peaks, A and B. C, study of the interconversion of the two forms; top, elution profiles obtained after injection of peak B concentrated at 0.4 and 0.8 mg/ml on Superdex 200 column; bottom, elution profiles obtained after injection of peak A on Superdex 200 column following successive 2-fold dilutions to final concentrations 3.5, 1.75, 0.88, 0.44, 0.22, and 0.11 mg/ml.
Interconversion between Forms A and B of DC-SIGN Neck
To answer the question if forms A and B are mutually interconvertible, 200 μl of form B were concentrated from 0.05 mg/ml to 0.4 and 0.8 mg/ml before being reloaded onto the calibrated Superdex 200 column. Most of the protein eluted at the position of form A with a minor peak at the position of form B (Fig. 2C, upper panel). The symmetrical experiment was also carried out: an aliquot of pool A (originally concentrated at 7 mg/ml) was reloaded onto the same column at decreasing concentrations 3.5, 1.75, 0.88, 0.44, 0.22, and 0.11 mg/ml (Fig. 2C, lower panel). No significant redistribution from form A to form B was observed upon dilution. Thus, under these buffer conditions, the different elution profiles show that form A corresponds to an oligomeric form of DC-SIGN Neck over a large concentration range. Whatever the concentration of form A, a very small quantity of form B is always seen to coexist within the solution.
Globally these data suggest the existence of equilibrium between two different oligomeric states, the equilibrium being strongly shifted toward form A under our experimental conditions. Elution volumes of both forms calibrated in Stokes radius with commercial globular proteins cannot be used to estimate the degree of oligomerization of both forms A and B because of the very elongated shape that could be expected from a succession of coiled-coil modules. However, based on the known oligomeric state of DC-SIGN ECD, form A of DC-SIGN Neck is assumed to be a tetramer in equilibrium with a monomeric form B. The association/dissociation process appears to be highly cooperative as no trace of any intermediate oligomer is ever detected, a feature to be expected from the multiple coiled-coil formation along the neck.
Effect of pH on DC-SIGN Neck and DC-SIGN ECD Oligomerization
The different pH conditions encountered by DC-SIGN within the different cell compartments can lead to titration of ionized groups within the protein with potential consequences at the structural and/or functional level. In particular, such changes may generate repulsive interactions between identical charges that could destabilize the oligomeric association or, more locally, secondary structure elements. To investigate the effect of pH on the oligomerization equilibrium of DC-SIGN Neck described above and on whole DC-SIGN ECD as well, SEC was used in conditions of decreasing pH. Fig. 3, A and B, shows elution profiles for DC-SIGN Neck and DC-SIGN ECD, respectively, at pH values ranging from 7.4 to 5.1. Not only DC-SIGN Neck but also DC-SIGN ECD was seen to elute under two distinct oligomeric states whose distribution exhibits a strong pH dependence. This was confirmed, in the case of DC-SIGN ECD, by the SDS-PAGE analysis of the two peaks obtained at lower pH, termed form A′ and B′, which reveals the presence of DC-SIGN ECD in both peaks (Fig. 3C). Both constructs exhibit an increase in the proportion of lower molecular mass form (B and B′) with pH decrease.
FIGURE 3.

Effect of pH on the oligomeric state of DC-SIGN ECD and Neck monitored by size exclusion chromatography. pH value from top to bottom: 7.4, 6.4, 5.9, and 5.1. A, DC-SIGN Neck. B, DC-SIGN ECD. C, SDS-PAGE analysis of the two peaks observed for DC-SIGN ECD at pH 5.9.
A closer examination of the SEC profiles shows that the pH corresponding to equivalent proportions of A and B (or A′ and B′) are different for the two proteins. This midpoint was reached at pH 6.4 for the DC-SIGN Neck and pH 5.9 for the DC-SIGN ECD. Similarly, at pH 5.1, both species still coexist for the ECD, whereas a complete disappearance of the tetrameric form was observed for the Neck. This shift in the equilibrium properties between the two proteins suggests that the CRD stabilizes the oligomeric form of the ECD construct through interactions within the tetramer.
Analytical Ultracentrifugation Analysis of DC-SIGN ECD
To complement the SEC measurements, we performed sedimentation velocity measurements at variable pH values. Previous experiments performed at pH 7.8 showed a unique tetramer peak at s20,w = 5.1 S. From the analysis of the boundary profiles in terms of s and diffusion coefficient, a molar mass of 148 kDa was obtained compatible with the tetramer. The derived frictional ratio of 1.9 indicates an elongated tetramer (29, 33). The present experiment at pH 8.9 revealed the same tetrameric species with s20,w = 5.2 ± 0.1 S (Fig. 4B). In the same conditions, DC-SIGN Neck migrates as a single peak of 3.4 S with a fitted f/f0 of 1.9 (Fig. 4C), corresponding to an association state of n = 3.85 (calculated according to Equation 1). This also corresponds to an elongated tetramer confirming that the sole Neck species present at that pH, form A, is a tetrameric assembly.
