

Abstract
Nicotinic receptors (AChRs) play key roles in synaptic transmission. We explored activation of neuronal α7 and mammalian muscle AChRs by morantel and oxantel. Our results revealed a novel action of morantel as a high efficacy and more potent agonist than ACh of α7 receptors. The EC50 for activation by morantel of both α7 and α7-5HT3A receptors is 7-fold lower than that determined for ACh. The minimum morantel concentration required to activate α7-5HT3A channels is 6-fold lower than that of ACh, and activation episodes are more prolonged than in the presence of ACh. By contrast, oxantel is a weak agonist of α7 and α7-5HT3A, and both drugs are very low efficacy agonists of muscle AChRs. The replacement of Gln57 in α7 by glycine, which is found in the equivalent position of the muscle AChR, decreases the efficacy for activation and turns morantel into a partial agonist. The reverse mutation in the muscle AChR (ϵG57Q) increases 7-fold the efficacy of morantel. The mutations do not affect activation by ACh or oxantel, indicating that this position is selective for morantel. In silico studies show that the tetrahydropyrimidinyl group, common to both drugs, is close to Trp149 of the principal face of the binding site, whereas the other cyclic group is proximal to Gln57 of the complementary face in morantel but not in oxantel. Thus, position 57 at the complementary face is a key determinant of the high selectivity of morantel for α7. These results provide new information for further progress in drug design.
Nicotinic acetylcholine receptors (AChRs),3 members of the Cys-loop receptor superfamily, are of fundamental importance in synaptic transmission throughout the nervous system in both vertebrates and invertebrates. They are implicated in a wide range of important pathologies and are targets of clinically relevant drugs. AChRs are pentameric proteins composed of highly homologous subunits (1, 2). Subunits are classified as either α, which contain a disulfide bridge formed by two adjacent cysteine residues important for acetylcholine (ACh) binding, or non-α subunits, which lack this motif (3).
AChRs assemble from five identical α subunits, forming homomeric receptors, such as neuronal α7 receptors, or from different α and non-α subunits, forming heteromeric receptors, such as the muscle AChR. Human adult muscle AChRs are composed of two α1, one β, one ϵ, and one δ subunits. The five homologous subunits are arranged as barrel staves around a central ion-conducting pore (4). Approximately half of each subunit is extracellular with the remainder comprising transmembrane domains M1–M4 and a large cytoplasmic domain spanning M3 and M4 (4). The neurotransmitter binding sites are formed within the extracellular domain at interfaces between subunits (4, 5). One of the sides, called the principal face, is formed by three discontinuous loops of the α subunit, whereas the complementary face is formed by three discontinuous β-strands of the adjacent subunit. Key residues of the principal face are grouped in regions called loop A (Trp86 and Tyr93), loop B (Trp149 and Gly153), and loop C (Tyr190, Cys192, Cys193, and Tyr198). The complementary face is formed by residues from α7 or δ or ϵ subunits in the adult muscle AChR. At this face of the muscle AChR, residues are clustered in loop D (Trp55), E (Leu109, Tyr111, Tyr117, and Leu119), and F (Asp174 and Glu176) (2, 5, 6). Residues of the principal face are highly conserved between α7 and α1 subunits, whereas less conservation is found in residues located at the complementary face (5, 7).
The anthelmintic agents levamisole, pyrantel, oxantel, and morantel are full agonists of nematode muscle AChRs, and exert their therapeutic actions by producing muscle paralysis (8). By contrast, levamisole and pyrantel have been shown to be low efficacy agonists of mammalian muscle AChRs (9). A few lines of experimental evidence suggest that these compounds also interact with some types of neuronal AChRs, but instead of acting as agonists, they act as modulators. Morantel and levamisole have been shown to allosterically potentiate responses of α3β2 and α3β4 receptors (10, 11). Thus, the actions of anthelmintic agents seem to be strongly dependent on the AChR subtype. Therefore, these compounds are useful tools for the identification of determinants of drug selectivity, which, in turn, is required for rational design of novel and more specific drugs.
We have here determined that, similarly to pyrantel and levamisole (9), morantel and oxantel are low efficacy agonists of mammalian muscle AChRs. However, whereas oxantel is also a weak agonist of α7, morantel is more potent than ACh. By site-directed mutagenesis we determined that position 57, located at the complementary face of the binding site, is involved in the differential selectivity of morantel for α7 and mammalian muscle AChR.
Neuronal α7 receptors may be involved in a range of neurological and psychiatric disorders that lead to cognitive impairment, including Alzheimer disease, attention deficit hyperactivity disorder, and schizophrenia (12). Given that its deficit is associated with cognitive impairment in these diseases, enhancement of its activity has recently emerged as a physiological and effective therapeutic strategy. Therefore, the characterization of the novel action of morantel as a potent agonist of α7 together with the identification of the structural basis of this high selectivity become of importance as they provide new information for further progress in drug design.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis and Expression of α7, Muscle AChR, and α7-5HT3A Receptors
Mutant subunits were constructed using the QuikChange Site-directed Mutagenesis kit (Strategene, Inc.) and were confirmed by sequencing. The high conductance form of α7-5HT3A receptor (13) was constructed as described before (14). Briefly, three arginine residues responsible for the low conductance of the serotonin type 3A receptor (5-HT3A) were mutated to glutamine, aspartic acid, and alanine (15). BOSC cells were transfected with cDNAs of mouse α, β, δ, and ϵ subunits, the high-conductance form of α7-5HT3A, or human α7 and the chaperone-protein Ric-3 (16), using calcium phosphate precipitation (17–19). A plasmid encoding green fluorescent protein was included in all transfections to allow identification of transfected cells under fluorescence optics. Cells were used for single-channel and macroscopic current measurements 1 or 2 days after transfection.
Single-channel Recordings and Analysis
Single-channel recordings were obtained in the cell-attached configuration (20) at a membrane potential of −70 mV and at 20 °C (14, 21). The bath and pipette solutions contained 140 mm KCl, 5.4 mm NaCl, 0.2 mm CaCl2, and 10 mm HEPES (pH 7.4) for α7-5HT3A and 140 mm KCl, 5.4 mm NaCl, 1.8 mm CaCl2, 1.7 mm MgCl2, and 10 mm HEPES (pH 7.4) for muscle and α7 receptors. Solutions free of magnesium and with low calcium were used to record channels from α7-5HT3A receptors to minimize channel block by divalent cations (14). Patch pipettes were pulled from 7052 capillary tubes (Garner Glass, CA) and coated with Sylgard (Dow Corning, Midland, MI). ACh and anthelmintic drugs were added to the pipette solution.
Single-channel currents were recorded using an Axopatch 200B patch clamp amplifier (Axon Instruments, Inc., CA), digitized at 5-μs intervals with the PCI-6111E interface (National Instruments, Austin, TX), recorded to the computer hard disk using the program Acquire (Bruxton Corporation, Seattle, WA), and detected by the half-amplitude threshold criterion using the program TAC 4.0.10 (Bruxton Corp.) at a final bandwidth of 10 kHz (18). Open and closed duration histograms were plotted using a logarithmic abscissa and a square root ordinate (22) and fitted to the sum of exponential functions by maximum likelihood using the program TACFit (Bruxton Corp.). Bursts of α7-5HT3A (14) or clusters of muscle AChR (18) were identified as a series of closely spaced events preceded and followed by closed intervals longer than a critical duration (tcrit). For α7-5HT3A, tcrit for defining bursts was taken as the point of intersection between the second and third closed components (14). For the muscle AChR, tcrit for defining clusters was taken as the point of intersection between the predominant closed time component, which is sensitive to ACh concentration, and the succeeding one in closed time histograms (18, 19).
For describing channel block by morantel and oxantel, the simple open-channel block model was used (9, 23),
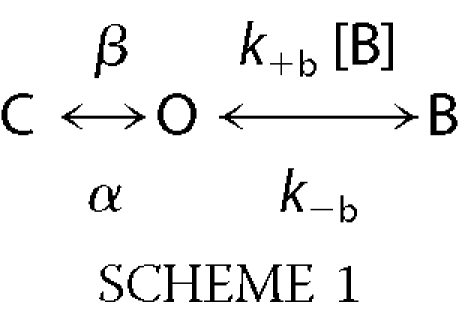 |
where C, O, and B are the closed, open, and blocked states, respectively. β and α are the apparent opening and closing rates, respectively. k+b is the blocking and k−b, the unblocking rate constants. k+b was determined from the slope of the relationship between the inverse of the mean open time and drug concentration (23). k−b was estimated by the inverse of the duration of the mean closed component corresponding to blocked periods (23). The dissociation constant for channel block (KB) was determined from the ratio k−b/k+b.
Macroscopic Current Recordings
Macroscopic current recordings of α7 and α7-5HT3A were obtained in the whole cell configuration. For the muscle AChR, currents were recorded in the outside-out patch configuration. A series of applications of extracellular solution containing ACh, morantel, or oxantel was applied to the cell as described before (24, 25). The pipette solution contained 140 mm KCl, 5 mm EGTA, 5 mm MgCl2, and 10 mm HEPES (pH 7.3). For recordings from α7 and muscle AChR, extracellular solution contained 150 mm NaCl, 1.8 mm CaCl2, 1 mm MgCl2, and 10 mm HEPES (pH 7.3). For recordings from α7-5HT3A receptors, extracellular solution contained 150 mm NaCl, 0.5 mm CaCl2, and 10 mm HEPES (pH 7.3). Macroscopic currents were filtered at 5 kHz, and digitized at 20 kHz. Data analysis was performed using the IgorPro software (WaveMetrics Inc., Lake Oswego, OR). The ensemble mean current was calculated for 5–10 individual current traces. Current records were aligned with each other at the point where the current had risen to 50% of its maximum level. Mean currents were fitted by a single exponential function: I(t) = I0 exp (−t/τd) + I∞, where I0 and I∞ are the peak and the steady state current values, respectively, and τd is the decay time constant. EC50 was calculated by non-linear regression analysis using the Hill equation: I/Imax = 1/[1 + (EC50/L)nH], where EC50 is the agonist concentration that elicits the half-maximal response, nH is the Hill coefficient, and L is the agonist concentration.