FIGURE 4.

Analytical ultracentrifugation analysis of the DC-SIGN-derived oligomeric species in solution as a function of pH. A, DC-SIGN ECD (7 μm) velocity sedimentation profiles obtained using absorbance optics. Superimposition of experimental sedimentation profiles and fitting curves from the c(s) analysis using SEDFIT (upper panel). B, DC-SIGN ECD c(s) velocity sedimentation analysis at different pH values using absorbance optics. All experiments were performed at 7 μm protein and 20 °C and the chosen pH values were 8.9, 6.4, and 5.9. c(s) distributions are displayed on the same scale. C, DC-SIGN Neck c(s) velocity sedimentation analysis of the profiles recorded in 25 mm Tris HCl buffer, pH 8.9, 150 mm NaCl, at 20 °C.
In measurements performed at pH 6.4 and 5.9 on DC-SIGN ECD solutions, two species are observed at 4.7 and 2.45 S (Fig. 4, A and B). Lowering the pH from 6.4 to 5.9 results in a significant decrease of the fractional concentration of the 4.7 S species and a concomitant increase (from 31 to 50%) of that of the lighter species (2.45 S). The peaks are at the same positions, only their proportions differ. Thus, over the time scale of the sedimentation experiment, the sample can be analyzed as a mixture of non-interacting species. To identify the oligomers corresponding to each species, we use c(s, f/f0) distribution analysis (50) that allows individual fitting of f/f0 for each species. For both pH values, the procedure produces a slight improvement of the root mean square deviation (3%). The distributions of f/f0 for each peak are broad (see Fig. S1 in supplementary material) but give consistent results at pH 6.4 and 5.9, with f/f0 values of 1.7 for the 2.45 S species (±0.3 at pH 6.4 and ±0.2 at pH 5.9) and of 2.2 for the 4.7 S species (±0.7 at pH 6.4 and ±0.8 at pH 5.9). The combination of f/f0 and s20,w values suggests that the smaller species corresponds to a monomer (calculated association state of n = 0.95) and the larger one to a tetramer (n = 4.04).
Tetramers are observed in both acidic and alkaline conditions. However, in the range of pH 5.9 to 6.5 the tetramer displays a smaller s value than that observed at pH 8.9 (4.7 S versus 5.2 S) together with a higher f/f0 value (f/f0 = 2.2 versus 1.9). This corresponds to an increase of the hydrodynamic radius of the tetramer of about 10% at lower pH values. Interestingly, careful examination of the SEC analysis (Fig. 3B) reveals a slight shift of the elution volume of the A′ form toward smaller elution volume when lowering the pH. This is in qualitative agreement with the increase of the hydrodynamic radius inferred from the sedimentation velocity experiments. The DC-SIGN ECD B′ form observed in SEC corresponds to the 2.45 S species and is identified as a monomer.
Analysis of pH-dependent Effects on the Secondary Structure by Circular Dichroism
CD spectroscopy was used to monitor the effect of pH on the secondary structure of DC-SIGN constructs and to correlate it with the results obtained from SEC and analytical ultracentrifugation. The CD spectra of DC-SIGN ECD seen in Fig. 5 display prominent minima at 208 and 222 nm, those of DC-SIGN Neck a minimum at 208 nm and a shoulder at 222 nm. At pH 7.4, the signal intensity at 222 nm, for ECD and Neck, indicates the presence of substantial helical structures as expected for coiled-coils. The CD spectra of both proteins exhibit reduced amplitude with decreasing pH showing that tetramer dissociation is accompanied by loss of secondary structure. However, the CD spectra of each protein present a distinct pH dependence: patterns of the Neck region show a gradual decrease in intensity, whereas the ECD spectra display a sharp 2-fold reduction in intensity when the pH decreases from 6.4 to 5.9 (Fig. 5, A and B). Again, this suggests that the CRD contributes some interaction stabilizing the tetramer.
FIGURE 5.

Circular dichroism spectra of DC-SIGN. Spectra were recorded at different pH values (7.4, 6.4 and 5.9). A, DC-SIGN Neck. B, DC-SIGN ECD. C, DC-SIGN CRD. The difference spectrum (DC-SIGN ECD–DC-SIGN Neck) at pH 7.4 is superimposed for comparison (dashed line).
CD spectra of CRD were recorded at various pH values (Fig. 5C). As a control, the CD spectrum of the Neck construct at pH 7.4 was subtracted from the corresponding spectrum of the ECD thereby yielding the spectrum of the CRD alone. This difference spectrum appears to be very close to the experimental one (Fig. 5C). Despite the presence of disulfide bridges, the CRD exhibits a loss of secondary structure with pH decrease that could explain the loss of binding capacity previously reported for the CRD upon acidification (32).