Docking of Morantel and Oxantel in α7 Homology Models
A homology model of the extracellular domain of the human α7 AChR was created based on the structure of the nicotine-bound Lymnaea stagnalis acetylcholine-binding protein (PDB code 1UW6). The amino acid sequences were first aligned using FUGUE (26) and then modeling was performed using the “model” routine of MODELLER 6, version 2 (27). Ten models were generated; of these, the one with the lowest energy and the smallest percentage of amino acids in the disallowed region of the Ramachandran plot (calculated using RAMPAGE; 28) was selected for docking studies. Protonated forms of morantel and oxantel were constructed in Chem3D Ultra 7.0 (CambridgeSoft, Cambridge, UK) and energy minimized using the MM2 and AM1 force fields. The ligands were docked into the ligand binding site of the α7 model, which was defined as being within 20 Å of Trp149. A hundred genetic algorithm runs were performed for each ligand.
RESULTS
Activation of α7 by Morantel and Oxantel
To evaluate activation of α7 by anthelmintic agents we first used the high conductance form of the α7-5HT3A receptor. This chimeric receptor has two advantages that make it an excellent model for pharmacological studies of α7: it shows, in contrast to wild-type α7, high surface expression in mammalian cells, and its single-channel kinetics have been described in detail (14, 29).
Macroscopic current recordings of α7-5HT3A receptors show that maximal responses elicited by oxantel are only 54.2 ± 2.5% of those elicited by ACh, indicating that this anthelmintic agent is a weak agonist of α7. By contrast, the maximal peak current elicited by morantel is similar to that elicited by ACh (Fig. 1A). From the relationship between the peak current and morantel concentration, an EC50 value of 29.0 ± 2.7 μm was obtained (Fig. 1B). This value is 7 times lower than that calculated for ACh activation (205 ± 10 μm; p < 0.05 n = 6; 14), indicating that morantel is a highly efficacious and potent agonist of α7 AChRs.
FIGURE 1.

Activation of α 7-5HT 3A (A and B) and human α 7 wild-type (C and D) receptors by ACh, morantel, and oxantel. A, macroscopic currents were recorded in the whole cell configuration from cells expressing α7-5HT3A chimeric receptors. Currents were elicited by 500 μm ACh, morantel, or oxantel. B, dose-response curves for ACh (●) and morantel (○) of α7-5HT3A chimeric receptors. C, macroscopic currents from human α7 in response to the application of 500 μm ACh, morantel, or oxantel in the whole cell configuration. Each trace represents the average of three to four applications of agonist. D, concentration-response curves for ACh (●) and morantel (○) of α7 AChRs. Each point is the average of three to five determinations, with the error bars representing the S.D. of the mean value. Curves are fits to the Hill equation. Membrane potential: −50 mV. The solid bar indicates the duration of the exposure to agonist (500 ms).
To confirm that morantel behaves as a potent agonist of wild-type α7 receptors we transfected cells with cDNAs encoding α7 and Ric-3, and recorded macroscopic currents in the whole cell configuration (Fig. 1C). As described for α7-5HT3A receptors, maximal peak currents are similar for α7 receptors activated by morantel and ACh. Moreover, the EC50 for activation is 6.5-fold lower for morantel (9.2 ± 0.4 μm) than for ACh (59.4 ± 3.0 μm) (Fig. 1D), as observed in α7-5HT3A receptors. Thus, our results confirm that morantel is a potent agonist of wild-type α7 receptors, and validate the use of the chimeric receptor as a model of α7.
To explore in more detail the activation of α7 by anthelmintic agents we performed single-channel recordings of α7-5HT3A. Single-channel currents of α7-5HT3A channels are observed at ACh concentrations higher than 30 μm (14). Channel currents appear either as isolated openings flanked by long closings or as bursts of several openings in quick succession (Fig. 2). Open time histograms at all ACh concentrations can be fitted by three components whose mean durations and relative areas are: τ1 = 6.30 ± 0.50 ms (0.44 ± 0.07), τ2 = 1.00 ± 0.40 ms (0.16 ± 0.10), and τ3 = 164 ± 10 μs (0.40 ± 0.10).
FIGURE 2.

Chimeric α 7–5HT 3A channels activated by ACh and anthelmintic agents. Channels activated by ACh, oxantel, or morantel at the indicated concentrations, were recorded from cells expressing the chimeric α7-5HT3A receptor. Left, traces of single-channel currents were displaced at a bandwidth of 10 kHz with channel openings as upward deflections. Right, open, closed, and burst duration histograms corresponding to each condition. Membrane potential, −70 mV.
In the presence of oxantel, single-channel currents are observed at concentrations higher than 50 μm, again indicating reduced potency with respect to ACh (Fig. 2). Open-time histograms are fitted by two components whose durations are briefer than those of ACh-activated channels (1.05 ± 0.15 ms (0.70 ± 0.05) and 145 ± 65 μs (0.30 ± 0.05)). Activation of α7-5HT3A by oxantel also appears in bursts but their durations are briefer than those of ACh-activated bursts (6.2 ± 0.8 ms; p < 0.05, n = 6).
Single-channel currents activated by morantel are detected at concentrations as low as 5 μm, thus confirming its high potency for α7 activation. Open time distributions of α7-5HT3A activated by 5 μm morantel show two main components whose durations and relative areas are 2.45 ± 0.15 ms (0.60 ± 0.05) and 80.0 ± 15.0 μs (0.40 ± 0.05) (Fig. 2). The mean burst duration at 5 μm morantel is more prolonged than that of ACh-activated channels (26.0 ± 3.0 ms).
Thus, macroscopic recordings reveal that morantel is a full agonist of α7, whereas oxantel is a partial one. The potency for activation, determined by both single-channel and macroscopic current recordings, is morantel > ACh > oxantel.
Block of α7-5HT3A by Morantel and Oxantel
Increasing morantel or oxantel concentrations produces a decrease in the duration of the mean open time of α7-5HT3A that can be explained by open-channel block. At concentrations higher than 5 μm morantel and 50 μm oxantel a flickering effect is observed (Fig. 2). The duration of the blocked periods can be estimated from the inverse of the duration of the briefest component of the closed duration histogram (79.7 ± 9.7 μs and 86.0 ± 6.8 μs for morantel and oxantel, respectively), whose area increases as a function of drug concentration (23). From the duration of the briefest closed component we estimated unblocking rates (k−b in Scheme 1) of 12,550 and 11,630 s−1 for morantel and oxantel, respectively. The forward blocking rate constant (k+b in Scheme 1), calculated by the slope of relationship between the inverse of the mean open time and the blocker concentration (23), is 36.5 × 106 and 4.3 × 106 m−1 s−1 for morantel and oxantel, respectively. The dissociation constants for channel block (KB) of the 5-HT3A pore at −70 mV membrane potential, calculated by k−b/k+b, are 340 μm and 2.8 mm for morantel and oxantel, respectively.
Oxantel and Morantel Are Low Efficacy Agonists of Mammalian Muscle AChRs
To determine the efficacy of activation of adult muscle AChRs by these anthelmintic drugs we first recorded macroscopic currents from outside-out patches. Fig. 3A shows ensemble currents obtained from a single outside-out patch exposed to applications of 100 μm ACh (control), 100 μm morantel, and 100 μm oxantel. The peak currents were 6.0 ± 1.2 and 15.5 ± 1.7% for morantel and oxantel, respectively, with respect to the peak current elicited by ACh. No significant changes in peak currents were observed when the anthelmintic concentration was increased to 300 μm, suggesting that these values correspond to the maximal responses. No clear currents were elicited by application of 60 μm morantel, thus impeding the determination of accurate EC50 values for morantel activation of muscle AChRs.
FIGURE 3.

Adult mammalian muscle AChRs activated by ACh, morantel, or oxantel. A, macroscopic currents were recorded from outside-out patches in response to rapid perfusion of 100 μm ACh, morantel, or oxantel. The solid bar indicates the duration of the exposure to the agonist. Membrane potential, −50 mV. B, single channels activated by 1 and 300 μm ACh, morantel, or oxantel were recorded from cells expressing adult muscle AChRs. Left, traces of currents are displaced at a bandwidth of 10 kHz with channel openings as upward deflections. Right, open and closed time histograms corresponding to each condition. Membrane potential, −70 mV.
We also recorded single-channel currents elicited by low and high concentrations of the drugs (Fig. 3B). AChR channels activated by morantel or oxantel are briefer than those activated by ACh. Open time distributions at 1 μm morantel or oxantel can be well fitted by a single component of ∼250 μs (Fig. 3B), whereas the duration of the main open component of ACh-activated muscle AChR channels is ∼1 ms (18).
At ACh concentrations higher than 10 μm, muscle AChRs open in clusters of well defined activation episodes (18; Fig. 3). Each activation episode begins with the transition of a single receptor from the desensitized to the activatable state and terminates by returning to the desensitized state. Increasing morantel concentration from 1 to 300 μm does not produce the typical clustering observed with ACh. Channel activity appears as isolated events at all morantel concentrations. For oxantel, clusters are observed but the closed times within clusters are more prolonged than for ACh (Fig. 3B). At 300 μm oxantel, the probability of channel opening within these clusters (Popen) is about 0.1, whereas it is 0.9 for ACh (18).
As described for α7-5HT3A receptors, morantel and oxantel produce flickering block of mammalian muscle AChRs. The dissociation constant (KB) for channel block is 8.2 μm and 59.0 μm for morantel and oxantel, respectively, revealing that these drugs are blockers more potent of the muscle AChR than of the 5-HT3A pore.
In conclusion, the brief duration openings, the lack of clustering of openings at high agonist concentrations, and the reduced peak currents reveal that morantel and oxantel are very low efficacy agonists of the mammalian muscle AChR. In addition, they act as potent open-channel blockers. Taken together, our results show that oxantel is a low efficacy agonist of both α7 and muscle AChRs, whereas morantel is a potent agonist of α7 but a low efficacy agonist of the muscle AChR.
Glutamine 57, Located at the Complementary Face of the Binding Site, Is a Key Determinant of the High Efficacy of Morantel for α7
To identify the structural basis of the contrasting actions of morantel at α7 and muscle AChRs we explored key residues located at the binding site but differentially conserved between both subtypes. Because residues at the principal face are highly conserved between α1 and α7, we studied the complementary face, which is formed by δ and ϵ subunits in the adult muscle AChR.