Effect of Ionic Strength on Tetramerization
Intrahelical or interhelical salt bridges are often involved in coiled-coil formation and stabilization (51, 52). It has been shown that salt can have a stabilizing or destabilizing effect depending on the nature of charged residues and ionic interactions present in the coiled-coil (53). To get some insight on the role of electrostatic interactions in the structural organization of DC-SIGN coiled-coil domain, the effect of increasing salt concentration (NaCl) on the oligomeric state has been investigated. Experiments were performed using the DC-SIGN Neck construct to simplify the system and thereby the interpretation of the results. Results in Fig. 6 show that at low ionic strength, below 50 mm NaCl, the tetramer, normally observed at pH 7.4, dissociates into monomers. Above 150 mm NaCl, all molecules elute as tetramers. The need for a minimum ionic strength to ensure complete tetramerization suggests that electrostatic repulsions occur between individual helices that destabilize the coiled-coil and that a minimal level of salt is required to neutralize this electrostatic effect. In addition, the resistance of tetramers to high salt concentration strongly suggests that hydrophobic interaction is the major driving force behind the coiled-coil formation.
FIGURE 6.

Effect of salt concentration on DC-SIGN Neck association. Size exclusion chromatography elution buffers: 25 mm Tris-HCl, pH 8.0, 4 mm CaCl2 with variable salt concentration. Thin line, no added salt; gray line, 50 mm; thick black line, superposition of identical elution profiles for 0.150, 0.3, 0.6, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, and 4.0 m.
Small Angle X-ray Scattering
Because scattering patterns recorded at 1 and 2 mg/ml were identical, further analysis is restricted to the latter curve. The pair distribution function P(r) derived from the scattering data exhibits a sharp maximum at small distances followed by a regular decrease out to large distances characteristic of an elongated cylinder-like particle (Fig. 7A). The maximum diameter of the protein is of the order of 320 ± 20 Å and the radius of gyration has a value of 98 ± 2 Å. The value of the molecular mass derived from the intensity at the origin agrees well with the theoretical mass of the tetrameric protein (M = 155 kDa), ruling out any significant aggregation of the protein in solution.
FIGURE 7.

Pair distribution function P(r) and ab initio model of DC-SIGN ECD. A, the pair (or distance) distribution function P(r) was computed from the scattering intensity I(q) using the program GNOM. The profile is characteristic of a very elongated object such as a long and thin cylinder. B, DC-SIGN model obtained using the ab initio modeling program GASBOR that represents the protein as a chain of dummy residues, one per amino acid residue in the protein, centered at the Cα positions. The volume of the spherical dummy residue is the average volume of amino acid residues. The 608 dummy residues corresponding to the 4 CRDs are colored black, whereas the 748 dummy residues describing the neck are colored gray. C, calculated scattering pattern of the GASBOR model shown above (gray continuous line) superimposed onto the experimental scattering intensity curve (black dots).
The overall conformation of the protein was investigated using both programs DAMMIN and GASBOR. Each program was run about 15 times and the various resulting conformations were compared using the program suite DAMAVER. Both ab initio approaches lead to identical conclusions: the protein adopts a very elongated shape in which a thicker end, the head, is followed by a thinner tail of considerable linear extension and correlatively low curvature. The head long-axis lies essentially parallel to the tail axis. All conformations are very similar with normalized spatial discrepancy values of 0.8 and 1.8 computed for DAMMIN and GASBOR models, respectively. The similarity is even higher than suggested by the normalized spatial discrepancy values, as these are very sensitive to minor, local differences between globally very similar tails. A typical example of a GASBOR model is shown in Fig. 7B, whereas the agreement between the calculated scattering curve of the model and the experimental data are shown in Fig. 7C (χ = 1.2). All models exhibit a tail with a length of 245–250 Å and a diameter of 25–30 Å, whereas the head has a length of 70–75 Å and transverse dimensions in the range of 50 to 80 Å.
We have attempted to position the known structure of DC-SIGN CRD (Protein Data Bank code 1k9i) within the head of the model. An “open flower”-like arrangement as found in DC-SIGNR tetramer (PDB code 1xar), another C-type lectin receptor closely related to DC-SIGN, is not compatible with our data. In the case of DC-SIGN, accommodating 4 CRDs within the head requires a compact arrangement of the domains with their longer axis in an orientation close to that of the tail. Such an orientation of the CRD is found in human mannose-binding protein (PDB code 1hup), which represents the prototype of the C-type family of lectin and for which an oligomeric structure is available. Mannose-binding protein also contains a coiled-coil repeat followed by a C-type CRD. DC-SIGN CRD (PDB code 1k9i) was superimposed on the mannose-binding protein CRD. Four copies of DC-SIGN CRD in this new orientation with respect to the helical repeat were then manually positioned within the SAXS envelope of the head.