We first replaced Gln57 in α7-5HT3A by glycine, which is found in the ϵ subunit. Macroscopic current recordings of the chimera carrying the Q57G mutation reveal a selective and significant decrease in the efficacy of morantel with respect to the control chimera (Fig. 4A). The peak current at saturating concentrations of morantel (500 μm) is 48.0 ± 8.2% of that elicited by 500 μm ACh, revealing that the maximal response to morantel is reduced in the mutant receptor. The EC50 for morantel increases 2-fold due to the Q57G mutation (54 ± 4.8 μm instead of 29 ± 2.7 μm in the control chimera) (Fig. 4B). Interestingly, the EC50 value does not change for ACh (220 ± 20 μm).
FIGURE 4.

Activation by morantel of α 7–5HT 3A receptors carrying the mutation Q57G. A, whole cell currents recorded from BOSC cells expressing Q57G mutants in response to 500 μm ACh or morantel. Membrane potential, −50 mV. B, dose-response curves for ACh (●) and morantel (○). Each point is an average of three to five determinations with the error bars representing the S.D. Curves are fits to the Hill equation. C, left, channels were recorded in the presence of 20 μm morantel. Right, open, closed, and burst duration histograms corresponding to wild-type and mutant α7-5HT3A. Membrane potential, −70 mV.
The reduced efficacy of morantel to activate the Q57G chimera is also revealed from single-channel current recordings (Fig. 4C). Single-channel openings are detected at concentrations higher than 20 μm, instead of 5 μm for the control chimera (Fig. 4C). The duration (0.85 ± 0.15 ms) and relative area of the slowest open component (0.70 ± 0.05) are similar to those of the control. However, the duration of bursts of openings decreases 4-fold in the mutant receptor (6.0 ± 0.1 and 22.3 ± 2.7 ms for mutant and control chimera, respectively, at 20 μm morantel). Such a reduction is accompanied by the reduction in the number of openings per burst (7.5 ± 1.0 and 22.0 ± 5.5 events per burst for Q57G and control chimera, respectively).
In contrast to the changes in the activation by morantel, no changes were observed in the activation of Q57G α7-5HT3A by oxantel. As in the control chimera, channels were detected at a minimum concentration of 50 μm oxantel. Neither the open time distributions (τ1, 1.00 ± 0.15 ms; τ2, 126 ± 70 μs) nor the mean burst duration (5.7 ± 0.6 ms) were affected by the Q57G mutation. No differences in the minimum concentration required to detect single-channel currents as well as in the mean open time, mean closed time, and mean burst duration are observed when α7-5HT3A carrying the Q57G mutation is activated by ACh with respect to those of the control chimera (7). Thus, the mutation Q57G selectively alters the activation by morantel.
Position 57 Is a Determinant of the Low Efficacy of Morantel at Muscle AChRs
Given that the replacement of glutamine 57 by glycine, which is found in the ϵ subunit, reduces the efficacy for activation of α7 receptors by morantel, we hypothesized that the reverse mutation in the muscle AChR should increase its efficacy. To test this hypothesis, we replaced in ϵ and δ subunits the residues at position 57 by glutamine, which is found in α7. We generated muscle AChRs containing mutant δD57Q and ϵG57Q subunits and evaluated channel activity elicited by morantel (Fig. 5).
FIGURE 5.

Morantel-activated channels from cells expressing wild-type and mutant muscle AChRs. A, left, single-channel currents recorded in response to 1 μm morantel. Traces of currents are displayed at a bandwidth of 10 kHz with channel openings as upward deflections. Membrane potential, −70 mV. Right, open and closed duration histograms corresponding to channels activated by 1 μm morantel. B, macroscopic currents activated by 100 μm ACh or 100 μm morantel of muscle AChRs carrying the ϵQ57G mutation. Membrane potential, −50 mV.
In δD57Q AChRs, neither the minimal concentration of morantel required to detect channel currents (1 μm) nor the single-channel properties are affected with respect to wild-type AChRs (Fig. 5A). Also, no changes in channel properties of this mutant AChR were detected in the presence of ACh or oxantel (data not shown).
By contrast, when Gly57 in ϵ is replaced by a glutamine residue as found in α7 (ϵG57Q) the efficacy of morantel increases significantly with respect to wild-type AChRs. Single-channel currents are detected at morantel concentrations as low as 0.25 μm, instead of 1.0 μm in the wild-type AChR. At low morantel concentrations (0.25–1 μm), open time distributions are described by two exponential components similar to those corresponding to wild-type receptors activated by ACh (1.00 ± 0.30 ms (relative area 0.35 ± 0.10) and 155 ± 50 (relative area 0.65 ± 0.15); Fig. 5A). In contrast to activation of wild-type receptors by morantel, clusters of opening events are clearly distinguished in the mutant receptors activated by 300 μm morantel, although the probability of channel opening within clusters is still low compared with that of ACh-elicited clusters (Popen < 0.1 and 0.9 for 300 μm morantel and ACh, respectively).
No differences in the channel properties of mutant ϵG57Q AChRs activated by ACh were observed with respect to wild-type receptors. At 1 μm ACh, the mean durations and relative areas of the open components were 0.94 ± 0.05 ms (0.50 ± 0.05) and 260 ± 0.02 μs (0.48 ± 0.02). Also, no changes were observed in the activation by oxantel of the ϵG57Q mutant. As in wild-type AChRs, open time histograms of mutant AChRs activated by 1 μm oxantel can be fitted by a single component of 230 ± 70 μs. These results reveal that the mutation ϵG57Q selectively enhances the activation of muscle AChR by morantel.