A model for each repeat was then built as follows: all 23 residues within a repeat were considered to adopt a helical conformation and the four neighboring helices within the tetramer were assumed to form a four-stranded coiled-coil. A 23-residue long helix was built using PyMOL (DeLano Scientific). Four copies of the helix were then arranged as the four α-helices observed in the structure of the tetramer of DC-SIGNR CRDs (PDB code 1xar). Clearly, this is only an approximation and it is most likely that several residues among the 23 amino acids do not adopt a stable helical conformation. However, the conformation of the neck derived from the program GASBOR imposes an elongated shape of the repeats, with a diameter of the order of 25–30 Å, very close to that of a four-stranded coiled-coil, which may therefore constitute a good approximation as regards to the calculation of the scattering pattern. Indeed, the latter is practically insensitive to small deviations in the residue positions with respect to the coiled-coil conformation. Seven identical modules were manually positioned in the neck part of the GASBOR model. Such a module also described the N-terminal domain; although it is not strictly equivalent to a complete repeat. However, secondary structure analyses indicate that the N-terminal residues have a strong helical propensity. Here again, the approximation can be considered as satisfactory as far as the calculation of the scattering pattern is concerned.
Finally, the 22 C-terminal residues of DC-SIGN CRD are absent from the crystal structure. To complete the protein description, they were modeled using MODLOOP (54) and the position of the four resulting C terminus ends was manually adjusted so as to yield a good fit of the calculated scattering pattern against the experimental data. The final arrangement is shown in Fig. 8A.
FIGURE 8.

DC-SIGN ECD ab initio model. A, superimposition of the CRDs tetramer (PDB code 1k9i) and neck modules assumed to be arranged in coiled-coil over the GASBOR model. Domains and modules were manually positioned. The oligosaccharide moiety found in PDB 1k9i is shown using an orange CPK representation, although it was not present in our samples, solely to help visualize the interaction sites of the protein with carbohydrate ligands. B, comparison of CRDs arrangement in the DC-SIGN model derived from our SAXS data and in the crystal structure of a fragment of DC-SIGNR containing the four CRDs and two neck repeats (PDB code 1xar).
We want to underline that our analysis cannot yield a description at the atomic level and that the arrangement of the various domains within the SAXS-derived model is only an approximation as is the four-stranded coiled-coil module used along the neck. However, this analysis gives reliable information regarding the CRD orientation within the head and the very extended conformation of the tail. We believe our model to be a correct, albeit low-resolution, description of DC-SIGN because it accounts for all our experimental observations. Indeed, its calculated scattering pattern is in very good agreement with the experimental curve (Fig. 7C) and the value of the sedimentation coefficient calculated using the program HYDROPRO is 5.2 S, identical to the experimentally measured value.
DISCUSSION
Structural Organization of the Neck Domain
In this work we report for the first time the recombinant production of the neck domain depleted of its C-terminal CRD. Using SEC, we show that the whole neck domain in a slightly alkaline solution at a few micromolar concentration is present in two co-existing oligomeric states, the higher order one in large excess with respect to the other one. Sedimentation velocity measurements showed that this major form is a tetramer, confirming that DC-SIGN ECD oligomerization is independent of the CRD. Moreover, we have shown by SEC that the second, monomeric form of DC-SIGN Neck associates to form a tetramer upon concentration increase by about 1 order of magnitude. No intermediate “dimeric state” could be detected, suggesting a highly cooperative association mechanism. Moreover, whereas both monomeric and tetrameric states are observed at low ionic strength, the tetrameric state is strongly stabilized by increasing ionic strength, suggesting a hydrophobically driven oligomerization.
When comparing analytical ultracentrifugation derived parameters, it is striking to see that the f/f0 value of 1.87 for the neck domain is very close to that for the entire ECD (1.95) but smaller if different at all. These values correspond to a very elongated shape. Assuming that the neck adopts essentially identical conformations in both constructs, a higher f/f0 ratio would be expected for the neck than for the ECD. Indeed, the tetramer of CRD heads represents a globular addition to the elongated neck. The constant value of f/f0 suggests that the neck domain C-terminal end may be somewhat frayed with individual chains splayed out that may be related to a decreased stability of the C-terminal end in the absence of the CRDs. In contrast, a stable tetrameric coiled-coil is observed up to the last repeat within the entire ECD, which implies that the CRD exerts a stabilizing effect on the last repeat, most likely through mutual interaction between CRDs. Finally the ab initio model determined by SAXS confirms that the neck domain, in the context of the ECD, is not organized as a branched structure but appears to be compatible with a canonical tetrameric coiled-coil organization.
The Neck Domain Is a pH-dependent Tetramer
Upon internalization, DC-SIGN-ligand complexes are successively passing through endosomal/lysosomal compartments of decreasing pH. In vivo studies showed that, when a pH around 6 is reached, DC-SIGN releases its ligands (23) through an unknown molecular mechanism. We investigated the effect of pH on ECD, Neck, and CRD constructs. In the case of the DC-SIGN neck construct, the pH decrease causes a tetramer dissociation into monomers that is complete around pH 5, with an approximate mid-transition point at pH 6.4. Under the same pH conditions, DC-SIGN ECD also dissociates into monomers, although the transition is shifted to lower pH values (approximate midpoint at pH 5.9) so that complete dissociation is never observed.
Analytical ultracentrifugation investigation of pH-induced dissociation of DC-SIGN-ECD reveals that the transition from mild alkaline pH (8.9 and 7.8), to slightly acidic pH (pH 6.5 and 5.9) does not only cause tetramer dissociation into monomers but also modification of the tetramer shape. Indeed, the tetramer observed under acidic conditions appears to be more extended than under mild alkaline conditions (4.7 versus 5.2 S corresponding to an increase in hydrodynamic radius of about 10%). This extended tetramer could thus correspond to a destabilized form of the assembled ECD.
Upon acidification, one or a few amino acid groups involved in stabilization of the neck tetramer must be protoned. From the size exclusion profile, an approximate pKa around 6 was found for this effect (slightly different for neck and ECD constructs). The sequence of 23 amino acids repeats composing the neck domain reads as follows: LQEIYQELTQLKAAVGELPEKSK (with a few point modifications between repeats). When examining this sequence, and in the absence of the histidine residue, the sole amino acid residue that can be expected to change its protonation state is a glutamic residue. Canonical pKa for glutamate is around 4.3, but it has been reported that, depending on the carboxylate environment within the protein, this value can increase dramatically (55). With decreasing pH, the corresponding glutamate(s) will be protonated thereby loosing their negative charge. Because pH decrease ends with the dissociation of the four helices assembly, it is tempting to speculate that this (or these) glutamate residue(s) are involved in stabilizing ionic interactions between different helices in the tetramer. When searching for plausible candidates, it is useful to write the repeats of DC-SIGN showing the heptads characteristic of coiled-coils forming sequence with hydrophobic residues in positions a and d (Fig. 9A).
FIGURE 9.

The 23-amino acid repeats composing the neck domain and their interchain salt bridge. A, positions a and d in the neck domain repeats occupied by hydrophobic amino acids are emphasized in gray. B, two repeats of the neck domain, the curved double arrow represents the putative I − i+5 interchain salt bridge between the glutamic acid (position g) of the first heptad and the lysine residue (position e) of the second one.
Upon examination, it appears that the second glutamic acid occupies position g and thus might be able to form an i-i+5 interchain salt bridge (Fig. 9, A and B) with lysine residue in position e of the following heptad as previously described in other coiled-coils (24). Protonation would lead to the loss of this interchain link, potentially destabilizing the 4-fold coiled-coil. However, this interpretation is questioned by our study of the effect of salt on the tetramer stability. Indeed, low ionic strength, lower or equal to 50 mm, at pH 7.4 causes partial dissociation of the tetramer of DC-SIGN Neck, whereas above 150 mm, all molecules are assembled. This is exactly the opposite of what the postulated Lys-Glu interchain model would predict. An alternative explanation must be found and we put forward the following proposal that could reconcile both observations. Indeed, glutamate protonation leads to the disappearance of all negative charges all along the neck, leaving only positive charges in fairly close proximity. This gives rise to electrostatic repulsions that could destabilize the helices and their coiled-coil assembly. The latter would be stable when repulsions are minimized through salt shielding or interactions with ionized carboxylates from glutamate residues. When ionic strength is too low or when carboxylate groups are protonated, repulsions between lysine side chains could destabilize the tetramer (for instance, between lysine residues in position g and e from the neighboring helix). This proposal will be put to an experimental test by investigating the structural consequences of key residue mutations.
The Arrangement of DC-SIGNR CRDs Tetramer Seen in the Crystal Is Not Compatible with Our DC-SIGN ECD Envelope
Our SAXS study gives a picture of the global conformation of complete DC-SIGN ECD (Figs. 8A and 10B). The size, 320 Å, and shape of this molecule immediately strike as a structure projecting its CRDs far above the cell surface, as a bait to trap potential pathogens and antigens. Another striking observation is the global orientation for the CRDs suggested by the model: CRDs are in a fairly closed arrangement with their long axis being roughly parallel to that of the neck (Fig. 8B). In DC-SIGNR, a closely related C-type lectin receptor, the CRDs are reported to be in a more open flower conformation as suggested by the two high resolution structures of the DC-SIGNR CRDs tetramer with one neck repeat (29). Such an arrangement is not compatible with our SAXS data. The difference between CRDs arrangement could be a consequence of crystal constraints. Alternatively, it may be associated with differences in the sequence of each protein. Despite very high sequence conservation between the necks of both lectins, the last repeat preceding the CRDs exhibits the highest level of divergence between the two proteins. This may cause some differences in the final presentation of the CRDs. A SAXS study of DC-SIGNR ECD in solution could shed some light on this issue by showing whether the CRDs arrangement seen in the crystal is preserved in solution. Finally, in the same paper reporting the crystal structure of the DC-SIGNR fragment, it was suggested, on the basis of the resistance of the neck to subtilisin and trypsin treatments, that there is little flexibility between repeats along the whole neck (31). This is in agreement with our very extended ab initio model suggesting that the neck is a fairly rigid structure that does not undergo sharp bending between repeats.