The minimum concentration of morantel required to detect single channels as well as the channel properties of the double δD57Q/ϵG57Q mutant receptors are the same as those of the single ϵG57Q mutant AChR (τ1, 1.0 ± 0.12 ms (relative area 0.26 ± 0.10); and τ2, 150 ± 20 μs (relative area 0.74 ± 0.10) at 1 μm morantel) (Fig. 5). No changes with respect to wild-type receptors were observed when the double mutant was activated by ACh or oxantel (data not shown).
We measured macroscopic currents from muscle AChRs carrying the ϵG57Q subunit (Fig. 5B). In the mutant receptor, the amplitude of the currents elicited by 100 μm morantel was 42.0 ± 5.0% of that of 100 μm ACh-evoked responses, thus revealing an 8-fold increase in the response with respect that of the wild-type receptor.
The overall results indicate that the amino acid at position 57, located at the complementary face of the binding site, is a main determinant of the differential activation of mammalian muscle and neuronal α7 AChRs by morantel, although other not yet identified residues may be involved in the selectivity.
Docking of Morantel and Oxantel in α7
To gain insights into how position 57 is involved in the efficacy of activation by anthelmintic drugs we performed docking of oxantel and morantel into homology models of the α7 extracellular domain. Docking of oxantel and morantel revealed 3 different orientations for each molecule. In orientation 1, the methyl-tetrahydropyrimidinyl group, common to both anthelmintic drugs (Fig. 6A), is located within 5 Å of Trp149 in binding loop B of the principal face. The other cyclic group of the molecules are a methyl-thienyl group in morantel and a hydroxy-phenyl group in oxantel (Fig. 6). In orientation 1, both groups are located near loop D and are <3 Å from Gln57 (Fig. 6, A and D). In orientation 2, these two latter groups are near loop E, although the methyl-tetrahydropyrimidinyl ring again is located near Trp149 (Fig. 6, B and E). In orientation 3, the anthelmintic molecules have significantly re-orientated such that the methyl-tetrahydropyrimidinyl ring of both is near loop E, whereas the methyl-thienyl and hydroxy-phenyl groups are near Trp149 (Fig. 6, C and F). In orientations 2 and 3 the anthelmintic molecules were >6 Å from Gln57.
FIGURE 6.

Docking of morantel and oxantel in α 7. Oxantel (left-hand panels: A, orientation 1; B, orientation 2; C, orientation 3) and morantel (right-hand panels: D, orientation 1; E, orientation 2; F, orientation 3) docked into a homology model of the α7 extracellular domain. The molecular structures of morantel and oxantel are shown, with colors indicating C (gray), H (white), N (blue), S (yellow), and O (red). Binding loops are colored orange (loop A), red (loop B), yellow (loop C), blue (loop D), and green (loop E). Residues Trp149 and Gln57 are depicted in red and blue, respectively.
The tetrahydropyrimidinyl ring carries a positively charged quaternary ammonium group (Fig. 6), and thus has the potential to form a cation-π interaction with Trp149, as has been shown for some other nicotinic ACh agonists (30). Given the relatively high efficacy of the anthelmintics for α7, it is likely that this interaction does occur; where it does not, e.g. with nicotine at the muscle AChR, there is no significant agonist activity (31). Thus the data best support docking orientations 1 and 2. The electrophysiological measurements show that morantel is more efficacious than oxantel for α7, and that the mutation at Gln57 affects morantel efficacy but not oxantel efficacy. These findings may be explained by different locations of the opposite cyclic group at the binding site. We propose that the configuration of morantel at the binding site is the one modeled as “orientation 1” (Fig. 6D) and that of oxantel is “orientation 2” (Fig. 6B). A strong interaction of the methyl-thienyl group of morantel with Gln57, as suggested by the model, can explain the high efficacy, and its sensitivity to the side chain of this residue.
DISCUSSION
Enhancement of activation of brain α7 AChRs has a broad therapeutic potential in central nervous system diseases related to cognitive dysfunction. Continuous efforts are being performed to identify novel drugs that can increase α7 activity. We here described a novel action of the anthelmintic drug morantel as a potent agonist of human α7 AChRs. We also show that morantel action is highly sensitive to the receptor subtype. It is a potent agonist of α7, a very weak agonist of mammalian muscle AChR, and it has been shown to act as an allosteric modulator of α3β2 AChRs (10). The broad spectrum of actions among different types of AChRs makes this molecule a potential tool for elucidating determinants of drug selectivity in which drug discovery relies.
The high efficacy and potency of morantel to activate α7 are revealed by dose-response curves showing a maximum value similar to that elicited by ACh but a 7-fold smaller EC50 in both wild-type α7 and α7-5HT3A receptors. At the single-channel level, the increased potency is revealed by a 6-fold decrease in the concentration required to detect single α7-5HT3A channels when compared with ACh. By contrast, oxantel is a weak agonist of α7 as revealed by: (i) briefer open durations than those of ACh-activated channels; (ii) reduced burst durations and reduced potency compared with ACh; and (iii) reduced maximal peak currents. Thus, although oxantel and morantel are both tetrahydropyrimidines (Fig. 6), we here determined that their actions at α7 receptors are very different.