FIGURE 10.

DC-SIGN cell-surface presentation and internalization model. A, a working hypothesis describing the dynamics of a DC-SIGN-ligand complex upon internalization and subsequent pH decrease. This model proposes that the DC-SIGN-ligand pathogen complex internalized at pH 7.4 resists to mild acidification at pH 6.4 (early endosomes) and subsequently dissociates at a pH around 5.9 (late endosomes) due to both DC-SIGN chain separation and CRD structure alteration. B, relative size comparison of the structure of the HIV gp120 (outlined in pink into black square) and of its different receptors (DC-SIGN is outlined in blue, CD4 in green, and CCR5 in red). Because no structure is available for CCR5, we used the molecular model (PDB code 1nd8).
Conclusions
From the observations made in this study a new vision of the DC-SIGN internalization dynamics emerges that is summarized in the schematic representation shown in Fig. 10A, providing a new working model to be challenged in future studies. Due to its very elongated shape, DC-SIGN is one of the first receptors, on the dendritic cell surface, able to interact with carbohydrate motifs at the surface of a potential pathogen as illustrated in Fig. 10B when compared with other HIV-binding receptors.
Upon internalization, pH decreases from the early endosome to lysosome. The data obtained suggests that this could trigger early ligand dissociation through two cumulative effects: CRD structural disorganization and dissociation of the ECD tetramer. This work has been based on the characterization of a recombinant ECD. In the cellular context, different effects may modify this phenomenon. Two elements could, for instance, introduce some additional stability and a delay in the pH dissociation of the neck: the transmembrane domain at the N terminus might participate to tetramer formation through self-association within the membrane, and the interaction of CRDs from the same tetramer with a common target may stabilize interactions between the ECD units. Similarly, ligand interaction could potentially stabilize and modulate the structural disorganization of the CRD upon acidification. Although many questions remain and have to be investigated in detail, this study offers a first insight into the molecular mechanism of ligand release at low pH as well as a picture of DC-SIGN ECD on the outside of the cell (Fig. 10B), pointing far from the membrane surface in search for surrounding pathogens.
Supplementary Material
Acknowledgments
We thank M. Roessle and D. Svergun for help with data collection on X33 (EMBL, Hamburg) and J. érez for access to the Swing instrument at the Soleil synchrotron. The sedimentation velocity measurements were performed using the analytical ultracentrifugation platform of the Institut de Biologie Structurale.
This work was supported by a post-doctoral grant from the B. & M. Gates Foundation (to M. T.) and the “Sidaction-Ensemble contre le Sida” (to F. F. and M. T.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- DC
- dendritic cells
- CD
- circular dichroïsm
- CRD
- carbohydrate-recognition domain
- DC-SIGN
- dendritic cell-specific ICAM3 grabbing non-integrin
- ECD
- extracellular domain
- Rg
- radius of gyration
- SAXS
- small angle x-ray scattering
- SEC
- size exclusion chromatography
- HIV-1
- human immunodeficiency virus, type 1
- PDB
- Protein Data Bank
- ICAM
- intercellular adhesion molecule.