The molecular structures of morantel and oxantel have in common the 1-methyl-1,4,5,6-tetrahydropyrimidine group and differ in the other ring, which is a 3-methyl-2-thienyl group in morantel and 3-hydroxy-phenyl in oxantel (Fig. 6). We have previously shown that pyrantel, an analog of morantel that lacks the methyl group on the thiophene ring, is also a full agonist of α7-5HT3A receptors (7). Interestingly, the EC50 of pyrantel at the chimeric receptor is about 2-fold higher than that of morantel. The comparison of the structure-function relationships suggests that the additional methyl group in the thienyl ring in morantel increases the potency of the compound as an agonist.
In contrast to their actions at α7 receptors, morantel and oxantel act as low efficacy agonists of mammalian muscle AChRs. We have previously shown that pyrantel (32) and levamisole (9) are also very weak agonists of this AChR type. Thus, all anthelmintic compounds characterized so far are weak agonists of mammalian muscle AChRs (7, 9, 32). On the other hand, most of these drugs act as full agonists of nematode muscle levamisole-sensitive AChRs, which is the basis for their therapeutic anthelmintic actions (33). Thus, understanding the determinants of selectivity of these drugs for AChR subtypes will also help to develop novel anthelmintic drugs.
Our study reveals that the residue at position 57 located in the complementary side of the binding pocket is a key site that determines the action of morantel at α7 AChRs. This position is differentially conserved among subunits. It is glycine in all mammalian ϵ subunits, glutamic or aspartic acid in δ subunits, and glutamine in all α7 subunits. If the glutamine residue in α7 is replaced by glycine the efficacy for activation by morantel decreases, and if the reverse mutation is performed in the ϵ subunit (ϵG57Q), the efficacy of morantel to activate muscle AChRs increases significantly. Although the results show that this position is an important determinant of selectivity, additional residues may be involved because the effects are not fully reversed by the mutations. We have previously shown that mutations at two residues of loop E (Asn111 and Gln117) affect similarly activation by pyrantel and ACh of α7-5HT3A receptors (7). Thus, these residues seem not to be determinants of selectivity for morantel, which agrees with orientation 1 for morantel. It is possible that the effects of mutations at loop E on activation may not be due to a direct interaction between the agonists and the receptor. The docking models will provide further information to identify additional determinants of morantel selectivity.
No changes were observed when the equivalent residue was mutated in the δ subunit (δD57Q). This result reveals that the drug binds differentially to both interfaces and that the strong activation is mediated mainly by the α-ϵ interface. It is also possible that morantel does not bind to the α-ϵ interface except in the presence of the ϵG57Q mutation. In this scenario, the brief openings would correspond to openings of single-liganded receptors, and by allowing binding of morantel to the second interface, the mutation would lead to more prolonged openings arising from biliganded receptors.
We also determined that position 57 is not involved in ACh or oxantel activation. However, it is involved in pyrantel activation (7), and in the sensitivity of α7 to neonicotinoid insecticides (34). Therefore, this residue appears to function as a determinant of selectivity for a broader spectrum of drugs. Because morantel enhances α3β2 activity (10), it would be interesting to test if position 57 is involved in this effect.
Cation-π interactions have been shown to be important in ACh binding and activation (30, 35). Aromatic amino acids at the agonist binding site provide a region of negative electrostatic potential for agonists with positive charge. By docking morantel and oxantel into the extracellular domain of α7 we determined that the methyl-tetrahydropyrimidinyl ring (present in both anthelmintic drugs) is located near Trp149 of loop B in the principal face, with which it could form a cation-π interaction. Previous studies revealed that αTrp149 forms such an interaction with the quaternary ammonium group of ACh in the bound state of the receptor (36), as does nicotine with Trp149 of loop B in brain receptors and AChBP (30, 31, 36). Our functional data support an orientation of morantel in the “horizontal” position (orientation 1 in Fig. 6), where the methyl-thienyl group has the potential to interact with Gln57 (<3 Å), and oxantel in the “vertical” position (orientation 2 in Fig. 6), where the hydroxy-phenyl group is located at a considerable distance from Gln57 (>6 Å). Further studies, ideally high resolution structures, are needed to confirm these molecular locations, but the considerable differences in orientations of these two similar molecules in the binding site can explain the significant differences in efficacy.
In conclusion, we have determined that morantel is a potent agonist of α7 and have identified a Gln residue at position 57 as a determinant for morantel selectivity among AChR subtypes. This information provides new insights for molecular modeling and drug screening, which, in turn, will contribute to the development of novel and more selective drugs.
Acknowledgment
We are grateful to Martin Lochner (University of Warwick, UK) for providing the structures of morantel and oxantel for docking.
This work was supported in part by grants from the Universidad Nacional del Sur, Agencia Nacional de Promocion Cientifica y Tecnologíca, Consejo Nacional de Investigaciones Científicas y Técnicas, Loreal-United Nations Educational, Scientific, and Cultural Organization, Fundación F. Fiorini (to C. B.), and the Wellcome Trust (to K. P. and S. C. R. L.).
- AChR
- acetylcholine receptor.