REFERENCES
- 1.Banchereau J., Steinman R. M. (1998) Nature 392, 245–252 [DOI] [PubMed] [Google Scholar]
- 2.van Kooyk Y., Geijtenbeek T. B. (2003) Nat. Rev. Immunol. 3, 697–709 [DOI] [PubMed] [Google Scholar]
- 3.Cambi A., de Lange F., van Maarseveen N. M., Nijhuis M., Joosten B., van Dijk E. M., de Bakker B. I., Fransen J. A., Bovee-Geurts P. H., van Leeuwen F. N., Van Hulst N. F., Figdor C. G. (2004) J. Cell Biol. 164, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Bakker B. I., de Lange F., Cambi A., Korterik J. P., van Dijk E. M., van Hulst N. F., Figdor C. G., Garcia-Parajo M. F. (2007) Chemphyschem. 8, 1473–1480 [DOI] [PubMed] [Google Scholar]
- 5.Geijtenbeek T. B., Kwon D. S., Torensma R., van Vliet S. J., van Duijnhoven G. C., Middel J., Cornelissen I. L., Nottet H. S., KewalRamani V. N., Littman D. R., Figdor C. G., van Kooyk Y. (2000) Cell 100, 587–597 [DOI] [PubMed] [Google Scholar]
- 6.Alvarez C. P., Lasala F., Carrillo J., Muñiz O., Corbí A. L., Delgado R. (2002) J. Virol. 76, 6841–6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lozach P. Y., Lortat-Jacob H., de Lacroix de Lavalette A., Staropoli I., Foung S., Amara A., Houles C., Fieschi F., Schwartz O., Virelizier J. L., Arenzana-Seisdedos F., Altmeyer R. (2003) J. Biol. Chem. 278, 20358–20366 [DOI] [PubMed] [Google Scholar]
- 8.Pöhlmann S., Zhang J., Baribaud F., Chen Z., Leslie G. J., Lin G., Granelli-Piperno A., Doms R. W., Rice C. M., McKeating J. A. (2003) J. Virol. 77, 4070–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navarro-Sanchez E., Altmeyer R., Amara A., Schwartz O., Fieschi F., Virelizier J. L., Arenzana-Seisdedos F., Desprès P. (2003) EMBO Rep. 4, 723–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serrano-Gómez D., Leal J. A., Corbí A. L. (2005) Immunobiology 210, 175–183 [DOI] [PubMed] [Google Scholar]
- 11.Cambi A., Gijzen K., de Vries J. M., Torensma R., Joosten B., Adema G. J., Netea M. G., Kullberg B. J., Romani L., Figdor C. G. (2003) Eur. J. Immunol. 33, 532–538 [DOI] [PubMed] [Google Scholar]
- 12.Steeghs L., van Vliet S. J., Uronen-Hansson H., van Mourik A., Engering A., Sanchez-Hernandez M., Klein N., Callard R., van Putten J. P., van der Ley P., van Kooyk Y., van de Winkel J. G. (2006) Cell Microbiol. 8, 316–325 [DOI] [PubMed] [Google Scholar]
- 13.Colmenares M., Puig-Kröger A., Pello O. M., Corbí A. L., Rivas L. (2002) J. Biol. Chem. 277, 36766–36769 [DOI] [PubMed] [Google Scholar]
- 14.van Die I., van Vliet S. J., Nyame A. K., Cummings R. D., Bank C. M., Appelmelk B., Geijtenbeek T. B., van Kooyk Y. (2003) Glycobiology 13, 471–478 [DOI] [PubMed] [Google Scholar]
- 15.Geijtenbeek T. B., Krooshoop D. J., Bleijs D. A., van Vliet S. J., van Duijnhoven G. C., Grabovsky V., Alon R., Figdor C. G., van Kooyk Y. (2000) Nat. Immunol. 1, 353–357 [DOI] [PubMed] [Google Scholar]
- 16.Geijtenbeek T. B., Torensma R., van Vliet S. J., van Duijnhoven G. C., Adema G. J., van Kooyk Y., Figdor C. G. (2000) Cell 100, 575–585 [DOI] [PubMed] [Google Scholar]
- 17.van Gisbergen K. P., Sanchez-Hernandez M., Geijtenbeek T. B., van Kooyk Y. (2005) J. Exp. Med. 201, 1281–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahnke K., Guo M., Lee S., Sepulveda H., Swain S. L., Nussenzweig M., Steinman R. M. (2000) J. Cell Biol. 151, 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon D. S., Gregorio G., Bitton N., Hendrickson W. A., Littman D. R. (2002) Immunity 16, 135–144 [DOI] [PubMed] [Google Scholar]
- 20.Turville S. G., Santos J. J., Frank I., Cameron P. U., Wilkinson J., Miranda-Saksena M., Dable J., Stössel H., Romani N., Piatak M., Jr., Lifson J. D., Pope M., Cunningham A. L. (2004) Blood 103, 2170–2179 [DOI] [PubMed] [Google Scholar]
- 21.Burleigh L., Lozach P. Y., Schiffer C., Staropoli I., Pezo V., Porrot F., Canque B., Virelizier J. L., Arenzana-Seisdedos F., Amara A. (2006) J. Virol. 80, 2949–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snyder G. A., Ford J., Torabi-Parizi P., Arthos J. A., Schuck P., Colonna M., Sun P. D. (2005) J. Virol. 79, 4589–4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gramberg T., Soilleux E., Fisch T., Lalor P. F., Hofmann H., Wheeldon S., Cotterill A., Wegele A., Winkler T., Adams D. H., Pöhlmann S. (2008) Virology 373, 189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLachlan A. D., Stewart M. (1975) J. Mol. Biol. 98, 293–304 [DOI] [PubMed] [Google Scholar]
- 25.Stetefeld J., Jenny M., Schulthess T., Landwehr R., Engel J., Kammerer R. A. (2000) Nat. Struct. Biol. 7, 772–776 [DOI] [PubMed] [Google Scholar]
- 26.Mitchell D. A., Fadden A. J., Drickamer K. (2001) J. Biol. Chem. 276, 28939–28945 [DOI] [PubMed] [Google Scholar]
- 27.Serrano-Gómez D., Sierra-Filardi E., Martínez-Nuñez R. T., Caparrós E., Delgado R., Muñoz-Fernández M. A., Abad M. A., Jimenez-Barbero J., Leal M., Corbí A. L. (2008) J. Biol. Chem. 283, 3889–3903 [DOI] [PubMed] [Google Scholar]
- 28.Feinberg H., Castelli R., Drickamer K., Seeberger P. H., Weis W. I. (2007) J. Biol. Chem. 282, 4202–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feinberg H., Guo Y., Mitchell D. A., Drickamer K., Weis W. I. (2005) J. Biol. Chem. 280, 1327–1335 [DOI] [PubMed] [Google Scholar]
- 30.Feinberg H., Mitchell D. A., Drickamer K., Weis W. I. (2001) Science 294, 2163–2166 [DOI] [PubMed] [Google Scholar]
- 31.Guo Y., Feinberg H., Conroy E., Mitchell D. A., Alvarez R., Blixt O., Taylor M. E., Weis W. I., Drickamer K. (2004) Nat. Struct. Mol. Biol. 11, 591–598 [DOI] [PubMed] [Google Scholar]
- 32.Snyder G. A., Colonna M., Sun P. D. (2005) J. Mol. Biol. 347, 979–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tabarani G., Reina J. J., Ebel C., Vivès C., Lortat-Jacob H., Rojo J., Fieschi F. (2006) FEBS Lett. 580, 2402–2408 [DOI] [PubMed] [Google Scholar]
- 34.Wang S. K., Liang P. H., Astronomo R. D., Hsu T. L., Hsieh S. L., Burton D. R., Wong C. H. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 3690–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reina J. J., Sattin S., Invernizzi D., Mari S., Martínez-Prats L., Tabarani G., Fieschi F., Delgado R., Nieto P. M., Rojo J., Bernardi A. (2007) Chem. Med. Chem. 2, 1030–1036 [DOI] [PubMed] [Google Scholar]
- 36.Timpano G., Tabarani G., Anderluh M., Invernizzi D., Vasile F., Potenza D., Nieto P. M., Rojo J., Fieschi F., Bernardi A. (2008) ChemBioChem 9, 1921–1930 [DOI] [PubMed] [Google Scholar]
- 37.Halary F., Amara A., Lortat-Jacob H., Messerle M., Delaunay T., Houlès C., Fieschi F., Arenzana-Seisdedos F., Moreau J. F., Déchanet-Merville J. (2002) Immunity 17, 653–664 [DOI] [PubMed] [Google Scholar]
- 38.Schuck P. (2000) Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.García De La Torre J., Huertas M. L., Carrasco B. (2000) Biophys. J. 78, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. (2007) J. Appl. Crystallogr. 40, s190–194 [Google Scholar]
- 41.Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 42.Guinier A., Fournet G. (1955) Small Angle Scattering of X-Rays, Wiley, New York [Google Scholar]
- 43.Svergun D. I. (1992) J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 44.Svergun D. I. (1999) Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Volkov V. V., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 860–864 [Google Scholar]
- 47.Kozin M. B., Svergun D. I. (2001) J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 48.Thépaut M., Vivès C., Pompidor G., Kahn R., Fieschi F. (2008) Acta Crystallogr. Sect. F Struct. Biol. Cryst. 64, 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thépaut M., Valladeau J., Nurisso A., Kahn R., Arnou B., Vivès C., Sealand S., Ebel C., Monnier C., Dezutter-Dambuyant C., Imberty A., Fieschi F. (2009) Biochemistry 48, 2684–2698 [DOI] [PubMed] [Google Scholar]
- 50.Brown P. H., Schuck P. (2006) Biophys. J. 90, 4651–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meier M., Lustig A., Aebi U., Burkhard P. (2002) J. Struct. Biol. 137, 65–72 [DOI] [PubMed] [Google Scholar]
- 52.Jelesarov I., Dürr E., Thomas R. M., Bosshard H. R. (1998) Biochemistry 37, 7539–7550 [DOI] [PubMed] [Google Scholar]
- 53.Kohn W. D., Kay C. M., Hodges R. S. (1997) J. Mol. Biol. 267, 1039–1052 [DOI] [PubMed] [Google Scholar]
- 54.Fiser A., Sali A. (2003) Bioinformatics 19, 2500–2501 [DOI] [PubMed] [Google Scholar]
- 55.Inoue M., Yamada H., Yasukochi T., Kuroki R., Miki T., Horiuchi T., Imoto T. (1992) Biochemistry 31, 5545–5553 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.