REFERENCES
- 1.Lester H. A., Dibas M. I., Dahan D. S., Leite J. F., Dougherty D. A. (2004) Trends Neurosci. 27, 329–336 [DOI] [PubMed] [Google Scholar]
- 2.Sine S. M., Engel A. G. (2006) Nature 440, 448–455 [DOI] [PubMed] [Google Scholar]
- 3.Sine S. M. (2002) J. Neurobiol. 53, 431–446 [DOI] [PubMed] [Google Scholar]
- 4.Unwin N. (2005) J. Mol. Biol. 346, 967–989 [DOI] [PubMed] [Google Scholar]
- 5.Brejc K., van Dijk W. J., Klaassen R. V., Schuurmans M., van Der Oost J., Smit A. B., Sixma T. K. (2001) Nature 411, 269–276 [DOI] [PubMed] [Google Scholar]
- 6.Changeux J. P., Taly A. (2008) Trends Mol. Med. 14, 93–102 [DOI] [PubMed] [Google Scholar]
- 7.Bartos M., Rayes D., Bouzat C. (2006) Mol. Pharmacol. 70, 1307–1318 [DOI] [PubMed] [Google Scholar]
- 8.Martin R. J. (1997) Vet. J. 154, 11–34 [DOI] [PubMed] [Google Scholar]
- 9.Rayes D., De Rosa M. J., Bartos M., Bouzat C. (2004) J. Biol. Chem. 279, 36372–36381 [DOI] [PubMed] [Google Scholar]
- 10.Wu T. Y., Smith C. M., Sine S. M., Levandoski M. M. (2008) Mol. Pharmacol. 74, 466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levandoski M. M., Piket B., Chang J. (2003) Eur. J. Pharmacol. 471, 9–20 [DOI] [PubMed] [Google Scholar]
- 12.Kalamida D., Poulas K., Avramopoulou V., Fostieri E., Lagoumintzis G., Lazaridis K., Sideri A., Zouridakis M., Tzartos S. J. (2007) FEBS J. 274, 3799–3845 [DOI] [PubMed] [Google Scholar]
- 13.Eiselé J. L., Bertrand S., Galzi J. L., Devillers-Thiéry A., Changeux J. P., Bertrand D. (1993) Nature 366, 479–483 [DOI] [PubMed] [Google Scholar]
- 14.Rayes D., Spitzmaul G., Sine S. M., Bouzat C. (2005) Mol. Pharmacol. 68, 1475–1483 [DOI] [PubMed] [Google Scholar]
- 15.Kelley S. P., Dunlop J. I., Kirkness E. F., Lambert J. J., Peters J. A. (2003) Nature 424, 321–324 [DOI] [PubMed] [Google Scholar]
- 16.Williams M. E., Burton B., Urrutia A., Shcherbatko A., Chavez-Noriega L. E., Cohen C. J., Aiyar J. (2005) J. Biol. Chem. 280, 1257–1263 [DOI] [PubMed] [Google Scholar]
- 17.Bouzat C., Bren N., Sine S. M. (1994) Neuron 13, 1395–1402 [DOI] [PubMed] [Google Scholar]
- 18.Bouzat C., Barrantes F., Sine S. (2000) J. Gen. Physiol. 115, 663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouzat C., Gumilar F., del Carmen Esandi M., Sine S. M. (2002) Biophys. J. 82, 1920–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981) Pflugers Arch. 391, 85–100 [DOI] [PubMed] [Google Scholar]
- 21.Bouzat C., Gumilar F., Spitzmaul G., Wang H. L., Rayes D., Hansen S. B., Taylor P., Sine S. M. (2004) Nature 430, 896–900 [DOI] [PubMed] [Google Scholar]
- 22.Sigworth F. J., Sine S. M. (1987) Biophys. J. 52, 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neher E., Steinbach J. H. (1978) J. Physiol. 277, 153–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y., Dilger J. P. (1991) Biophys. J. 60, 424–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spitzmaul G., Dilger J. P., Bouzat C. (2001) Mol. Pharmacol. 60, 235–243 [DOI] [PubMed] [Google Scholar]
- 26.Shi J., Blundell T. L., Mizuguchi K. (2001) J. Mol. Biol. 310, 243–257 [DOI] [PubMed] [Google Scholar]
- 27.Sali A., Blundell T. L. (1993) J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 28.Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 29.Bouzat C., Bartos M., Corradi J., Sine S. M. (2008) J. Neurosci. 28, 7808–7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dougherty D. A. (2008) Chem. Rev. 108, 1642–1653 [DOI] [PubMed] [Google Scholar]
- 31.Xiu X., Puskar N. L., Shanata J. A., Lester H. A., Dougherty D. A. (2009) Nature 458, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rayes D., De Rosa M. J., Spitzmaul G., Bouzat C. (2001) Neuropharmacology 41, 238–245 [DOI] [PubMed] [Google Scholar]
- 33.Rayes D., Flamini M., Hernando G., Bouzat C. (2007) Mol. Pharmacol. 71, 1407–1415 [DOI] [PubMed] [Google Scholar]
- 34.Shimomura M., Okuda H., Matsuda K., Komai K., Akamatsu M., Sattelle D. B. (2002) Br. J. Pharmacol. 137, 162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong W., Gallivan J. P., Zhang Y., Li L., Lester H. A., Dougherty D. A. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 12088–12093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Celie P. H., van Rossum-Fikkert S. E., van Dijk W. J., Brejc K., Smit A. B., Sixma T. K. (2004) Neuron 41, 907–914 [DOI] [PubMed] [Google Scholar]